
Abstract
Aims/hypothesis
Sirtuin (SIRT)3 is a mitochondrial protein deacetylase that regulates reactive oxygen species (ROS) production and exerts anti-inflammatory effects. As chronic inflammation and mitochondrial dysfunction are key factors mediating pancreatic beta cell impairment in type 2 diabetes, we investigated the role of SIRT3 in the maintenance of beta cell function and mass in type 2 diabetes.
Methods
We analysed changes in SIRT3 expression in experimental models of type 2 diabetes and in human islets isolated from type 2 diabetic patients. We also determined the effects of SIRT3 knockdown on beta cell function and mass in INS1 cells.
Results
SIRT3 expression was markedly decreased in islets isolated from type 2 diabetes patients, as well as in mouse islets or INS1 cells incubated with IL1β and TNFα. SIRT3 knockdown in INS1 cells resulted in lowered insulin secretion, increased beta cell apoptosis and reduced expression of key beta cell genes. SIRT3 knockdown also blocked the protective effects of nicotinamide mononucleotide on pro-inflammatory cytokines in beta cells. The deleterious effects of SIRT3 knockdown were mediated by increased levels of cellular ROS and IL1β.
Conclusions/interpretation
Decreased beta cell SIRT3 levels could be a key step in the onset of beta cell dysfunction, occurring via abnormal elevation of ROS levels and amplification of beta cell IL1β synthesis. Strategies to increase the activity or levels of SIRT3 could generate attractive therapies for type 2 diabetes.
Similar content being viewed by others


Introduction
Type 2 diabetes develops when pancreatic beta cells lose the ability, through loss of function and mass, to compensate for insulin resistance [1–4]. This leads to hyperglycaemia, glucose intolerance and onset of type 2 diabetes [1–4]. Hence, an understanding of the mechanisms responsible for reduced beta cell function and mass is essential for the development of strategies to prevent the onset of overt type 2 diabetes.
The precise mechanisms responsible for beta cell failure in type 2 diabetes are complex and have yet to be fully described, but chronic inflammation is increasingly implicated. Type 2 diabetes is a progressive inflammatory state, characterised by abnormally elevated islet levels of pro-inflammatory cytokines, such as IL1β and TNFα, which have been demonstrated to suppress beta cell function and mass [5–9].
Our recent studies expanded this concept by describing a central role for the NAD+ synthesising enzyme, nicotinamide phosphoribosyltransferase (NAMPT), in protecting mouse islets from the deleterious effects of pro-inflammatory cytokines [10]. NAMPT catalyses the conversion of nicotinamide to nicotinamide mononucleotide (NMN), which is in turn converted to NAD+ by NMN-acetyltransferases [11]. NAMPT exists in intra- and extracellular forms; however intracellular NAMPT abundance is low in mouse islets and rat INS1 cells [12] (Caton and Sugden, unpublished observations), suggesting a reliance on circulating extracellular NAMPT for maintenance of adequate NAD+ levels. Consistent with this idea, Nampt +/− mice develop impaired islet function, in association with lowered plasma NMN levels, while exogenous administration of NMN restored islet function in these mice (12). In addition, our recent work demonstrated that exogenous NMN exerts anti-inflammatory effects on islets in a mouse model of type 2 diabetes and in isolated mouse islets exposed to pro-inflammatory cytokines [10].
The mechanism of action of NMN in islets is unclear. NAD+ is an essential cofactor for the activation of sirtuin (SIRT) enzymes (SIRT1 to 7), which control a number of metabolically important processes, predominantly through their function as protein deacetylases [13]. SIRT1 is reported to improve islet function [14–16], in part through anti-inflammatory mechanisms [17]. Interestingly, we have previously reported that a SIRT1 inhibitor (EX527) only partially blocked the effects of NMN in mouse islets, suggesting the potential involvement of additional members of the SIRT family in mediating the effects of NMN [10]. However, little is known about the role of the remaining SIRTs in islet beta cell function. Recent studies have highlighted a central role for mitochondrial dysfunction in pancreatic beta cell dysfunction [18], while other reports have demonstrated that mitochondrial proteins undergo significant levels of acetylation [19] and that stringent control of relevant acetylase and deacetylase activities is required to maintain appropriate mitochondrial function. To further elucidate the mechanisms of action of NMN, we assessed the role of the major mitochondrial SIRT, SIRT3 [20], in beta cell function. SIRT3 has not been previously reported to have functions in islets or beta cells; however, in other tissues, SIRT3 reportedly provides protection against inflammation [21] and apoptosis [22], in part by lowering reactive oxygen species (ROS) [21, 23], thus indicating that SIRT3 may exert a protective role in beta cells.
Methods
Cell culture and transfection
INS1 beta cells were cultured in RPMI 1640 medium containing 10% (vol./vol.) FBS, 10 mmol/l HEPES, 2 mmol/l l-glutamine, 1 mmol/l sodium pyruvate and 50 μmol/l β-mercaptoethanol. Cells were transfected with SIRT3-specific small interfering (si) RNA (siSirt3; 72 h; 75–150 nmol/l; Dharmacon, Lafayette, CO, USA) or with scrambled siRNA control (Ambion, Paisley, UK) using OligofectAMINE (Invitrogen, Paisley, UK). Alternatively, INS1 cells were incubated for 48 h with combinations of IL1β (2.5–5.0 ng/ml), TNFα (5–10 ng/ml) (both R+D Systems, Abingdon, UK) or NMN (100 μmol/l; Sigma-Aldrich, Poole, UK), and for 2 h with BAY-7082 (20 μmol/l; Cayman Chemical, Ann Arbor, MI, USA) or UO126 (10 μmol/l; Cell Signaling Technology, Danvers, MA, USA). After incubation, cells were either lysed for protein and gene expression analysis, or incubated with leucine (3 h; 20 mmol/l) in Hanks' balanced salt solution containing 17 mmol/l glucose, 10 mmol/l HEPES (pH 7.4) and 0.2% BSA (wt/vol.) to induce insulin secretion, or they were processed for measurement of caspase 3 activity or ROS levels.
Experimental animals
Male C57Bl/6 mice (7 weeks old) (Charles River, Margate, UK) were fed a high-fat, high-carbohydrate ‘Western diet’ (HFD) (5TJN; TestDiets, Richmond, IN, USA) or a low-fat control diet (LFD) (5TJS; TestDiets) for 16 weeks. Separately, 7-week-old C57Bl/6 mice previously maintained on a standard rodent diet were fasted for 24 h prior to tissue sampling. Animal experiments were conducted in accordance with the Home Office regulations on the Operation of Animals (Scientific Procedures) Act 1986, published by Her Majesty’s Stationery Office (London, UK), and the study was conducted in accordance with the Principles of Laboratory Care. Animals were maintained on a 12 h light and 12 h dark cycle (light from 07:00 h).
Quantitative RT-PCR and immunoblotting of mouse and INS1 samples
Gene expression and protein levels were measured as previously described [10]. See electronic supplementary material (ESM) Methods for further details.
Mouse pancreatic islet isolation and ex vivo insulin secretion
Mouse pancreases were isolated as previously described [10]. Insulin secretion assays were conducted in batches of eight to ten size-matched islets, as previously described [10].
Measurement of apoptosis
Levels of apoptosis in INS1 cells were estimated by fluorescence measurement of caspase 3 activity (Cayman Chemical).
Cellular ROS measurement
Intracellular ROS levels were measured by a fluorescence method using the fluorescent probe 5-(and-6)-chloromethyl-2,7-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA; Molecular Probes, Paisley, UK). Full details are provided in the ESM Methods.
Real-time quantitative PCR of human samples
Pancreases were obtained with the approval of the local ethics committee and consent by donors' relatives. Experiments were performed as previously described [24]. Total RNA (1 μg) was extracted from human islets removed from five non-diabetic and four type 2 diabetic individuals (ESM Table 1, ESM Methods), and reverse-transcribed with a kit (iScript cDNA Synthesis; Bio-Rad, Segrate, Italy) The mRNA expression of SIRT3 (Eurogentec, Southampton, UK) was quantified and normalised against β-actin, as previously described, in a bioanalyser (Applied Biosystem 7700; Applied Biosystems, Monza, Italy). Gene expression was determined by \( {{2}^{{ - \Delta \Delta {{{\rm{C}}}_{{\rm{t}}}}}}} \) methodology, normalised against the reference gene β-actin. Changes in gene expression are represented as fold change relative to one, where control equals one.
Ex vivo insulin secretion from human islets
The details of these experiments have been provided elsewhere [24–26]. Briefly, islet samples were kept for 45 min at 37°C in KRB, 0.5% (vol./vol.) albumin, pH 7.4, containing 3.3 mmol/l glucose (washing phase). Then the medium was replaced with KRB containing 3.3 mmol/l glucose to assess basal insulin secretion for 45 min, after which the islets were challenged with 16.7 mmol/l glucose to assess glucose-stimulated insulin release. Insulin was quantified using an immunoradiometric assay (Pantec Forniture Biomediche, Turin, Italy).
Immunohistochemistry of human pancreatic sections
Pancreases were obtained with the approval of the local ethics committee and consent by donors' relatives. Serial sections (4 μm), fixed in buffered formalin and paraffin-embedded, were cut from each case and mounted on glass slides coated in (3-aminopropyl)-triethoxysilane (Sigma). Sections were processed and labelled using a standard immunoperoxidase technique for paraffin sections. Antigens were unmasked by heat-induced epitope retrieval in 10 mmol/l citrate buffer, pH 6.0. Primary antibody (SIRT3; Millipore, Watford, UK) was applied overnight at 4°C (1:100). A detection system (REAL Envision; Dako, Cambridge, UK) was used for antigen detection. For details of human donors [27–29], see ESM Methods and ESM Table 2.
Immunofluorescence of human and mouse pancreatic sections
To examine the islet cell subtypes producing SIRT3, double immunofluorescence staining was performed on sections that had been fixed in buffered formalin and paraffin-embedded. Rabbit antiserum with specificity for SIRT3 was incubated overnight at 4°C at 1:50 (human) or 1:100 (mouse) dilution, and SIRT3 was detected with goat anti-rabbit AlexaFluor 568 (1:400; human) or goat anti-rabbit AlexaFluor 488 (1:1,000; mouse) conjugated secondary antibody (Invitrogen). Sections were then washed and stained (1 h) for islet hormones: guinea pig anti-insulin (Dako) or mouse anti-glucagon (Abcam, Cambridge, UK) for human pancreas and guinea pig anti-insulin (Abcam) for mouse sections. Immunopositivity was detected with an appropriate goat secondary antibody conjugated with AlexaFluor 488 (1:400; human) or AlexaFluor 647 (1:1,000; mouse). DAPI (1:1,000, Invitrogen) was included in the final incubation step to stain cell nuclei. Sections were mounted in a hard-set mounting medium (Vectashield; Vector Laboratories, Peterborough, UK) under glass coverslips. Human pancreatic sections were analysed using a microscope (Eclipse 80i; Nikon, Kingston upon Thames, UK) and images overlaid using NIS-Elements BR 3.0 software (Nikon). Mouse pancreatic sections were analysed using an epi-fluorescent microscope (DM5000; Leica, Milton Keynes, UK) and Leica Application Suite software.
Statistical analysis
Results are expressed as mean ± SEM. Statistical comparisons were obtained using GraphPad (GraphPad Software, San Diego, CA, USA). Statistical differences were calculated using a paired t test or one-way ANOVA followed by Bonferroni’s post hoc test where appropriate.
Results
SIRT3 protein is present in mouse islets and rat INS1 beta cells
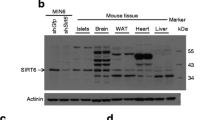
The presence and function of SIRT3 has not been previously examined in pancreatic islets. To address this, we initially sought to confirm the expression of this SIRT in normal mouse islets and rat INS1 cells. Using mouse liver as a positive control, we confirmed by immunoblotting that SIRT3 is present in mouse pancreatic islets (Fig. 1a). We next sought to identify the islet cellular localisation of SIRT3. Immunoblotting also confirmed that SIRT3 is present in the rat beta cell line, INS1 (Fig. 1b). Immunofluorescence analysis of mouse pancreatic sections demonstrated that SIRT3 co-localised with insulin, indicating that SIRT3 is present in beta cells of mouse islets (Fig. 1c). To examine SIRT3 in human islets, we used pancreas sections obtained from non-diabetic human donors. Immunohistochemical analysis confirmed that SIRT3 localised to endocrine cells within human islets, but was absent from non-endocrine pancreas (Fig. 1d). However, in contrast to observations in mouse islets, immunofluorescence analysis suggested that alpha cell staining was most intense in human islets (Fig. 1e), although clear immunostaining for SIRT3 was also present in insulin-positive beta cells (Fig. 1f).

(a) SIRT3 protein is produced in mouse islets and (b) in INS1 cells. (c) Double immunofluorescence images of mouse islet SIRT3 (green), insulin (red) and SIRT3/insulin overlay. (d) Immunohistochemistry image of SIRT3 in human pancreas (scale bar, 10 μm), and double immunofluorescence images of SIRT3 and insulin (e) or glucagon (f). Images are representative (n = 4)
Beta cell SIRT3 levels are suppressed by pro-inflammatory cytokines
Culture of INS1 cells with IL1β (2.5–5.0 ng/ml) and TNFα (5–10 ng/ml) for 48 h resulted in significant reductions of SIRT3 levels (Fig. 2a) and Sirt3 mRNA expression (Fig. 2b). Moreover, 48 h incubation with TNFα and IL1β significantly suppressed Sirt3 mRNA in isolated mouse islets in culture (Fig. 2c). Our previous studies described a protective effect of NMN in mouse islets exposed to chronic inflammatory conditions (10). Consistent with these findings, co-incubation of INS1 cells with NMN blocked the suppressive effects of IL1β and TNFα on SIRT3 levels, restoring Sirt3 mRNA and SIRT3 to basal levels (Fig. 2d, e). Taken together, these data show that in INS1 cells, SIRT3 protein and Sirt3 gene are suppressed by exposure to pro-inflammatory cytokines and that these effects are attenuated by NMN.

SIRT3 levels are suppressed by pro-inflammatory cytokines. (a) INS1 cells were incubated with TNFα and IL1β as indicated. After 48 h incubation, SIRT3 protein levels were measured by immunoblot. (b) INS1 cells were incubated with TNFα (10 ng/ml) and IL1β (5 ng/ml), and after 24 or 48 h incubation, Sirt3 mRNA expression was measured by quantitative RT-PCR. (c) Islets isolated from control C57Bl/6 mice were incubated for 48 h with TNFα and IL1β as indicated, prior to measurement of Sirt3 mRNA by quantitative RT-PCR. (d) INS1 cells were incubated for 48 h with TNFα, and IL1β, with or without NMN (dosages as indicated), and SIRT3 levels or (e) Sirt3 mRNA expression were determined. CON, control; CK, cytokine. Data are expressed as mean ± SEM. (a) ***p < 0.001 vs untreated cells; (b) **p < 0.01 vs time 0; (c, e) *p < 0.05, **p < 0.01 and ***p < 0.001
Islet expression of SIRT3 mRNA is decreased in type 2 diabetic humans
To examine the relevance of SIRT3 in humans, we next measured SIRT3 mRNA expression in islets isolated from human patients with type 2 diabetes, an average disease duration of 8 years and blood glucose values of 10.4 mmol/l (full details, see ESM Table 1). Our findings demonstrated significant decreases in SIRT3 mRNA (~60%) in type 2 diabetic compared with non-diabetic control islets (Fig. 3a). Reductions in SIRT3 were associated with suppressed insulin secretion in these islets (Fig. 3b, c), consistent with a role for SIRT3 in the pathophysiology of beta cell dysfunction in human type 2 diabetes.

Changes in SIRT3 levels in islets from human type 2 diabetes donors and in fasted or HFD mice. Islets were isolated from type 2 diabetic (T2DM; black bars) and non-diabetic (ND; white bars) human participants; (a) islet mRNA expression of SIRT3, (b) insulin secretion in response to 3 and 17 mmol/l glucose, and (c) insulin stimulation index were determined. (d) C57Bl/6 mice were fasted for 24 h, and islet mRNA expression of Sirt3 and (e) ex vivo islet insulin secretion in response to 1 h glucose (17 mmol/l) and to (f) leucine (20 mmol/l) stimulation were determined. (g–i) A separate group of C57Bl/6 mice was maintained on an HFD (black bars) or LFD (white bars) for 16 weeks, after which (i) islet mRNA expression of Sirt3 and (g) ex vivo islet insulin secretion in response to 1 h glucose (3 and 17 mmol/l) and (h) to leucine (2 and 20 mmol/l) stimulation were determined. Data are expressed as mean ± SEM. *p < 0.05 and **p < 0.01
Fasting decreases islet levels of SIRT3
To assess changes in SIRT3 levels in response to changes in nutritional status, islets were isolated from C57Bl/6 mice that had been fasted for 24 h after previously being maintained on a standard rodent diet. Expression of Sirt3 was markedly decreased in islets isolated from fasted mice compared with those isolated from fed mice (Fig. 3d). Lower Sirt3 expression was associated with decreased ex vivo insulin secretion in response to glucose (17 mmol/l) (Fig. 3e) and leucine (20 mmol/l) (Fig. 3f), together with decreased plasma insulin and glucose levels (ESM Fig. 1a, b). Thus, lower Sirt3 expression is associated with altered regulation of islet insulin secretion and whole-body insulin levels in response to nutritional deprivation.
Islet expression of Sirt3 is altered in HFD-fed mice and may mediate islet compensation
We next examined changes in Sirt3 expression in islets isolated from an HFD-mouse model of metabolic disease. HFD mice displayed: (1) increased levels of plasma insulin and glucose (ESM Fig. 1c, d); and (2) parallel decreases in liver and skeletal muscle abundance of phospho(Ser473)-Akt (ESM Fig. 1e), both of which are indicative of insulin resistance. However, ex vivo insulin secretion was unchanged in HFD mice compared with LFD controls (Fig. 3g, h). Since insulin resistance would normally be expected to result in islet compensation and increased insulin secretion [1–4], these results, taken together, suggest partially impaired islet function. Interestingly, in this model, islet Sirt3 mRNA expression was mildly but non-significantly increased in HFD animals (Fig. 3i), which may be indicative of a role for SIRT3 in mediating islet compensation for insulin resistance.
SIRT3 knockdown impairs beta cell function and increases beta cell apoptosis
To determine specific effects of SIRT3 on beta cell function, we used specific Sirt3 siRNA (siSirt3). SIRT3 levels and Sirt3 mRNA expression were decreased by ~75% and ~62%, respectively, in INS1 beta cells incubated with siSirt3 (72 h; 75–150 nmol/l) (Fig. 4a, b). These changes are of a similar magnitude to those observed following cytokine-incubation (Fig. 2), and thus potentially represent pathophysiological conditions. SIRT3 knockdown did not significantly affect the mRNA expression of other SIRTs (ESM Fig. 2). SIRT3 knockdown in INS1 cells impaired leucine-stimulated insulin secretion by ~32% (Fig. 4c) and increased caspase 3 activity by 5.9-fold, suggesting a rise in apoptosis (Fig. 4d). However, it is not possible to definitively determine from these data whether the suppressed insulin secretion occurred as a consequence of decreased cell viability or via a primary defect in stimulus–secretion coupling.

SIRT3 knockdown impairs beta cell function. (a) INS1 cells were incubated for 72 h with siSirt3 as indicated or with scrambled siRNA (Sc) control (150 nmol/l), and SIRT3 protein (representative western blot, n = 4), (b) Sirt3 mRNA, (c) leucine-stimulated insulin secretion, (d) caspase 3 activity and (e) cellular ROS levels were determined. (f–h) Separately, INS1 cells were incubated with siSirt3 (150 nmol/l; 72 h). After 24 h, pro-inflammatory cytokines (TNFα 10 ng/ml, IL1β 5 ng/ml) and NMN (100 μmol/l) were added to the cells for the remaining 48 h, and (f) insulin secretion, (g) caspase 3 activity and (h) cellular ROS levels were measured. Fold changes (e, h) are relative to control, where control is 1. Data are expressed as mean ± SEM; *p < 0.05, **p < 0.01 and ***p < 0.01 for differences between treatments. U, untreated
Consistent with previous studies in other tissues [23], lowering of SIRT3 levels led to a marked increase in cellular levels of ROS in INS1 cells (Fig. 4e). ROS exerts a variety of toxic effects on pancreatic beta cells [18] and may provide a mechanism for the observed beta cell defects in response to SIRT3 suppression.
SIRT3 knockdown blocks the beneficial actions of NMN in beta cells
We next sought to determine the role of SIRT3 in mediating the beneficial effects of NMN on beta cells. As expected, the incubation of INS1 cells with IL1β and TNFα led to a marked suppression of insulin secretion, which was reversed by co-incubation with NMN. Crucially, these effects of NMN were almost completely blocked when SIRT3 was knocked down by siSirt3 (Fig. 4f). Pro-inflammatory cytokines can induce apoptosis, leading to reduced beta cell mass [8]. Consistent with this, the incubation of INS1 cells with TNFα and IL1β led to a marked increase in caspase 3 activity, which was blocked by NMN. The effect of NMN on caspase 3 activity was prevented when SIRT3 levels were lowered with siSirt3 (Fig. 4g). Similarly, NMN blocked pro-inflammatory cytokine-mediated increases in cellular ROS levels, these affects being reversed by SIRT3 knockdown (Fig. 4h). Taken together, these data show that SIRT3 plays an important role in mediating the protective effects of NMN on beta cells exposed to chronic inflammatory conditions.
Effects of SIRT3 knockdown on beta cell gene expression
Our previous studies reported that, in mouse islets, NMN functions in part by correcting pro-inflammatory cytokine-mediated suppression of genes that are crucial for beta cell function [10]. We showed here that incubation of INS1 cells with Sirt3 siRNA or pro-inflammatory cytokines leads to a marked decrease in mRNA expression of Pdx1, Mafa, Ins1 and Slc2a2 (Fig. 5a–d). Pdx1 and Mafa are crucial for beta cell development and also function as transcription factors for other important genes, including insulin (Ins1) and Slc2a2 (essential for glucose uptake into the beta cell) [30–34]. For all these genes, incubation with Sirt3 siRNA caused a similar level of suppression as incubation with TNFα and IL1β. Similar to previous observations in mouse islets [10], NMN blocked the effects of TNFα and IL1β on beta cell gene expression, restoring mRNA expression to basal levels. Importantly, the lowering of SIRT3 levels with siRNA blocked these beneficial effects of NMN, with beta cell gene expression remaining at levels similar to those observed following incubation with TNFα and IL1β alone. These data imply that SIRT3 mediates the effects of NMN on beta cell gene expression, and that suppressed levels of SIRT3 are likely to lead to poor beta cell development and lower levels of insulin expression and secretion.

SIRT3 knockdown alters beta cell gene expression. (a) INS1 cells were incubated with siSirt3 or scrambled siRNA (150 nmol/l; 72 h). After 24 h, pro-inflammatory cytokines (TNFα 10 ng/ml, IL1β 5 ng/ml) and NMN (100 μmol/l) were added to the cells for the remaining 48 h, and expression of Pdx1, (b) Mafa, (c) Ins1 and (d) Slc2a2 was quantified. Data are mean ± SEM; n = 9; *p < 0.05 and ***p < 0.001 for difference between treatments
SIRT3 knockdown leads to increased beta cell levels of IL1β and mitogen-activated protein kinases
We next sought to determine the mechanism of action by which SIRT3 affects beta cell function. We have reported above that cellular ROS levels were significantly increased in INS1 cells treated with Sirt3 siRNA (Fig. 4). High ROS levels can induce increased IL1β levels through activation of mitogen-activated protein kinases (MAPKs) and nuclear factor κB (NFκB) [21, 35, 36]. We demonstrate here that SIRT3 knockdown increased beta cell levels of the activated MAPKs, phospho-extracellular signal-regulated kinase (ERK) and phospho-c-JUN N-terminal regulated kinase (JNK) (Fig. 6a). These changes occurred in parallel with increased levels of the mature form of IL1β (Fig. 6a). To investigate whether these changes mediate the effect of SIRT3 knockdown, we investigated the effects on beta cell function of inhibiting MAPK and NFκB (NFκB reportedly mediates signalling between ROS, MAPKs and IL1β) [21, 36]. The inhibition of ERK signalling with UO126 (10 μmol/l; 2 h) did not reverse Sirt3 siRNA-mediated suppression of insulin secretion from INS1 cells (Fig. 6b) or on the blocking of NMN action by Sirt3 siRNA (Fig. 6c). However, ERK inhibition did have some effects on the blocking of cytokine-mediated impairment of insulin secretion (Fig. 6d). Thus MAPK activation can be induced in response to reduced SIRT3 levels, but appears not to be obligatory for the effects of SIRT3 knockdown in this system. In contrast, NFκB inhibition with BAY-7082 (20 μmol/l; 2 h) prevented Sirt3 siRNA from lowering insulin secretion, and from influencing the actions of exogenous NMN (Fig. 6b, c) and pro-inflammatory cytokines (Fig. 6d). Thus, signalling through NFκB, which may occur as a result of increased ROS production, provides a likely signalling mechanism to mediate the effects of SIRT3 knockdown in beta cells.

SIRT3 knockdown increases IL1β and activated MAPK levels, and inhibition of NFκB blocks the effects of lowered SIRT3 levels. (a) INS1 cells were incubated for 72 h with siSirt3 (150 nmol/l) or scrambled siRNA (Sc) control (150 nmol/l), and levels of phospho-(Thr183/Tyr185)-JNK and phospho-(Thr202/Tyr204)-ERK and IL1β were determined (representative western blot, n = 4). (b–d) Changes in insulin secretion following co-incubation of INS1 cells with siSirt3 (150 nmol/l; 72 h), scrambled siRNA (150 nmol/l; 72 h), pro-inflammatory cytokines (TNFα 10 ng/ml, IL1β 5 ng/ml), U0126 (10 μmol/l; 2 h) and BAY-1072 (10 μmol/l; 2 h) as indicated. (e) Schematic representation of proposed pathway mediated by SIRT3 knockdown. Chronic inflammation lowers SIRT3 levels, leading to increased cellular ROS levels, which in turn activates MAPK and NFκB, resulting in elevated beta cell IL1β synthesis and reduced beta cell function and mass. (b–d) Data are mean ± SEM; n = 6; *p < 0.05, **p < 0.01 and ***p < 0.001 for differences between treatments. U, untreated
Discussion
Mitochondrial dysfunction is a primary contributor to beta cell dysfunction in type 2 diabetes (18). SIRT3 is a key regulator of mitochondrial protein acetylation status [20, 37], but the precise regulatory role of this enzyme has not been examined in pancreatic islets or beta cells. We report here that SIRT3 mRNA expression is suppressed in human islets isolated from type 2 diabetes patients (compared with islets from non-diabetic controls), as well as in mouse islets and INS1 cells exposed to chronic inflammatory conditions. These findings, together with results from our previous studies [10] and a recent report showing decreased SIRT3 levels in high-passage MIN6 cells [38], prompted us to investigate the specific effects of SIRT3 knockdown in beta cells.
We report that SIRT3 plays a key role in the regulation of pancreatic beta cell function and in their protection from apoptotic cell death. Moreover, SIRT3 mediates many of the protective effects of NMN on beta cells exposed to chronic inflammatory conditions. Previous studies in other tissues have revealed that SIRT3 regulates ROS production, with lower SIRT3 levels associated with increased cellular ROS levels [21, 23, 39]. In agreement with this, we report that SIRT3 knockdown led to increased cellular ROS levels in INS1 cells. We also show that NMN can protect against pro-inflammatory cytokine-mediated increases in ROS levels and that this effect of NMN was also blocked upon SIRT3 knockdown.
ROS can exert toxic effects on beta cells [18] and loss of SIRT3-mediated regulation of ROS production could be a mechanism driving the beta cell dysfunction observed in this model. However, despite a likely role for increased ROS production, it is not possible from the present study to determine whether impaired insulin secretion following SIRT3 knockdown occurred as a result of a primary defect in insulin secretion or as a consequence of increased beta cell apoptosis. The precise mechanisms governing increased ROS production in INS1 cells are unclear; however, in different tissues, SIRT3 has been reported to alter the acetylation status of a number of proteins linked to ROS production [20, 23, 40–43].
Through exposure to pro-inflammatory cytokines and other stimuli, beta cells can synthesise IL1β and TNFα [7, 9]. This occurs in part through ROS-mediated activation of MAPKs (ERK, JNK and p38), which in turn activate NFκB with subsequent induction of IL1β. In the present study, SIRT3 knockdown increased levels of activated ERK and JNK, and, crucially, increased IL1β levels. This pathway was directly related to impaired beta cell function, since inhibition of NFκB, a key mediator of this pathway [21, 35, 36], blocked the effects of Sirt3 siRNA on insulin secretion. We therefore propose that, during progression to type 2 diabetes, pro-inflammatory cytokines suppress SIRT3 production. This in turn leads to increased ROS-mediated induction of IL1β, and decreased beta cell function and mass (Fig. 6e). In particular, IL1β has been reported to suppress expression of the key beta cell genes Pdx1 and Mafa [44, 45], which play a crucial role in beta cell development and also function as transcription factors for other important genes, including Ins1 and Slc2a2 [31, 33, 34]. Consistent with a role for SIRT3 in the regulation of beta cell IL1β levels, knockdown of SIRT3 markedly reduced the expression of Pdx1, Mafa, Ins1 and Slc2a2, while the ability of NMN to reverse TNFα- and IL1β-mediated suppression of these genes was also blocked when SIRT3 levels were lowered. However, the situation in human islets may be further complicated by a significant SIRT3 presence in alpha cells as well as in beta cells. Further research is required to elucidate the precise role of SIRT3 in the alpha cell.
In addition to amplifying IL1β and ROS signalling, SIRT3 may also influence beta cell function and mass by other mechanisms, including deacetylation of Ku70 [46] and glutamate dehydrogenase [20, 47], and the regulation of ATP production [41].
Our previous studies have shown that, in mouse islets, SIRT1 appeared to account for about 50% of the protective effects of NMN [10]. We demonstrate here that SIRT3 also plays a key role in mediating the effects of NMN. Interestingly, the present study demonstrates that SIRT3 mediates the majority of NMN effects, as opposed to the 50% or less that would have been expected by directly extrapolating from the mouse islet study. This discrepancy may arise from the use of INS1 cells in this study, which are particularly rich in mitochondria, or also because the use of a chemical SIRT1 modulator in the previous study may have resulted in non-specific effects. Alternatively, the differences in responses to NMN may be due to as yet undetermined effects of SIRTs and NMN on other non-beta cell endocrine cells in the islet. Further studies are required to address the precise regulatory role of SIRT3 in islets and primary beta cells. However, given the similar qualitative effects of pro-inflammatory cytokines and NMN on mouse islets and INS1 cells, the present results certainly support a physiologically relevant role for SIRT3.
Interestingly, Sirt3 expression was marginally increased in islets isolated from HFD-fed insulin-resistant mice with mildly impaired islet function. Thus, SIRT3 may also play a role in mediating islet compensation of insulin resistance, such that only when SIRT3 levels decrease can impaired islet function and type 2 diabetes occur. Increased islet SIRT3 levels are consistent with other studies in skeletal muscle and liver, which reported increases in mitochondrial proteins, including SIRT3, in earlier stages of high-fat feeding [48, 49]. We also demonstrated marked reductions in SIRT3 levels in islets isolated from animals that had been fasted for 24 h. This is at odds with findings in liver, where SIRT3 levels increased following a 24 h fast [50, 51]. However, lower fasting SIRT3 levels in islets would be consistent with a co-ordinated response to fasting, whereby SIRT3 induces catabolic processes in liver, while lower SIRT3-mediated insulin secretion has a limiting effect on the insulin-mediated anabolic process.
To summarise, we propose that the decreased SIRT3 mRNA expression observed in islets isolated from human type 2 diabetic patients, and in mouse islets or INS1 cells exposed to chronic inflammatory conditions, may play a crucial role in beta cell dysfunction and type 2 diabetes, via increased cellular ROS levels, beta cell IL1β production and the onset of beta cell dysfunction. In addition, we show that many of the protective actions of exogenous NMN in beta cells are mediated by SIRT3. Thus, strategies to increase the enzymatic activity or expression of SIRT3 could lead to attractive therapies for type 2 diabetes.
Abbreviations
- ERK:
-
Extracellular signal-regulated kinase
- HFD:
-
High-fat, high-carbohydrate ‘Western diet’
- JNK:
-
c-JUN N-terminal regulated kinase
- LFD:
-
Low-fat control diet
- MAPK:
-
Mitogen-activated protein kinase
- NAMPT:
-
Nicotinamide phosphoribosyltransferase
- NFκB:
-
Nuclear factor κB
- NMN:
-
Nicotinamide mononucleotide
- ROS:
-
Reactive oxygen species
- si:
-
Small interfering
- SIRT:
-
Sirtuin
References
Kahn SE, Hull RL, Utzschneider KM (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444:840–846
Henquin JC, Cerasi E, Efendic S, Steiner DF, Boitard C (2008) Pancreatic beta-cell mass or beta-cell function? That is the question! Diabetes, Obesity & Metabolism 10(Suppl 4):1–4
Prentki M, Nolan CJ (2006) Islet beta cell failure in type 2 diabetes. J Clin Investig 116:1802–1812
Polonsky KS (2000) Dynamics of insulin secretion in obesity and diabetes. International Journal of Obesity and Related Metabolic Disorders: Journal of the International Association for the Study of Obesity 24(Suppl 2):S29–S31
Dinarello CA, Donath MY, Mandrup-Poulsen T (2010) Role of IL-1beta in type 2 diabetes. Current Opinion in Endocrinology, Diabetes, and Obesity 17:314–321
Donath MY, Boni-Schnetzler M (2010) IL-1beta activation as a response to metabolic disturbances. Cell Metabolism 12:427–428
Donath MY, Boni-Schnetzler M, Ellingsgaard H, Halban PA, Ehses JA (2010) Cytokine production by islets in health and diabetes: cellular origin, regulation and function. Trends in Endocrinology and Metabolism: TEM 21:261–267
Chou DH, Bodycombe NE, Carrinski HA et al (2010) Small-molecule suppressors of cytokine-induced beta-cell apoptosis. ACS Chem Biol 5:729–734
Lawrence MC, Naziruddin B, Levy MF, Jackson A, McGlynn K (2011) Calcineurin/nuclear factor of activated T cells and MAPK signaling induce TNF-{alpha} gene expression in pancreatic islet endocrine cells. J Biol Chem 286:1025–1036
Caton PW, Kieswich J, Yaqoob MM, Holness MJ, Sugden MC (2011) Nicotinamide mononucleotide protects against pro-inflammatory cytokine-mediated impairment of mouse islet function. Diabetologia 54:3083–3092
Imai S (2009) Nicotinamide phosphoribosyltransferase (Nampt): a link between NAD biology, metabolism, and diseases. Curr Pharm Des 15:20–28
Revollo JR, Grimm AA, Imai S (2007) The regulation of nicotinamide adenine dinucleotide biosynthesis by Nampt/PBEF/visfatin in mammals. Curr Opin Gastroenterol 23:164–170
Haigis MC, Sinclair DA (2010) Mammalian sirtuins: biological insights and disease relevance. Annual Review of Pathology 5:253–295
Ramsey KM, Mills KF, Satoh A, Imai S (2008) Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in beta cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell 7:78–88
Bordone L, Motta MC, Picard F et al (2006) Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biology 4:e31
Vetterli L, Brun T, Giovannoni L, Bosco D, Maechler P (2011) Resveratrol potentiates glucose-stimulated insulin secretion in INS-1E beta-cells and human islets through a SIRT1-dependent mechanism. J Biol Chem 286:6049–6060
Lee JH, Song MY, Song EK et al (2009) Overexpression of SIRT1 protects pancreatic beta-cells against cytokine toxicity by suppressing the nuclear factor-kappaB signaling pathway. Diabetes 58:344–351
Ma ZA, Zhao Z, Turk J (2012) Mitochondrial dysfunction and beta-cell failure in type 2 diabetes mellitus. Experimental Diabetes Research 2012:703538
Kim SC, Sprung R, Chen Y et al (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Molecular Cell 23:607–618
Lombard DB, Alt FW, Cheng HL et al (2007) Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol 27:8807–8814
Koyama T, Kume S, Koya D et al (2011) SIRT3 attenuates palmitate-induced ROS production and inflammation in proximal tubular cells. Free Radic Biol Med 51:1258–1267
Yang H, Yang T, Baur JA et al (2007) Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130:1095–1107
Jing E, Emanuelli B, Hirschey MD et al (2011) Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc Natl Acad Sci U S A 108:14608–14613
Masini M, Bugliani M, Lupi R et al (2009) Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 52:1083–1086
Bugliani M, Masini M, Liechti R et al (2009) The direct effects of tacrolimus and cyclosporin A on isolated human islets: a functional, survival and gene expression study. Islets 1:106–110
Marchetti P, Bugliani M, Lupi R et al (2007) The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia 50:2486–2494
Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG (2009) Islet-associated macrophages in type 2 diabetes. Diabetologia 52:1686–1688
Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG (2009) The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia 52:1143–1151
Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG (2010) Evidence of increased islet cell proliferation in patients with recent-onset type 1 diabetes. Diabetologia 53:2020–2028
Zhang C, Moriguchi T, Kajihara M et al (2005) MafA is a key regulator of glucose-stimulated insulin secretion. Mol Cell Biol 25:4969–4976
Butler AE, Robertson RP, Hernandez R, Matveyenko AV, Gurlo T, Butler PC (2012) Beta cell nuclear musculoaponeurotic fibrosarcoma oncogene family A (MafA) is deficient in type 2 diabetes. Diabetologia 55(11):2985–2988
Oliver-Krasinski JM, Stoffers DA (2008) On the origin of the beta cell. Genes Dev 22:1998–2021
Waeber G, Thompson N, Nicod P, Bonny C (1996) Transcriptional activation of the GLUT2 gene by the IPF-1/STF-1/IDX-1 homeobox factor. Mol Endocrinol 10:1327–1334
Fujimoto K, Polonsky KS (2009) Pdx1 and other factors that regulate pancreatic beta-cell survival. Diabetes, Obesity & Metabolism 11(Suppl 4):30–37
Donath MY, Shoelson SE (2011) Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 11:98–107
Bubici C, Papa S, Dean K, Franzoso G (2006) Mutual cross-talk between reactive oxygen species and nuclear factor-kappa B: molecular basis and biological significance. Oncogene 25:6731–6748
Giralt A, Villarroya F (2012) SIRT3, a pivotal actor in mitochondrial functions: metabolism, cell death and aging. Biochem J 444:1–10
Cheng K, Delghingaro-Augusto V, Nolan CJ, Turner N, Hallahan N, Andrikopoulos S, Gunton JE (2012) High passage MIN6 cells have impaired insulin secretion with impaired glucose and lipid oxidation. PLoS One 7:e40868
Bell EL, Guarente L (2011) The SirT3 divining rod points to oxidative stress. Molecular Cell 42:561–568
Yu W, Dittenhafer-Reed KE, Denu JM (2012) SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem 287:14078–14086
Ahn BH, Kim HS, Song S et al (2008) A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A 105:14447–14452
Qiu X, Brown K, Hirschey MD, Verdin E, Chen D (2010) Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metabolism 12:662–667
Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CF, Steegborn C (2008) Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J Mol Biol 382:790–801
Moore F, Naamane N, Colli ML et al (2011) STAT1 is a master regulator of pancreatic {beta}-cell apoptosis and islet inflammation. J Biol Chem 286:929–941
Shao C, Lawrence MC, Cobb MH (2010) Regulation of CCAAT/enhancer-binding protein homologous protein (CHOP) expression by interleukin-1 beta in pancreatic beta cells. J Biol Chem 285:19710–19719
Sundaresan NR, Samant SA, Pillai VB, Rajamohan SB, Gupta MP (2008) SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol Cell Biol 28:6384–6401
Fahie LA, Macdonald MJ (2011) The complex mechanism of glutamate dehydrogenase in insulin secretion. Diabetes 60:2450–2454
Hirschey MD, Shimazu T, Jing E et al (2011) SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Molecular Cell 44:177–190
Muoio DM, Neufer PD (2012) Lipid-induced mitochondrial stress and insulin action in muscle. Cell Metabolism 15:595–605
Caton PW, Holness MJ, Bishop-Bailey D, Sugden MC (2011) PPARalpha-LXR as a novel metabolostatic signalling axis in skeletal muscle that acts to optimize substrate selection in response to nutrient status. Biochem J 437:521–530
Hirschey MD, Shimazu T, Goetzman E et al (2010) SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464:121–125
Acknowledgements
We thank T. Maffucci and M. Turner (both Centre for Diabetes, Blizard Institute, Queen Mary University of London, London, UK) for providing INS1 cells. We also thank A. Foulis (School of Medicine, University of Glasgow, Glasgow, UK) for access to human pancreas sections and V. Rakyan (Centre for Diabetes, Blizard Institute) for providing mouse pancreas for immunofluorescence.
Funding
This research was supported by: Diabetes UK grants BDA:RD08/0003665, BDA:RD06/0003424 and BDA:RD03/0002725 (to M.J. Holness, M.C. Sugden); the European Union (project Naimit in Framework Programme 7 of the European Community, to M. Bugliani and P. Marchetti); project PEVNET (FP7/2007-2013) under grant agreement number 261441 (to N.G. Morgan); and the Northcott Devon Medical Foundation. P.W. Caton is supported by a European Foundation for the Study of Diabetes/Eli Lilly research fellowship. S.J. Richardson is in receipt of a non-clinical research fellowship from the Diabetes Research and Wellness Foundation.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
All authors contributed to the study conception and design, or analysis and interpretation of data. PWC, SJR, JK, MB, MLH, MJH and MCS contributed to the experimental work. All authors contributed to the drafting of the article or revising it critically, and to the final approval of the version to be published.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 97 kb)
ESM Table 1
(PDF 178 kb)
ESM Table 2
(PDF 171 kb)
ESM Fig. 1
Plasma glucose and insulin changes following fasting or HFD in mice. C57Bl/6 mice were fasted for 24 h; (a) plasma insulin (b) plasma glucose. A separate group of C57Bl/6 mice were maintained on a high-fat, high-carbohydrate ‘Western diet’ (HFD) or low-fat (LFD) for 16 weeks; (c) plasma insulin (d) plasma glucose and (e) liver and skeletal muscle levels of phospho(Ser473)-Akt protein. Western blots are representative. Data are expressed as mean ± SEM. Statistically significant differences between groups are indicated by: * P < 0.05, ** P < 0.01, *** P < 0.01. (PDF 60 kb)
ESM Fig. 2
The effect of exposure to SIRT3 siRNA on mRNA levels of SIRTs 1 – 7. INS1 cells were incubated with siSIRT3 (150 nM; 72 h); SIRT1, 2, 4 – 7 mRNA levels were measured by qPCR. Data are expressed as mean ± SEM (n = 4). (PDF 14 kb)
Rights and permissions
About this article
Cite this article
Caton, P.W., Richardson, S.J., Kieswich, J. et al. Sirtuin 3 regulates mouse pancreatic beta cell function and is suppressed in pancreatic islets isolated from human type 2 diabetic patients. Diabetologia 56, 1068–1077 (2013). https://doi.org/10.1007/s00125-013-2851-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-2851-y