
Abstract
Aims/hypothesis
The stress-activated nuclear protein transcription regulator 1 (NUPR1) is induced in response to glucose and TNF-α, both of which are elevated in type 2 diabetes, and Nupr1 has been implicated in cell proliferation and apoptosis cascades. We used Nupr1 −/− mice to study the role of Nupr1 in glucose homeostasis under normal conditions and following maintenance on a high-fat diet (HFD).
Methods
Glucose homeostasis in vivo was determined by measuring glucose tolerance, insulin sensitivity and insulin secretion. Islet number, morphology and beta cell area were assessed by immunofluorescence and morphometric analysis, and islet cell proliferation was quantified by analysis of BrdU incorporation. Islet gene expression was measured by gene arrays and quantitative RT-PCR, and gene promoter activities were monitored by measuring luciferase activity.
Results
Nupr1 −/− mice had increased beta cell mass as a consequence of enhanced islet cell proliferation. Nupr1-dependent suppression of beta cell Ccna2 and Tcf19 promoter activities was identified as a mechanism through which Nupr1 may regulate beta cell cycle progression. Nupr1 −/− mice maintained on a normal diet were mildly insulin resistant, but were normoglycaemic with normal glucose tolerance because of compensatory increases in basal and glucose-induced insulin secretion. Nupr1 deletion was protective against HFD-induced obesity, insulin resistance and glucose intolerance.
Conclusions/interpretation
Inhibition of NUPR1 expression or activity has the potential to protect against the metabolic defects associated with obesity and type 2 diabetes.
Similar content being viewed by others
Introduction
Nuclear protein transcription regulator 1 (NUPR1)/P8 is a stress-activated protein that was first identified in the pancreas after the onset of experimentally induced pancreatitis [1]. Upregulation of NUPR1 expression was associated with improved exocrine pancreas function [2]. The Nupr1 gene was subsequently shown to be induced in response to a variety of stressful stimuli [3], suggesting that it is part of a general defence mechanism against cellular injury. Consistent with this defensive role, targeted disruption of the murine Nupr1 gene, though not lethal, resulted in higher mortality in the lipopolysaccharide-injection model of sepsis [4]. NUPR1 shares structural characteristics with high-mobility group (HMG) proteins: it is a nuclear helix–loop–helix protein that possesses DNA-binding properties [5] and it is involved in the molecular mechanisms regulating DNA repair, apoptosis, proliferation and development [1, 3, 6–9].
There is some evidence that Nupr1 may be involved in beta cell responses to the development of type 2 diabetes. Thus, its expression is upregulated by glucose and TNF-α [2], both of which are elevated in type 2 diabetes [10], and INS-1 insulinoma cells showed glucose-dependent elevations in Nupr1 gene expression [9]. The forced overexpression of Nupr1 in human islets enhanced cumulative insulin secretion in vitro and increased the insulin content of islets transplanted below the renal capsule of diabetic mice in vivo [11], consistent with beneficial effects of Nupr1 overexpression on glycaemic control. We therefore hypothesised that Nupr1 deficiency would result in reduced beta cell mass and the progressive development of type 2 diabetes, and tested this using wild-type and Nupr1 −/− C57BL/6 mice [8]. In contrast, the Nupr1 −/− phenotype was associated with mild insulin resistance but with maintained normoglycaemia because of hyperinsulinaemia driven by increases in islet number and beta cell mass as a consequence of the loss of NUPR1-mediated repression of Ccna2 and Tcf19, genes that regulate the later stages of the cell cycle.
Methods
Materials
Cell culture reagents and anti-cyclin antibody were obtained from Sigma Aldrich (Poole, UK). Molecular biology reagents were obtained from Invitrogen (Paisley, UK) and Promega (Southampton, UK). SYBR Green and BrdU proliferation kits were from Roche (Burgess Hill, UK). Antibodies against insulin, glucagon and somatostatin were from Dako UK (Ely, UK). FITC- and tetramethylrhodamine isothiocyanate (TRITC)-labelled secondary antibodies were from Jackson Immunoresearch (Newmarket, UK). Anti-transcription factor 19 (TCF19) antibody was from Santa Cruz Biotechnology (Wembley, UK). Metafectene Pro was from Biontex Laboratories (Martinsried/Planegg, Germany). Non-interfering RNAs and small interfering (si)RNAs were from Thermo Scientific (Surrey, UK). Ultrasensitive mouse insulin ELISA kits were obtained from Mercodia (Uppsala, Sweden).
Generation of Nupr1-knockout mice
The Nupr1 −/− mouse model was created as described previously [8], and a colony was established at King’s College London in accordance with UK Home Office standards. Mice were fed ad libitum with a standard normal-fat diet (NFD; 4% fat); for some experiments they were fed a high-fat diet (HFD; 55% fat), starting at 5 weeks of age, for 16 weeks.
Metabolic studies
After i.p. administration of 2 g/kg glucose, glucose tolerance tests were performed by quantification of blood glucose concentrations with a glucose meter, and serum insulin concentrations were determined using an ultrasensitive ELISA. Insulin tolerance tests were performed following i.p. administration of insulin (0.75 U/kg body weight). Weight gain was recorded weekly until the day the mice were killed. Pancreases, perigonadal fat pads and gastrocnemius muscles were retrieved on the day the mice were killed, and were weighed.
Mouse islet isolation and maintenance
Islets were isolated from 3-month-old Nupr1 −/− mice and their wild-type littermates by collagenase digestion as previously described [12] and maintained in culture for at least 24 h before use. Islet insulin and double-stranded (ds)DNA contents were quantified by RIA [13] and PicoGreen fluorescence, respectively, and the insulin per dsDNA ratios were calculated.
Immunohistochemistry and morphometric analysis
Fixed pancreases were cut into 5 μm sections and morphometric analyses were performed on every tenth section throughout each pancreas. For quantification of islet size and number and alpha, beta and delta cell areas, sections were incubated with antibodies against glucagon, insulin and somatostatin, and then with FITC- and TRITC-conjugated secondary antibodies. Islet size and the area of glucagon-, insulin- and somatostatin-labelled cells on each section were quantified using ImageJ software (ImageJ 1.45: http://rsbweb.nih.gov/ij/download.html).
Mouse islet beta cell proliferation
Mouse islet cell proliferation in vivo was estimated by immunohistochemical analysis of fixed pancreases using a similar approach to that described above. Briefly, 1 mg/ml BrdU was administered to mice in drinking water for 7 days, pancreases were removed, fixed for 16 h in 4% paraformaldehyde, sectioned and analysed by double staining with guinea pig anti-insulin and mouse anti-BrdU antibodies.
Plasmids
CCNA2 (encoding cyclin A2) and Tcf19 promoter activities were assessed using −3,200/+245 human CCNA2 [14] and −1,498/−1 mouse Tcf19 promoter-driven luciferase expression cloned into pGL2. The Tcf19 promoter sequence was produced by PCR amplification using a C57BL/6 mouse islet cDNA library following introduction of SacI and HindIII restriction sites. The PCR product was purified, digested, inserted into the pGL2 luciferase reporter vector and sequenced. Experiments in which NUPR1 was overexpressed were performed using the pcDNA3.1 plasmid coding for the full-length human NUPR1 gene, as previously reported [7]. The pcDNA3.1 expression vector encoding the full-length mouse Tcf19 gene was obtained by PCR amplification of a C57BL/6 mouse islet cDNA library and the amplicon, flanked with HindIII and EcoRI restriction sites, was purified, digested, inserted into pcDNA3.1 and sequenced.
NUPR1 knockdown in human islets
Human islets were isolated from pancreases retrieved from organ donors at the King’s College Hospital Islet Transplantation Unit, with appropriate ethical approval [15]. NUPR1 was downregulated by transient transfection with siRNAs (150 nmol/l) directed against human NUPR1 using previously described protein-knockdown protocols [16]. Human islets transfected with non-interfering RNAs (150 nmol/l) were used as controls. The transfected islets were maintained in culture for 48 h, after which quantitative RT-PCR (qPCR) was carried out to confirm NUPR1 knockdown, and CCNA2 and Tcf19 mRNAs were also quantified by qPCR.
Western blotting
Protein extracts (70 μg) of islets isolated from 3-month-old Nupr1 −/− mice and their wild-type littermates were fractionated by electrophoresis on 12% NuPAGE Bis-Tris mini gels, transferred to polyvinylidene difluoride membranes and immunoprobed with an anti-cyclin A antibody (1:500 dilution), or an anti-TCF19 antibody (1:200 dilution). Equal loading was confirmed by immunoprobing with an anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (1:2,000 dilution).
MIN6 beta cell transfection
For gene reporter assay studies, 5 × 106 MIN6 beta cells were transiently transfected (4 μg of DNA of interest) by electroporation using a Nucleofector II device as previously described [17]. The transfected cells were maintained in culture overnight at 37°C. Promoter activity levels were then determined by bioluminescence using luciferase assay kits, as described by the manufacturer. Promoter activity levels were normalised to β-galactosidase expression, which was obtained by co-transfecting MIN6 beta cells with pSV-β-Gal. For proliferation experiments, MIN6 beta cells were transfected twice [18] over 36 h using 250 ng DNA and a 1:1 DNA:Metafectene Pro ratio and maintained in culture for an additional 24 h at 37°C in DMEM supplemented with 2.5–25 mmol/l glucose and 10% fetal bovine serum. Cell proliferation was quantified by measuring the levels of BrdU incorporation into newly synthesised DNA, essentially as previously described [19].
RNA isolation and qPCR
Total RNA was isolated from 5 × 105 MIN6 beta cells, groups of 100–150 wild-type or Nupr1 −/− mouse islets or 150 human islets exposed to non-interfering RNAs or Nupr1 siRNAs using Qiagen RNeasy minikits. Quantifications of mRNAs encoding NUPR1, Tcf19, Ccna2 and β-actin were performed using the Roche Lightcycler LC480 system, and the expression level of each amplified gene was normalised to β-actin. The specificities of amplifications were verified by melting-curve analyses and by size characterisation of the amplification products on 2% agarose gels. The primers used in this study are shown in electronic supplementary material (ESM) Table 1.
cRNA preparation and array hybridisation
Approximately 3 μg of total RNA purified from wild-type or Nupr1 −/− mouse islets were used to prepare cDNA templates for biotinylated cRNAs production. The labelled cRNAs were produced using a T7 high-yield transcription kit according to the manufacturer’s instructions, then fragmented and hybridised onto Affymetrix Mouse 430A 2.0 arrays. Statistical expression analysis between two arrays was performed using the GeneChip Operating Software from Affymetrix.
Data analysis
Numerical data are expressed as means ± SEM. Student’s t tests were used for statistical analysis of the differences between two groups, and the statistical significance of differences among multiple groups was determined by ANOVA followed by the Tukey honestly significant difference (HSD) test. For statistical analysis of glucose and insulin tolerance tests, the data were also expressed and compared as areas under the curves. Differences between groups were considered significant at p < 0.05.
Results
Metabolic characterisation of Nupr1 −/− mice
We compared growth, glycaemic control and insulin sensitivity of male and female Nupr1 −/− mice and their wild-type littermates to determine whether complete deletion of Nupr1 promoted a diabetogenic phenotype. Growth (Fig. 1a, b) and fertility (data not shown) were not significantly different between wild-type and Nupr1 −/− mice, whether male or female, during 26-week-investigation periods. Similarly, perigonadal fat deposition (Fig. 1c, d) and gastrocnemius muscle mass (Fig. 1e, f) were not significantly different between wild-type and Nupr1 −/− mice, suggesting that Nupr1 expression is not required for normal growth and reproduction. Nupr1 deletion had no detectable effects on fasting or random plasma glucose levels, as shown in Fig. 2 (male, Fig. 2a, b; female, Fig. 2c, d). Consistent with this, both male (Fig. 2e, f) and female (Fig. 2g, h) Nupr1 −/− mice showed normal glucose tolerance to an i.p. glucose load at 3 months (Fig. 2e, g) and 6 months (2f, h). Because no significant metabolic differences were observed between 3- and 6-month-old mice, further studies used only 3-month-old mice.

Weight variables. (a, b) Body weight profiles of wild-type (solid lines) and Nupr1 null (dashed lines) male (a) and female (b) C57BL/6 mice measured from the 5th to 25th week of age. Perigonadal fat (c, d) and gastrocnemius muscle (e, f) weights of 3-month-old wild-type (black bars) and Nupr1 −/− (white bars) male (c, e) and female (d, f) C57BL/6 mice. Perigonadal fat and gastrocnemius muscle masses are expressed as percentages of total body weight. Data are mean ± SEM, (a, b) n = 10–23, (c, d) n = 13–22 and (e, f) n = 10–23

Glucose metabolism variables. (a–d) Blood glucose concentrations were quantified after an overnight fast or randomly during the day in male (a, b) and female (c, d) wild-type (black bars) and Nupr1-null (white bars) mice at the age of 3 months (a, c) and 6 months (b, d); n = 13–20. (e–h) Glucose tolerance curves obtained from wild-type (solid lines) and Nupr1 −/− (dashed lines) 3-month-old (e, g) and 6-month-old (f, h) male (e, f) and female (g, h) mice. Data are mean ± SEM, n = 8–16
Nupr1-knockout mice are hyperinsulinaemic because of increased beta cell mass
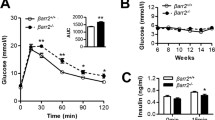
Both male and female Nupr1 −/− mice were less sensitive than their wild-type littermates to i.p. administration of insulin (Fig. 3a, c). Nupr1 −/− mice showed significantly lower insulin-dependent reductions in blood glucose as assessed by area under curve measurements (Fig. 3b, d), consistent with the development of mild insulin resistance in the absence of Nupr1. Insulin-resistant Nupr1 −/− mice remained normoglycaemic (Fig. 2) by secreting more insulin, with a 40 ± 9% increase in fasting serum insulin levels (Fig. 4a), and significantly higher insulin secretory responses to an i.p. glucose challenge (Fig. 4b).

Insulin-sensitivity tests. Non-fasted 3-month-old male (a) and female (c) wild-type (solid lines) and Nupr1 −/− (dashed lines) mice were injected i.p. with insulin, 0.75 U/kg body weight. Blood glucose concentrations were monitored every 15 min for 1 h and expressed as % of their initial levels. (b, d) The corresponding areas under the curves of glucose levels in wild-type (black bars) and Nupr1 −/− (white bars) mice are shown. Data are mean ± SEM, n = 10–19, * p < 0.05

Basal and glucose-induced insulin secretion. (a, b) Serum insulin levels in 3-month-old wild-type (black bar and solid line) and Nupr1 −/− (white bar and dashed line) male mice at fast (a) or at time 0, 30 and 60 min after i.p. administration of glucose (b). Data are mean ± SEM, n = 8–12, * p < 0.05
To determine whether the hyperinsulinaemia observed in Nupr1 −/− mice reflected changes in beta cell mass we examined pancreases retrieved from Nupr1 −/− mice and wild-type littermates. There was no significant difference in pancreas weights (wild type, 385 ± 37.5 mg; Nupr1 −/−; 348 ± 23.5 mg; n = 10; p > 0.2), but Nupr1 −/− mice showed significant increases in islet number per mm2 pancreas (Fig. 5a), together with increased beta cell mass (Fig. 5b, c), giving an overall significant increase in islet area (Table 1). The islet architecture was similar between Nupr1 −/− and control mice and the ratios of beta to alpha, beta to delta and alpha to delta cells did not change significantly, suggesting that the increased islet area arose from expansion of all three endocrine cell types (Table 1). Consistent with this, the insulin content of Nupr1 −/− islets was not different from that of control islets when normalised against DNA content to account for increased islet cell number (male wild-type, 5.8 ± 0.19 ng/ng DNA; male Nupr1 −/−, 6.1 ± 0.39 ng/ng DNA; female wild-type: 5.5 ± 0.45ng/ng DNA; female Nupr1 −/−, 5.4 ± 0.46 ng/ng DNA; n = 8–10; NS).

Nupr1 regulates islet mass expansion. Pancreases isolated from 3-month-old wild-type and Nupr1 −/− mice were used for immunohistochemistry and morphometric studies to characterise the effects of Nupr1 deletion on islet number, islet mass and beta cell proliferation. (a) Islet density expressed as islet number per mm2, (b) beta cell mass, (c) insulin (green), glucagon (red) and somatostatin (green) immunostaining of representative pancreas sections, (d) representative insulin (green) and BrdU (red) immunostained pancreases to assess beta cell proliferation, (e) number of BrdU-positive beta cells per islet and (f) number of BrdU-positive beta cells as a proportion of the total number of beta cells. Data are mean ± SEM, wild-type: black bars; Nupr1 −/−: white bars; (a) n = 6, (b) n = 4, * p < 0.05, ** p < 0.01 and *** p < 0.001
The number of BrdU-positive beta cells per islet was significantly increased in Nupr1 −/− islets (Fig. 5d, e), resulting in a significant increase in the percentage of total beta cells labelled with BrdU (Fig. 5f). This direct assessment of cell replication by BrdU incorporation suggests that the increased islet mass in Nupr1 −/− mice was caused by increased islet cell proliferation.
Nupr1 influences beta cell mass expansion via inhibition of Tcf19 and cyclin A expression and promoter activities
To investigate the molecular mechanisms by which Nupr1 may regulate beta cell proliferation, microarray analysis was used to compare gene-expression patterns of Nupr1 −/− and wild-type mouse islets. Of the 14,000 genes and expressed sequence tags (ESTs) for which the expression levels were compared, 1,076 islet genes showed significant differences in expression at p < 0.0002 following Nupr1 deletion. Of these genes, 304 had expression levels altered by 1.5-fold or more and these have been grouped into sub-families according to their predicted roles in cell function (ESM Table 2). Given the increased islet mass with Nupr1 deletion, attention was focused on genes known to regulate the cell cycle. Messenger RNAs for cyclin A2, which has been identified as a target in the regulation of beta cell mass [20], and TCF19, which has been implicated in regulating genes involved in the later stages of cell cycle progression [21], emerged as possible candidates. Our array analysis indicated that Ccna2 and Tcf19 mRNAs were increased by 1.6-fold (p < 0.0004) and 1.9-fold (p = 0.00002), respectively, in Nupr1 −/− islets. These observations were confirmed by qPCR amplification demonstrating that Tcf19 and Ccna2 mRNA levels were significantly increased in islets isolated from Nupr1 −/− mice (Fig. 6a, b, respectively). In addition, western blotting indicated that cyclin A and TCF19 protein levels were also elevated in islets following Nupr1 deletion (Fig. 6c). In these experiments, densitometric quantification of immunoreactive proteins indicated that cyclin A and TCF19 expression levels were increased by 47.5 ± 5.3% and 56.0 ± 4.8%, respectively, in Nupr1 −/− islets (n = 3, p < 0.05). Furthermore, treatment of human islets with siRNAs, which induced significant knockdown of NUPR1 (64.0 ± 16.8% reduction after 48 h), was associated with significant increases in TCF19 and CCNA2 mRNA levels (Fig. 6d, e).

Nupr1 represses Tcf19 and Ccna2 expression and inhibits beta cell proliferation in vitro. (a) Tcf19 and (b) Ccna2 mRNA expression in wild-type (black bars) and Nupr1 null islets (white bars) and (c) their corresponding protein levels. (d) TCF19 and (e) CCNA2 mRNA levels in human islets treated with non-interfering RNAs (black bars) and those treated with NUPR1 siRNAs (white bars). (f) Transfection efficiency following MIN6 cell transfection either with pcDNA3.1/NUPR1 (white bars) or the empty vector (black bars) as verified by qPCR. (g) Effects of NUPR1 overexpression on MIN6 cell proliferation expressed as % of control (pcDNA3.1-transfected MIN6 cells at 2.5 mmol/l glucose). (h) Transfection efficiency following MIN6 cell transfection with either pcDNA3.1/Tcf19 (white bars) or the empty vector (black bars) as verified by qPCR. (i) Effects of Tcf19 overexpression on proliferation (absorbance at 450 nm) and (j) Ccna2 mRNA levels expressed as % of control (pcDNA3.1-transfected MIN6 cells). (k, l) −3,200/+245 bp CCNA2 (k) and −1,498/−1 bp Tcf19 (l) promoter activities in response to Tcf19 and NUPR1 overexpression in MIN6 cells expressed as % of control (CCNA2/pr or Tcf19/pr). Numerical data are mean ± SEM, (a, b) n = 4 and (c) n = 3 for each genotype, (d–j) n = 3, (k) n = 4, (l) n = 3, * p < 0.05, ** p < 0.01 and *** p < 0.001
Potential functional links between Nupr1, Tcf19 and Ccna2 were assessed in vitro using expression and reporter vectors in the readily transfectable MIN6 mouse beta cell line, as shown in Fig. 6f–l. MIN6 beta cells were transiently transfected either with pcDNA3.1/NUPR1 or pcDNA3.1/Tcf19 constructs that expressed full-length forms of human NUPR1 or murine Tcf19, respectively. Overexpression of NUPR1 (Fig. 6f) reduced basal MIN6 beta cell proliferation (2.5mmol/l glucose) and suppressed glucose-dependent proliferation (12 and 25mmol/l glucose) (Fig. 6g). In contrast, overexpression of Tcf19 (Fig. 6h) resulted in a significant stimulation of MIN6 beta cell proliferation (Fig. 6i), which was associated with enhanced Ccna2 mRNA expression (Fig. 6j). These data are consistent with a direct effect of Nupr1 in beta cells to suppress proliferation via the repression of Ccna2 and/or Tcf19 expression at the promoter level. This was confirmed using gene reporter assays in transiently transfected MIN6 beta cells in which luciferase activity was driven by CCNA2 (Fig. 6k) and Tcf19 (Fig. 6l). Both promoter activities were reduced by 45 ± 13.2% and 27.1 ± 6.9%, respectively, in NUPR1-overexpressing MIN6 beta cells, and cells in which Tcf19 was overexpressed in the presence of the −3,200/+245 bp CCNA2 promoter sequence indicated that Tcf19 upregulated CCNA2 promoter activity (Fig. 6k).
Nupr1-knockout mice are protected against metabolic consequences of an HFD
As Nupr1 deletion induced an upregulation of beta cell proliferation that enabled insulin-resistant Nupr1 −/− mice to maintain normal glucose homeostasis (Fig. 2), we assessed the effects of a diabetogenic HFD (55% fat) in this mouse model. As expected, wild-type mice fed the HFD showed increased body weight compared with age-matched wild-type mice fed the NFD (4% fat). This was already evident after the first week and continued to increase throughout the 16 weeks of the dietary intervention (Fig. 7a, c). The weight gain of the HFD-fed wild-type animals was accompanied by increased perigonadal fat deposition (Fig. 7d), but no change in gastrocnemius muscle mass (Fig. 7e). The HFD induced perturbations in glucose handling in wild-type animals, with a significant increase in fasting blood glucose concentrations (Fig. 7f), and impaired tolerance to i.p. administration of glucose (Fig. 7g, h). Again, as expected, wild-type mice maintained on the HFD became progressively insulin resistant (Fig. 8a) and developed hyperinsulinaemia, with significantly elevated serum insulin levels under fasting conditions (Fig. 8b) and after i.p. injection of glucose (Fig. 8c) when compared with mice maintained on the NFD.

Weight and glucose metabolism variables during an HFD challenge. Five-week-old mice were fed ad libitum for 16 weeks with a standard 4% fat (solid lines and black bars) or a 55% fat diet (dashed lines and white bars), and weight and glucose metabolism variables were monitored. (a, b) Body weight gains recorded for 16 weeks in wild-type (a) and Nupr1 −/− (b) mice, and (c) the final body weight gains recorded 16 weeks after the start of the diets are expressed as percentage increases over initial weights. (d) Perigonadal fat deposition and (e) gastrocnemius muscle mass expressed as a proportion of total body weight. (f, g) Blood glucose concentrations measured at fast (f) and for 90 min (g) after an i.p. glucose challenge. NFD, solid lines; HFD, dashed lines; wild-type mice, thin lines; Nupr1 −/− mice, thick lines. (h) Areas under the curves were calculated from the glucose tolerance curves. Data are mean ± SEM of repeated measures, n = 3 for each genotype and diet condition. * p < 0.05 and ** p < 0.01

Insulin sensitivity and serum insulin levels during an HFD challenge. Five-week-old wild-type and Nupr1 −/− mice were fed ad libitum for 16 weeks with a standard 4% fat diet (solid lines, black bars) or a 55% fat diet (white bars, dashed lines). Insulin tolerance tests were performed and serum insulin levels monitored. (a) Blood glucose levels (as % of initial glucose concentrations) measured for 60 min following an insulin challenge. (b, c) Serum insulin concentrations measured at fast (b) and after i.p. administration of glucose (c). (a, c) NFD, solid lines; HFD, dashed lines; wild-type mice, thin lines; Nupr1 −/− mice, thick lines. Data are mean ± SEM of repeated measures, n = 3 for each genotype and diet condition, * p < 0.05
Global deletion of Nupr1 was protective against some of the HFD-induced changes. Thus, Nupr1 −/− mice showed similar weight gains whether maintained on the NFD or the HFD, with HFD-fed Nupr1 −/− mice gaining approximately 40% less weight than HFD-fed wild-type mice at week 16 (Fig. 7b, c). This was associated with lower perigonadal fat accumulation by HFD-fed Nupr1 −/− compared with wild-type mice (Fig. 7d), and no effect on gastrocnemius muscle mass (Fig. 7e). Similarly, Nupr1 deletion was protective against HFD-induced glucose perturbations, with no significant effect on basal plasma glucose (Fig. 7f), and no impairment of glucose tolerance (Fig. 7g, h). Nupr1 deletion also protected against HFD-induced insulin resistance, with HFD-fed Nupr1 −/− mice being significantly more sensitive to insulin-stimulated glucose clearance than HFD-fed wild-type mice (Fig. 8a). These data suggest that Nupr1 is involved in metabolic responses of peripheral tissues to a diet enriched in fat, such that Nupr1 deletion reduces many of the peripheral responses to chronic exposure to HFD. In contrast, HFD-fed Nupr1 −/− mice had higher serum insulin levels at fast (Fig. 8b) or after an i.p. glucose challenge (Fig. 8c) when compared with wild-type mice maintained on an HFD, consistent with the effects of Nupr1 deletion to enhance basal and glucose-induced insulin secretion in vivo (Fig. 4b), and to increase beta cell mass (Fig. 5).
Discussion
The role of Nupr1 in the exocrine pancreas has been investigated previously in the Nupr1 −/− mouse model used in the current studies. These earlier experiments demonstrated that the exocrine pancreas was not altered following Nupr1 deletion, but that acute pancreatitis was more severe in Nupr1 −/− mice [1, 22]. However, the in vivo function of Nupr1 in the regulation of glucose homeostasis and islet function has not yet been established. Experimental overexpression in vitro of Nupr1 in INS-1 beta cells and human islets was associated with enhanced proliferation and increased insulin content, respectively [11], suggesting that Nupr1 deletion may lead to metabolic dysregulation. However, our data using the Nupr1 −/− mouse suggest that suppression of NUPR1 expression and/or activity is associated with phenotypic effects which would be predicted to be beneficial in obesity-related type 2 diabetes. Thus, Nupr1 deletion resulted in a significant increase in both total number of islets and average islet size, resulting in an overall increase in islet mass, which was associated with enhanced basal and glucose-induced insulin secretory responses. The Nupr1 −/−-associated increase in functional beta cell mass is unlikely to be a compensatory response secondary to hyperglycaemia or severe insulin resistance, as occurs during progression towards type 2 diabetes [23–26]. The Nupr1 −/− mice showed normal glucose tolerance, with their mild insulin resistance being accommodated by increased insulin secretion. In addition, the increased islet mass in Nupr1 −/− mice resulted from a concomitant expansion of alpha, beta and delta cells, again suggesting a primary effect of NUPR1 on regulating islet cell proliferation rather than a compensatory expansion of beta cells in response to metabolic demand.
This conclusion is further supported by our demonstration that transient overexpression of NUPR1 reduced the rate of MIN6 beta cell proliferation in parallel with inhibiting expression of the cell cycle regulators Ccna2 and Tcf19 through effects to suppress their promoter activities in beta cells. The Ccna2 gene regulates progression at both the G1/S and G2/M checkpoints of the cell cycle [27, 28] and lentiviral-mediated infection of islets to overexpress Ccna2 stimulates beta cell self-replication [20]. The TCF19 protein contains a sequence exhibiting characteristics of transcription factors and its expression is regulated in the late stages of the cell cycle [21], consistent with it playing a regulatory role in the expression of genes necessary for cell cycle progression. It is not known how NUPR1 regulates Ccna2 and Tcf19 promoter activities, but it is unlikely to act as a direct repressor as previous reports have demonstrated that it plays important roles in coordinating protein complexes that regulate the expression of various genes involved in cell proliferation and differentiation. Thus, it stimulates the glucagon gene promoter activity through its interactions with E1A binding protein (P300), paired box gene (PAX)2A, PAX2B and PAX2 trans-activation domain-interacting protein [29], and it promotes myogenic differentiation and inhibits cell proliferation via its interactions with myogenic differentiation 1 (MyoD), P300 and P68 [30]. Moreover, as increased Nupr1 expression has been associated with enhanced transcription of genes that are downstream of ATF-4 [31], Nupr1 might also regulate the transcriptional effects of activating transcription factor (ATF) proteins. Thus, as both Tcf19 and Ccna2 promoter regions contain response elements for ATF proteins, P300, MyoD and/or cAMP response element-binding protein (CREB), it is possible that NUPR1 inhibits Ccna2 and Tcf19 promoter activities by modulating the association and function of these transcription factors.
Although it is mainly nuclear, NUPR1 is also expressed in the cytosol and its functions depend on its location within cells and post-translational modifications [32]. Transcriptional regulation of Nupr1 is fairly well defined, with evidence that its promoter is stimulated in response to stress via a range of transcription factors, including nuclear factor of κ light polypeptide gene enhancer in B cells 1 (NFκ-B) [33], ATF-4 [34, 35], SMAD proteins [36] and CCAAT/enhancer binding protein (C/EBP)α/β [7]. In contrast, very little is known about the molecular processes that control NUPR1 production at the protein level. In mesangial cells, it has been suggested that glycogen synthase kinase (GSK)3-mediated phosphorylation stabilises NUPR1, while the vasoactive peptide endothelin-1 is reported to induce cell hypertrophy by stimulating NUPR1 degradation through the ubiquitin/proteasome system [37]. Nothing is known about Nupr1 regulation in islets, but our preliminary results suggest that the calcium/calmodulin-dependent kinase 4 (CaMK4) inhibits beta cell Nupr1 expression, as transfection of MIN6 beta cells with a constitutively active Camk4 (ΔCamk4) repressed Nupr1 promoter activity, while expression of a dominant negative construct (ΔK75ECamk4) had the opposite effect (ESM Fig. 1). Therefore, as Camk4 promotes beta cell survival and proliferation [17, 38], repressing Nupr1 expression via CaMK4 stimulation might be part of a transduction cascade regulating beta cell mass expansion.
The reasons for the discrepancies between our current results and earlier studies in which Nupr1 was experimentally upregulated [9, 11] are not immediately obvious, but could result from differences in experimental approaches and/or interpretation. For example, the increased Nupr1 expression observed in response to glucose [9] may have been stress induced, as has been reported in similar studies [29]. Indeed, NUPR1 is a stress-activated protein [3] that was first reported to be stimulated in the pancreas after the onset of experimentally induced pancreatitis [1]. Our observations are consistent with similar studies showing that proliferation is significantly increased in fibroblasts isolated from Nupr1 −/− mice [8] and that Nupr1 silencing in pancreatic cell lines in vitro is associated with increased proliferation [39]. Taken together, our results suggest that Nupr1 plays an important role as a molecular brake for beta cell expansion, tonically inhibiting beta cell proliferation by repressing expression of genes involved in regulating the late stages of the cell cycle, notably Ccna2 and Tcf19. Our demonstration that NUPR1 knockdown in human islets also enhanced TCF19 and CCNA2 mRNA expression indicates that the repressive effects of NUPR1 on these mRNAs are not confined to mouse islets, but further work is required to confirm these observations at the protein level in human beta cells.
Nupr1 is widely expressed in a range of tissues [30, 40] and our studies demonstrated that global deletion of Nupr1 had profound effects on metabolic responses to high-fat feeding by protecting mice against HFD-induced weight gain, perigonadal fat accumulation, insulin resistance and glucose intolerance, all of which were evident in the wild-type littermates maintained on the HFD. However, the expected effects of the HFD to enhance fasting and glucose-stimulated insulin secretion [41, 42] were observed in both wild-type and Nupr1 −/− mice, with the HFD further increasing the already enhanced basal and glucose-stimulated insulin secretion in Nupr1 −/− mice. These observations are consistent with the effects of Nupr1 deletion to increase beta cell mass and suggest that the protective effects of Nupr1 deletion on the responses of peripheral tissues to high-fat feeding are independent of its effects on the functional beta cell mass. The precise mechanism(s) through which Nupr1 regulates peripheral metabolic responses is the subject of further study, but the protective effects of Nupr1 deletion against the deleterious effects of an HFD offers another potential therapeutic target for type 2 diabetes.
In summary, the results from this study demonstrate a key role for NUPR1 in regulating glucose homeostasis through two separate mechanisms. Nupr1 deletion increases the functional beta cell mass by removing a tonic inhibition of beta cell proliferation, and it also provides protection against HFD-induced obesity, glucose intolerance and insulin resistance. Both mechanisms offer the potential for therapeutic intervention in type 2 diabetes.
Abbreviations
- ATF:
-
Activating transcription factor
- HFD:
-
High-fat diet
- MyoD:
-
Myogenic differentiation 1
- NFD:
-
Normal-fat diet
- NUPR1:
-
Nuclear protein transcription regulator 1
- P300:
-
E1A-binding protein
- PAX:
-
Paired box gene
- qPCR:
-
Quantitative RT-PCR
- si:
-
Small interfering
- TCF19:
-
Transcription factor 19
- TRITC:
-
Tetramethylrhodamine isothiocyanate
References
Mallo GV, Fiedler F, Calvo EL et al (1997) Cloning and expression of the rat p8 cDNA, a new gene activated in pancreas during the acute phase of pancreatitis, pancreatic development, and regeneration, and which promotes cellular growth. J Biol Chem 272:32360–32369
Vasseur S, Folch-Puy E, Hlouschek V et al (2004) p8 improves pancreatic response to acute pancreatitis by enhancing the expression of the anti-inflammatory protein pancreatitis-associated protein I. J Biol Chem 279:7199–7207
Jiang YF, Vaccaro MI, Fiedler F, Calvo EL, Iovanna JL (1999) Lipopolysaccharides induce p8 mRNA expression in vivo and in vitro. Biochem Biophys Res Commun 260:686–690
Vasseur S, Hoffmeister A, Garcia-Montero A et al (2003) Mice with targeted disruption of p8 gene show increased sensitivity to lipopolysaccharide and DNA microarray analysis of livers reveals an aberrant gene expression response. BMC Gastroenterol 3:25
Encinar JA, Mallo GV, Mizyrycki C et al (2001) Human p8 is a HMG-I/Y-like protein with DNA binding activity enhanced by phosphorylation. J Biol Chem 276:2742–2751
Malicet C, Giroux V, Vasseur S, Dagorn JC, Neira JL, Iovanna JL (2006) Regulation of apoptosis by the p8/prothymosin alpha complex. Proc Natl Acad Sci USA 103:2671–2676
Vasseur S, Vidal Mallo G, Fiedler F et al (1999) Cloning and expression of the human p8, a nuclear protein with mitogenic activity. Eur J Biochem 259:670–675
Vasseur S, Hoffmeister A, Garcia-Montero A et al (2002) p8-deficient fibroblasts grow more rapidly and are more resistant to adriamycin-induced apoptosis. Oncogene 21:1685–1694
Päth G, Opel A, Knoll A, Seufert J (2004) Nuclear protein p8 is associated with glucose-induced pancreatic beta-cell growth. Diabetes 53(Suppl 1):S82–S85
Pellerito O, Calvaruso G, Portanova P et al (2010) The synthetic cannabinoid WIN 55,212-2 sensitizes hepatocellular carcinoma cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by activating p8/CCAAT/enhancer binding protein homologous protein (CHOP)/death receptor 5 (DR5) axis. Mol Pharmacol 77:854–863
Päth G, Opel A, Gehlen M et al (2006) Glucose-dependent expansion of pancreatic beta-cells by the protein p8 in vitro and in vivo. Am J Physiol Endocrinol Metab 291:E1168–E1176
Papadimitriou A, King AJF, Jones PM, Persaud SJ (2007) Anti-apoptotic effects of arachidonic acid and prostaglandin E2 in pancreatic beta-cells. Cell Physiol Biochem 20:607–616
Jones PM, Salmon DM, Howell SL (1988) Protein phosphorylation in electrically permeabilized islets of Langerhans. Effects of Ca2+, cyclic AMP, a phorbol ester and noradrenaline. Biochem J 254:397–403
Yoshizumi M, Wang H, Hsieh C-M, Sibinga NES, Perrella MA, Lee M-E (1997) Down-regulation of the cyclin a promoter by transforming growth factor-β1 is associated with a reduction in phosphorylated activating transcription factor-1 and cyclic AMP-responsive element-binding protein. J Biol Chem 272:22259–22264
Huang GC, Zhao M, Jones P et al (2004) The development of new density gradient media for purifying human islets and islet-quality assessments. Transplantation 77:143–145
Muller D, Huang GC, Amiel S, Jones PM, Persaud SJ (2006) Identification of insulin signaling elements in human beta-cells: autocrine regulation of insulin gene expression. Diabetes:2835–2842
Persaud SJ, Liu B, Sampaio HB, Jones PM, Muller DS (2011) Calcium/calmodulin-dependent kinase IV controls glucose-induced Irs2 expression in mouse beta cells via activation of cAMP response element-binding protein. Diabetologia 54:1109–1120
Muller, D S, Jones, P M, Persaud, SJ (2007) Expression of a potent anti-apoptotic protein in MIN6 b-cells using METAFECTENE PRO [Internet]. Biontex. Available from: www.biontex.com/con_4_6_4/cms/upload/pdf/Muller_MP_en.pdf. Accessed 12 July 2013
Muller D, Jones PM, Persaud SJ (2006) Autocrine anti-apoptotic and proliferative effects of insulin in pancreatic beta-cells. FEBS Lett 580:6977–6980
Song W-J, Schreiber WE, Zhong E et al (2008) Exendin-4 stimulation of cyclin A2 in beta-cell proliferation. Diabetes 57:2371–2381
Ku DH, Chang CD, Koniecki J et al (1991) A new growth-regulated complementary DNA with the sequence of a putative trans-activating factor. Cell Growth Differ 2:179–186
Saltiel AR (2001) New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell 104:517–529
Butler AE, Janson J, Soeller WC, Butler PC (2003) Increased beta-cell apoptosis prevents adaptive increase in beta-cell mass in mouse model of type 2 diabetes: evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes 52:2304–2314
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52:102–110
Plachot C, Movassat J, Portha B (2001) Impaired beta-cell regeneration after partial pancreatectomy in the adult Goto-Kakizaki rat, a spontaneous model of type II diabetes. Histochem Cell Biol 116:131–139
Portha B (2005) Programmed disorders of beta-cell development and function as one cause for type 2 diabetes? The GK rat paradigm. Diabetes Metab Res Rev 21:495–504
Sherr CJ (1995) Mammalian G1 cyclins and cell cycle progression. Proc Assoc Am Physicians 107:181–186
Sherr CJ, Roberts JM (1995) Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev 9:1149–1163
Garcia-Montero A, Vasseur S, Mallo GV, Soubeyran P, Dagorn JC, Iovanna JL (2001) Expression of the stress-induced p8 mRNA is transiently activated after culture medium change. Eur J Cell Biol 80:720–725
Taïeb D, Malicet C, Garcia S et al (2005) Inactivation of stress protein p8 increases murine carbon tetrachloride hepatotoxicity via preserved CYP2E1 activity. Hepatology 42:176–182
Carracedo A, Egia A, Guzmán M, Velasco G (2006) p8 Upregulation sensitizes astrocytes to oxidative stress. FEBS Lett 580:1571–1575
Valacco MP, Varone C, Malicet C, Cánepa E, Iovanna JL, Moreno S (2006) Cell growth-dependent subcellular localization of p8. J Cell Biochem 97:1066–1079
Kallwellis K, Grempler R, Günther S, Päth G, Walther R (2006) Tumor necrosis factor alpha induces the expression of the nuclear protein p8 via a novel NF kappaB binding site within the promoter. Horm Metab Res 38:570–574
Passe CMM, Cooper G, Quirk CC (2006) The murine p8 gene promoter is activated by activating transcription factor 4 (ATF4) in the gonadotrope-derived LbetaT2 cell line. Endocrine 30:81–91
Averous J, Lambert-Langlais S, Cherasse Y et al (2011) Amino acid deprivation regulates the stress-inducible gene p8 via the GCN2/ATF4 pathway. Biochem Biophys Res Commun 413:24–29
Pommier RM, Gout J, Vincent DF et al (2012) The human NUPR1/P8 gene is transcriptionally activated by transforming growth factor β via the SMAD signalling pathway. Biochem J 445(2):285–293
Goruppi S, Kyriakis JM (2004) The pro-hypertrophic basic helix-loop-helix protein p8 is degraded by the ubiquitin/proteasome system in a protein kinase B/Akt- and glycogen synthase kinase-3-dependent manner, whereas endothelin induction of p8 mRNA and renal mesangial cell hypertrophy require NFAT4. J Biol Chem 279:20950–20958
Liu B, Barbosa-Sampaio H, Jones PM, Persaud SJ, Muller DS (2012) The CaMK4/CREB/IRS-2 cascade stimulates proliferation and inhibits apoptosis of β-cells. PLoS One 7(9):e45711
Malicet C, Lesavre N, Vasseur S, Iovanna JL (2003) p8 inhibits the growth of human pancreatic cancer cells and its expression is induced through pathways involved in growth inhibition and repressed by factors promoting cell growth. Mol Cancer 2:37
Sambasivan R, Cheedipudi S, Pasupuleti N, Saleh A, Pavlath GK, Dhawan J (2009) The small chromatin-binding protein p8 coordinates the association of anti-proliferative and pro-myogenic proteins at the myogenin promoter. J Cell Sci 122(Pt 19):3481–3491
Saltiel AR, Kahn CR (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414:799–806
Kahn SE, Hull RL, Utzschneider KM (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444:840–846
Acknowledgements
We are grateful to Junichi I. Miyazaki (University of Osaka, Japan) for the provision of MIN6 beta cells and to Mu E. Lee (Harvard School of Public Health, USA) for the human CCNA2 promoter-driven pGL2 expression vector. We thank the families of donors for making available pancreases and the islet isolation unit at King’s College Hospital (Guo Cai Huang and Stephanie Amiel) for providing human islets for some of the experiments.
Funding
We gratefully acknowledge grant support from Diabetes UK (ref: 06/0003387, 10/0003995 and 08/0003706), the European Foundation for the Study of Diabetes (EFSD)/Merck Sharp & Dohme (MSD) programme and the Society for Endocrinology (Early Career Grant).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
HCB-S contributed to the acquisition, analysis and interpretation of data and to the drafting/revision of the article. BL, RD, AMRdL, AJK, JEB, CM and JLI substantially contributed to the acquisition of data and to the revision of the manuscript. PMJ contributed to the conception and design of the study and to drafting the article. SJP and DSM contributed to the conception and design of the study, the acquisition, analysis and interpretation of data and to drafting/revising the manuscript. All authors have approved the final version of the article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Shanta J. Persaud and Dany S. Muller are joint senior authors of this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
(PDF 33 kb)
ESM Table 2
(PDF 64 kb)
ESM Fig. 1
Regulation of Nupr1 promoter activity by calcium/calmodulin-dependent kinase 4 (CaMK4). MIN6 beta cells were transfected with the CAT vector whose expression was under control of the -1,471/+37bp Nupr1 promoter sequence (control). Promoter activity was monitored in response to expression of the truncated and constitutively active (Δ) or dominant negative (ΔK75E) forms of CaMK4. Data are mean ± sem, *p<0.05. (PDF 17 kb)
Rights and permissions
About this article
Cite this article
Barbosa-Sampaio, H.C., Liu, B., Drynda, R. et al. Nupr1 deletion protects against glucose intolerance by increasing beta cell mass. Diabetologia 56, 2477–2486 (2013). https://doi.org/10.1007/s00125-013-3006-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-3006-x