
Abstract
Aims/hypothesis
Muscle insulin resistance, one of the earliest defects associated with type 2 diabetes, involves changes in the phosphoinositide 3-kinase/Akt network. The relative contribution of obesity vs insulin resistance to perturbations in this pathway is poorly understood.
Methods
We used phosphospecific antibodies against targets in the Akt signalling network to study insulin action in muscle from lean, overweight/obese and type 2 diabetic individuals before and during a hyperinsulinaemic–euglycaemic clamp.
Results
Insulin-stimulated Akt phosphorylation at Thr309 and Ser474 was highly correlated with whole-body insulin sensitivity. In contrast, impaired phosphorylation of Akt substrate of 160 kDa (AS160; also known as TBC1D4) was associated with adiposity, but not insulin sensitivity. Neither insulin sensitivity nor obesity was associated with defective insulin-dependent phosphorylation of forkhead box O (FOXO) transcription factor. In view of the resultant basal hyperinsulinaemia, we predicted that this selective response within the Akt pathway might lead to hyperactivation of those processes that were spared. Indeed, the expression of genes targeted by FOXO was downregulated in insulin-resistant individuals.
Conclusions/interpretation
These results highlight non-linearity in Akt signalling and suggest that: (1) the pathway from Akt to glucose transport is complex; and (2) pathways, particularly FOXO, that are not insulin-resistant, are likely to be hyperactivated in response to hyperinsulinaemia. This facet of Akt signalling may contribute to multiple features of the metabolic syndrome.
Similar content being viewed by others


Introduction
Insulin resistance is an early pathological feature of type 2 diabetes and is due to defective insulin action in muscle and liver [1–3]. Insulin resistance does not affect all the downstream actions of insulin equally and may be specific to carbohydrate metabolism [4]. Liver from a mouse model of type 2 diabetes exhibits impaired insulin regulation of gluconeogenesis but not lipogenesis [5]. This selective insulin resistance, combined with concomitant hyperinsulinaemia caused by hyperglycaemia, may potentiate hypertriglyceridaemia and hepatic steatosis, important components of the metabolic syndrome [5].
Impaired insulin regulation of glucose transport in human muscle represents a major pathological factor contributing to insulin resistance [3, 6, 7]. Defective regulation of glycogen synthesis by insulin is an additional feature of insulin-resistant muscle [8, 9]. However, insulin controls many processes in muscle. It remains unclear whether insulin resistance perturbs all of the metabolic actions of insulin in muscle or, as in liver, whether there is evidence for selective insulin resistance.
Phosphoinositide 3-kinase and Akt, two major nodes downstream of insulin receptor substrate 1 (IRS1), have been implicated in many of the metabolic actions of insulin [2]. Disruptions in this pathway have been demonstrated in human insulin resistance and type 2 diabetes [9–18]. Akt phosphorylates numerous regulatory molecules pivotal to metabolic regulation [19]. For example, phosphorylation of proline-rich Akt substrate of 40 kDa (PRAS40) is involved in protein synthesis [20, 21]; phosphorylation of Akt substrate of 160 kDa (AS160; also known as TBC1D4) on Thr642 in GLUT4 translocation [22, 23]; forkhead box O (FOXO) phosphorylation in its transcriptional activity [24]; and glycogen synthase kinase 3 (GSK3) phosphorylation in glycogen synthesis [2, 25]. There are reports that the phosphorylation of some of these substrates is defective in insulin resistance, but it is not known whether insulin resistance is universal for all of the actions of Akt in muscle.
A caveat in studying human insulin resistance is that obesity is closely associated with insulin resistance and segregating these two variables can be difficult. For example, visceral adiposity is highly correlated with insulin resistance, but it also correlates with hepatic lipid levels [26]. By separating obese individuals based on visceral adiposity vs ectopic fat depots, others have shown that hepatic lipid [26] and other ectopic fat such as intramyocellular lipid [27] are the best correlates of whole-body insulin sensitivity.
In this study, we examined insulin signalling in muscle of individuals spanning the full range of insulin sensitivity and adiposity. We hypothesised that impaired phosphorylation of Akt in insulin resistance might not uniformly affect downstream substrates of Akt, and that different pathways of insulin signalling would be differentially affected.
Methods
Participants
Volunteers responded to local advertisements. We screened 1,032 individuals by telephone and, of these, 81 were included in the current study. Those without known diabetes were screened according to BMI, HOMA of insulin resistance (HOMA-IR) [28] and a 75 g OGTT. Participants were classified into the following groups: lean insulin-sensitive controls (BMI ≤ 25 kg/m2, HOMA-IR < 2.0, n = 23), overweight/obese non-diabetic insulin-resistant individuals (Obresistant; BMI > 25 kg/m2, HOMA-IR > 3.0, n = 21), those with type 2 diabetes (BMI > 25 kg/m2, n = 21 [seven of whom were newly diagnosed]) and overweight/obese non-diabetic insulin-sensitive individuals (Obsensitive; BMI > 25 kg/m2, HOMA-IR < 1.5, n = 16). In participants not known to have diabetes, diabetes was defined as fasting blood glucose ≥7.0 mmol/l and/or a 2 h post-challenge blood glucose level ≥11.1 mmol/l. Impaired fasting glucose (IFG) was defined as a fasting blood glucose level of 5.6–6.9 mmol/l, and impaired glucose tolerance (IGT) as a 2 h post-challenge blood glucose level of 7.8–11.0 mmol/l. All those in the lean control group had normal fasting glucose levels and normal glucose tolerance. Of the 21 individuals in the Obresistant group, two demonstrated IFG, six had IGT and one had both. Two individuals in the Obsensitive group showed IGT.
Participants with known diabetes were recruited if HbA1c was <9% (75 mmol/mol), they had no clinical evidence of diabetes-related complications, had been diagnosed with diabetes for less than 5 years and were not taking medication for diabetes, except for metformin or a sulfonylurea. Metformin was stopped 2 weeks prior to the study and a sulfonylurea was added if fasting glucose readings were consistently over 7.0 mmol/l on fingerprick testing (n = 2; ceasing on the day prior to the studies). All participants smoked fewer than ten cigarettes per day, consumed less than 20 g (women) or 40 g (men) of alcohol per day, were taking no medication affecting carbohydrate metabolism, did not participate in competitive level physical activity and had had <5% change in body weight in the 3 months preceding the study. A subset of patients was taking statins; there was no difference in clamp insulin sensitivity between statin users and non-users in each group (data not shown). Menstruating women (n = 9) were studied in the follicular phase of their menstrual cycle.
Ethical approval was obtained from the St Vincent’s Hospital Health Research Ethics Committee, Sydney. Informed written consent was obtained from all participants.
Hyperinsulinaemic–euglycaemic clamp
In the 48 h prior to the study, participants were asked not to exercise or consume alcohol. Following an overnight fast, participants underwent a 2.5 h hyperinsulinaemic (80 mUm−2 min−1)–euglycaemic (5 mmol/l) clamp. The glucose infusion rate (GIR) was determined during the last 30 min of the clamp. This was normalised to fat-free mass (FFM) as measured by dual energy X-ray absorptiometry (DEXA) scanning.
Muscle biopsies
Percutaneous needle biopsies of vastus lateralis muscle were performed at baseline and 30 min and 145 min after the start of the clamp (last biopsy optional; performed in 83%, 95%, 81% and 88% of the lean control, Obresistant, type 2 diabetes and Obsensitive groups, respectively). As shown in the video (www.garvan.org.au/news-events/human-muscle-biopsy-video), by exchanging the syringe used for suction with one filled with ice-cold saline, ice-cold saline was used to gently expel muscle fragments from the Bergström needle, allowing simultaneous rapid chilling and lavage of blood. While still ice-cold, fat and connective tissue were rapidly removed and the sample was snap frozen in liquid nitrogen. Biopsies were performed through three separate incisions 5 cm apart. Muscle samples from six participants were considered unsuitable for analysis for technical reasons. Hence, muscle data are reported for 22 lean participants, 18 Obresistant participants, 19 participants with type 2 diabetes, and 16 Obsensitive participants.
Indirect calorimetry
Participants underwent indirect calorimetry (ParvoMedics, Sandy, UT, USA) at baseline after a 15 min rest and during the last 30 min of the clamp [29].
Body fat determination
Participants underwent DEXA scanning to calculate total body fat, FFM and central fat, computed tomography (CT) at the T12/L1 level to determine liver density (in Hounsfield units [HU]) as an indicator of liver fat (HU value being inversely proportional to amount of liver fat), and CT scanning at the L2/L3 and L4/L5 levels to determine abdominal visceral and subcutaneous fat areas. The mean visceral fat areas at L2/L3 and L4/L5 and the mean subcutaneous fat areas at L2/L3 and L4/L5 were used in all the analyses.
Assays and antibodies
Blood glucose was analysed using a glucose oxidase electrode (YSI Life Sciences, Yellow Springs, OH, USA). Screening insulin levels were analysed by the Advia Centaur immunoassay (Walpole, MA, USA). Clamp serum insulin levels were measured using radioimmunoassay (Linco Research, St Charles, MO, USA; intra-assay and interassay CVs 3.1–4.4% and 2.9–6.0%, respectively). NEFA were assayed using an enzymatic calorimetric method (Wako, Osaka, Japan; intra-assay and interassay CVs 0.61–0.75% and 0.75–4.91%, respectively).
Serum adiponectin, fatty acid-binding protein 4 (FABP4), lipocalin-2 and fibroblast growth factor 21 (FGF21) were measured by sandwich ELISA [30]). The intra-assay and interassay CVs for total adiponectin, FABP4, lipocalin-2 and FGF21 were, respectively: 4.4–6.9% and 3.9–7.1% [31]; 3.7–6.4% and 2.6–5.3%; 3.8–6.0% and 3.1–5.2%; and 4.2–5.6% and 5.8–7.3% [32].
Polyclonal antibodies used are described in detail in the electronic supplementary material (ESM) Methods.
Tissue processing
This is described in detail in the ESM Methods. Briefly, muscle tissue was homogenised in ice-cold homogenising buffer using a Dounce homogeniser (Sigma Aldrich, St Louis, MO, USA) and solubilised for 1 h at 4°C. Insoluble material was removed by centrifugation, and 10 μg of protein lysate was resolved by SDS-PAGE. Membranes were incubated in 5% powdered milk in Tris-buffered saline and immunoblotted with antibodies overnight at 4°C. Membranes were washed, incubated with horseradish peroxidase-labelled secondary antibodies and detected by Supersignal West Pico chemiluminescent substrate for phospho (p)AS160, pGSK and pFOXO (Millipore Corporation, Billerica, MA, USA). For Akt, pAkt474 and pAkt309, GSK, AS160, PRAS40, pPRAS40, infrared dye 700- or 800-conjugated secondary antibodies were used. Blots were quantified using Odyssey infrared imaging system software v2.0 (LI-COR Biosciences, Lincoln, NE, USA). There was no difference in muscle signalling phosphoproteins between sexes; hence, data for males and females were pooled.
Gene expression analysis
Fasting-state muscle from a subset of sex- and age-matched participants from all groups were selected for gene expression analysis. A more detailed description of analysis is available in the ESM Methods. Briefly, RNA was prepared using RNeasy protocol (Qiagen, Valencia, CA, USA). All samples were labelled with Cy5 and hybridised against Cy3-labelled universal human reference (Stratagene, Palo Alto, CA, USA). Gene expression profiles were generated by hybridising labelled samples to the Agilent’s Whole Human Genome 4 × 44 K arrays (Agilent Technologies, Santa Clara, CA, USA). Agilent’s Feature Extraction software 10.7 was used to analyse acquired array images.
All data analysis for the gene expression data was performed in the statistical analysis environment R (www.r-project.org) [33]. Data from Agilent result files were pre-processed by within-array print-tip loess normalisation [34]. Tests for a change in gene expression between two groups were performed using the moderated t test [35]. The subset of interest is a list of FOXO downstream target genes [36]. As some genes are upregulated by FOXO and some downregulated, the t values used for the Gene Set Test were multiplied by the direction of FOXO regulation so that we could test for an impairment of the FOXO pathway.
Statistical analysis
Data are reported as means ± SE, unless stated otherwise. One-way ANOVA and repeated measures ANOVA were used to compare groups; post-hoc comparisons were performed using the Tukey honestly significant difference test. Correlation analyses were carried out using the Pearson correlation measure. Where data were missing, the datasets were reduced. The minimum number of participants used for the individual pair-wise correlation measures was 64, with an average of 74. Stepwise multilinear regression was performed using a null initial model, comparing the explanatory power of incrementally larger and smaller models. Analyses were performed using SPSS v18.0 (SPSS Inc, Chicago, IL, USA) and Matlab v7.10 (MathWorksInc, Natick, MA, USA). Two-tailed p < 0.05 was taken to be statistically significant.
Results
Segregation of individuals based on BMI and HOMA
Volunteers were assigned to four metabolic groups based on diabetes status, BMI and insulin sensitivity, designated by HOMA-IR (Table 1). There was no difference between the groups in age. Obresistant and type 2 diabetic individuals had a higher BMI, waist circumference and WHR ratio than lean controls. A major strength of our study was the inclusion of a fourth group of Obsensitive individuals, who were as insulin-sensitive (HOMA-IR 1.2 ± 0.3) as the lean group (HOMA-IR 1.2 ± 0.4).
Hyperinsulinaemic–euglycaemic clamps were performed on all participants, generating insulin levels during clamp steady state of 1,632 ± 288, 2,004 ± 564, 2,148 ± 720 and 1,938 ± 612 pmol/l in lean controls, Obresistant, type 2 diabetic and Obsensitive participants, respectively. These insulin concentrations have been shown to suppress hepatic glucose output fully in non-obese healthy and insulin-resistant individuals and those with IGT, and to suppress hepatic glucose production by 75% in individuals with poorly controlled type 2 diabetes [37, 38]. As diabetes was well controlled in the current study (HbA1c 6.1 ± 0.6%, or 43 ± 7 mmol/mol), hepatic glucose production is likely to have been fully suppressed during the clamp. Hence, the GIR at steady state is a measure of predominantly peripheral (muscle) insulin sensitivity.
GIR (normalised to FFM) was significantly lower in Obresistant and type 2 diabetic participants compared with lean controls (Fig. 1a). Obsensitive individuals had similar GIRs to lean controls, despite having almost double the amount of total and central fat.

(a) GIR corrected for FFM, (b) visceral fat, (c) liver density and (d) subcutaneous abdominal fat. Black bars, lean insulin-sensitive individuals; light grey bars, overweight/obese insulin-resistant individuals; white bars, individuals with type 2 diabetes; dark grey bars, overweight/obese insulin-sensitive individuals. Horizontal lines linking bars indicate statistically significant (p < 0.05) differences between the groups. HU values are inversely related to amount of liver fat present. Data are means ± SE
Obsensitive individuals have reduced hepatic lipid levels
All measures of total and central fat were higher in Obresistant, type 2 diabetic and Obsensitive individuals compared with lean controls (Table 1, Fig. 1b, d). Although Obresistant participants had a higher BMI than type 2 diabetic and Obsensitive participants, these three groups were matched for percentage total body fat by DEXA (Table 1). There was no difference in subcutaneous fat (by CT) between the Obresistant and Obsensitive groups (Table 1, Fig. 1d). Obresistant individuals had significantly more visceral fat than Obsensitive individuals, who had similar visceral fat content to those in the group with type 2 diabetes (Fig. 1b). In contrast, liver density in HU (an inverse measurement of hepatic fat content) was significantly higher in Obsensitive than type 2 diabetic or Obresistant individuals and was similar to that observed in lean controls (Fig. 1c).
Insulin-stimulated Akt phosphorylation correlates with whole-body insulin sensitivity
Phosphorylation of Akt at Ser474 and Thr309 coincides with full kinase activation [39], and this is frequently used as a surrogate for Akt activity. In lean individuals, Akt phosphorylation at Ser474 was increased nine- and 14-fold over basal levels at 30 min and 145 min, respectively, after commencement of the clamp. Corresponding increases for Thr309 phosphorylation were eight- and 13-fold, respectively. Insulin-induced Akt phosphorylation at Ser474 was reduced by 30% and 41% at 145 min in Obresistant and type 2 diabetic individuals, respectively, compared with lean controls (Fig. 2). This was not observed in the Obsensitive group (Fig. 2). An even greater impairment was observed in insulin-stimulated Akt Thr309 phosphorylation in Obresistant and type 2 diabetic participants (Fig. 2), but no defect was observed in the Obsensitive group. There was no difference in total Akt levels (Fig. 3a, c) or the loading control (14-3-3β, Fig. 3b) among the groups. The ratio of phosphorylated to total protein levels resulted in similar findings, albeit with less statistical significance as a result of the combined error of both measurements (data not shown).

(a–f) Phosphorylation of insulin signalling intermediates at baseline, 30 min and 145 min after commencement of insulin. Black bars, lean insulin-sensitive individuals; light grey bars, overweight/obese insulin-resistant individuals; white bars, individuals with type 2 diabetes; dark grey bars, overweight/obese insulin-sensitive individuals. Horizontal lines linking bars indicate statistically significant (p < 0.05) differences between the groups. Data are means ± SE. Fold changes are in comparison to lean baseline levels. (g–l) Representative western blots

(a) Total levels of insulin signalling intermediates at baseline. Black bars, lean insulin-sensitive individuals; light grey bars, overweight/obese insulin-resistant individuals; white bars, individuals with type 2 diabetes; dark grey bars, overweight/obese insulin-sensitive individuals. (b–f) Representative 14-3-3β and total western blots
Insulin-stimulated phosphorylation of Akt at Ser474 and Thr309 correlated strongly with GIR/FFM across the entire cohort (Fig. 4a, ESM Table 1, ESM Fig. 1). Significant correlations were also found between insulin-stimulated phosphorylation of Akt and liver fat content and visceral fat (Fig. 4a, ESM Table 1). Correlations were similar when men and women were analysed separately (Fig. 4b).

Pearson correlation analyses between metabolic variables. The levels of correlation between the different pairs of variables are indicated as positive (direct) relationships in red and inverse correlations in green. Only those relationships which had significant correlations between the two variables (p < 0.05) are shown. (a) All participants; (b) males alone (upper triangles) and females alone (lower triangles). HU values were inversely related to amount of liver fat present
Lack of concordance between Akt phosphorylation and Akt substrate phosphorylation
We observed robust changes in GSK3, PRAS40, FOXO and AS160/TBC1D4 phosphorylation in lean individuals during the clamp (Fig. 2). There was little correspondence between insulin-dependent phosphorylation of Akt substrates and Akt itself across the different groups.
AS160 phosphorylation was significantly reduced in both Obresistant and Obsensitive individuals at the 145 min time point, and non-significantly reduced in type 2 diabetic individuals (Fig. 2). The pattern of GSK3 and PRAS40 phosphorylation mirrored that of pAS160. GSK3 phosphorylation at 145 min and PRAS40 phosphorylation at 30 min were significantly reduced in the Obresistant group only (Fig. 2). In contrast, no defect was observed in FOXO phosphorylation in any of the groups (Fig. 2). There was no difference in the total level of any of the Akt substrates examined between any of the groups at baseline (Fig. 3) or during insulin stimulation (data not shown).
Insulin-stimulated AS160 and PRAS40 phosphorylation were not related to GIR/FFM, visceral fat or hepatic fat (Fig. 4a, ESM Table 1). However, insulin-stimulated AS160 was inversely associated, albeit of borderline statistical significance, with subcutaneous fat. GSK3 phosphorylation at 145 min was positively correlated with GIR/FFM and inversely correlated with visceral fat (Fig. 4a, ESM Table 1). Insulin-stimulated FOXO phosphorylation at 145 min correlated directly with visceral fat, but was not related to GIR/FFM or other adiposity variables (Fig. 4a, ESM Table 1).
Visceral fat correlated directly with baseline FOXO phosphorylation only. Fasting insulin was directly related to baseline FOXO and PRAS40 phosphorylation, but not baseline AS160 or GSK3 phosphorylation.
FOXO gene expression
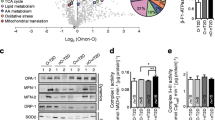
To test our hypothesis that compensatory hyperinsulinaemia may result in hyperphosphorylation of FOXO and inhibition of its activity, we examined the expression of FOXO-regulated genes [36] in muscle biopsies. We combined data from the Obresistant and type 2 diabetic individuals (insulin-resistant [IR] group) and compared this with lean and Obsensitive individuals (insulin-sensitive [IS] group). Consistent with our hypothesis, basal FOXO phosphorylation was 1.5-fold higher in the IR group (p = 0.03) (Fig. 5a) who exhibited relative basal hyperinsulinaemia.

Basal FOXO phosphorylation is increased and FOXO target genes are dysregulated in Obresistant/type 2 diabetic compared with Obsensitive/lean individuals. (a) Basal FOXO phosphorylation is increased 1.54-fold in Obresistant/type 2 diabetic compared with Obsensitive/lean individuals (p = 0.031). (b) Gene set enrichment analysis of basal FOXO target gene expression in Obresistant/type 2 diabetic vs Obsensitive/lean individuals determined that these genes were significantly dysregulated, with a Gene Set Test (p = 0.037). The 15 most dysregulated FOXO target genes are shown. The y-axis represents the moderated t statistics, which represents a measure of the fold change divided by the standard error. Ten of the 15 genes are associated with insulin action (EIF4EBP1, INSR, PPP2R4, PIK3CA), autophagy (MAP1LC3A, ATG9B), oxidative stress response (PRDX3, SOD2) and metabolism (CITED2, PPARGC1A). Error bars represent 1 SD. AU, arbitrary units; OIS, Obsensitive; OIR, Obresistant
The expression of genes in the FOXO target set was significantly dysregulated (p = 0.037) in the IR group based on a Gene Set Test. Ten out of the 15 most dysregulated genes (Fig. 5b) are associated with insulin action, autophagy, oxidative stress response or metabolism. The expression of the FOXO target gene 4EBP1, an insulin-regulated phosphoprotein, was reduced by 28%. This gene was one of only 18 genes significantly modified in insulin-resistant individuals when we analysed changes at a single-gene level. These data indicate that FOXO function is chronically repressed in the skeletal muscle of individuals with insulin resistance and/or type 2 diabetes.
Circulating factors
Compared with lean individuals, adiponectin levels were lower in Obresistant and type 2 diabetic individuals, yet similar in Obsensitive individuals (Fig. 6a). FGF21 levels were higher in diabetic participants than controls at baseline (but not during the clamp) (Fig. 6b). Lipocalin 2 levels were similar across the groups, with the exception of lower lipocalin 2 in Obsensitive compared with diabetic participants at baseline (Fig. 6c). In contrast, FABP4 was higher in the three overweight/obese groups compared with the lean controls (Fig. 6d). Only FGF21 levels increased with insulin (Fig. 6).

Adipokines at baseline and steady state of the hyperinsulinaemic–euglycaemic clamp. Black bars, lean insulin-sensitive individuals; light grey bars, overweight/obese insulin-resistant individuals; white bars, individuals with type 2 diabetes; dark grey bars, overweight/obese insulin-sensitive individuals. Horizontal lines linking bars indicate statistically significant (p < 0.05) differences between the groups. Data are means ± SE
Both fasting adiponectin and FGF21 levels correlated strongly with GIR/FFM, liver fat and visceral fat, and with each other (all p < 0.001), but not with subcutaneous fat (Fig. 4a). When these relationships were examined in men and women separately, the correlation between GIR/FFM and adiponectin was limited to females (Fig. 4b). Lipocalin 2 correlated, albeit weakly, with subcutaneous fat (p = 0.04), but not with GIR/FFM, liver fat or visceral fat. FABP4 correlated strongly with both visceral and subcutaneous fat (p < 0.001), but not with GIR/FFM.
Stepwise multilinear regression
Stepwise multilinear regression was performed in the whole cohort, inclusive of the variables that were significantly correlated with GIR/FFM in univariate analysis (liver fat, visceral fat, baseline FOXO phosphorylation, insulin-stimulated phosphorylation of Akt309, Akt474 and GSK3 at 145 min, NEFA level at 30 min and fasting adiponectin and FGF21 levels [Fig. 3]). GIR/FFM was best predicted by insulin-stimulated phosphorylation of Akt309 at 145 min (β = 3.3511 ± 0.9746, p = 0.0011), NEFA levels at 30 min into the clamp (β = −183.5030 ± 62.5, p = 0.0048), fasting adiponectin (β = 0.7037 ± 0.2894, p = 0.0182) and fasting FGF21 (β = −0.0667 ± 0.0301, p = 0.0306) (model adjusted R 2 0.49, F 15.8511, p < 0.0001). Visceral adipose tissue and liver fat were not significantly related to GIR/FFM when accounting for the effect of the other variables.
Discussion
Dissecting the molecular mechanisms that contribute to insulin resistance is challenging. First, insulin resistance is closely linked to many factors, including obesity, liver lipid and low-grade inflammation, and it is difficult to disentangle their individual contributions to metabolic disease. Second, while it is likely that insulin resistance stems from a defect in the insulin signalling pathway, this pathway is a complex network. For example, although the Akt pathway controls lipid and carbohydrate metabolism in the liver, a defect in Akt signalling in type 2 diabetes selectively targets carbohydrate but not lipid metabolism [4, 5]. In the current study, inclusion of a group of obese insulin-sensitive individuals enabled us to show that the defect in insulin-stimulated Akt phosphorylation in skeletal muscle was related to whole-body insulin sensitivity (ESM Fig. 1) and not obesity. Strikingly, the Akt defect was not accompanied by a similar defect in the insulin-dependent phosphorylation of four major Akt substrates known to control essential downstream actions of insulin. Defective phosphorylation of GSK3 and AS160 was observed, but this correlated with obesity rather than insulin sensitivity. Conversely, no defect was observed in FOXO phosphorylation.
The defect in insulin-dependent Akt phosphorylation in muscle of insulin-resistant or type 2 diabetic humans is consistent with previous studies [9–17]. Some studies have failed to report defective Akt phosphorylation [12, 13, 40], perhaps due to differences in experimental design. Notably, in the development of the current study, we observed considerable variation in Akt phosphorylation depending upon the method of tissue collection and processing used, which required considerable optimisation to overcome.
Few studies have examined phosphorylation of downstream Akt substrates in insulin-resistant and type 2 diabetic individuals. We have examined the phosphorylation of four different Akt substrates that more comprehensively encapsulate a range of important Akt functions. Of note, the dynamic range of insulin-stimulated phosphorylation of these substrates was very robust in our study compared with other reports (e.g. a sixfold rise in AS160 [9, 15, 17]), which may explain the variation in results between these different studies [9, 14, 15, 41]. While we observed defective insulin-dependent AS160 phosphorylation in insulin-resistant individuals (Fig. 2), a similar defect was also observed in obese insulin-sensitive individuals. This is intriguing because reduced insulin-regulated glucose transport is one of the major metabolic defects in skeletal muscle from insulin-resistant and type 2 diabetic individuals, and Akt-dependent AS160 phosphorylation plays a key role in insulin-regulated glucose transport in muscle [42, 43].
Others have found that insulin resistance may be contributed to by defects in glycogen synthesis [44]. We found that non-oxidative glucose disposal, a reflection of glycogen synthesis, was significantly lower in those with insulin resistance (Table 1), and that phosphorylation of GSK3 correlated with insulin sensitivity measured by clamp (Pearson R 0.26, p = 0.03; ESM Table 1). However, in the Obsensitive group there is a discordance between Akt phosphorylation (as high as the lean sensitive group) and GSK3 phosphorylation (as low as the type 2 diabetes group), making it difficult to draw conclusions on the role of defective GSK3 phosphorylation in the development of insulin resistance. This may be because glycogen synthase activity may be regulated by non-insulin mediated mechanisms such as exercise [45] or allosteric changes independent of GSK3 [46].
FOXO phosphorylation is apparently preserved despite the clear defect in Akt phosphorylation, perhaps because FOXO phosphorylation occurs at submaximal levels of Akt phosphorylation [47, 48], and particularly if FOXO phosphorylation remains more sensitive to insulin stimulation than other Akt pathways. We measured phosphorylation of FOXO1 on Ser256, a site predominantly phosphorylated by Akt, and to a lesser extent serum/glucocorticoid inducible kinase. Other kinases are known to phosphorylate FOXO at alternate sites, including mammalian sterile 20-like kinase (MST), cyclin-dependent kinases, AMP-activated protein kinase (AMPK), extracellular signal-related kinase (ERK) and IκB kinase (IKK) [49]. In contrast, under fasting conditions, where insulin levels were significantly elevated in insulin-resistant and type 2 diabetic individuals, there was a concomitant increase in basal FOXO phosphorylation. This behaviour was not seen for the other Akt substrates, suggesting that the sensitivity of FOXO to insulin (and hyperinsulinaemia) is unimpaired.
Insulin-dependent FOXO phosphorylation leads to the accumulation of FOXO in the cytosol, concomitant with reduced transcriptional activity. We hypothesised that a non-defective pathway under clamp conditions would be hyperphosphorylated due to the compensatory hyperinsulinaemia [48]. FOXO hyperphosphorylation would then be accompanied by the impaired expression of FOXO target genes, which we found in gene expression analysis. The elevated insulin response of FOXO compared with other Akt substrates suggests that there is selective insulin resistance, even within the Akt pathway itself, as previously reported in other pathways [4, 47, 50]. Selective insulin resistance in the face of persistent hyperinsulinemia is likely to result in previously unappreciated demands on metabolic control as well as having implications for the treatment of type 2 diabetes. For example, hyperphosphorylation of FOXO will result in dysregulation of FOXO target genes, many of which are stress-response genes, including SOD2 and LC3. In the long term, this is likely to impair stress defence pathways. In these circumstances, treatments aimed at improving insulin secretion without correcting the selective insulin resistance may be ill advised.
A similar discordance between pFOXO, Akt and GLUT4 translocation has been reported in 3T3-L1 adipocytes [47]. The mechanism may be related to the differential dose response relationship between pAkt and its various downstream substrates [48], or the temporal pattern of the insulin signal shown in modelling studies [51].
Insulin sensitivity by clamp was highly correlated with liver fat, adiponectin and Akt phosphorylation (R 0.53, 0.55 and 0.54, respectively [ESM Table 1]). However, known downstream targets of Akt did not follow the same pattern. Liver fat was a key distinguishing feature between Obresistant and Obsensitive participants. Whether it is a cause of insulin resistance or a consequence of secondary hyperinsulinaemia is unknown. Apart from hepatic fat, other depots of ectopic adiposity may also be contributing to the difference seen between Obresistant and Obsensitive individuals, although these were not measured in the current study.
In conclusion, this study highlights the striking non-linearity in the Akt signalling pathway in muscle. These findings suggest that, in insulin resistance, some non-carbohydrate related insulin responses are unimpeded, and may even be hyperstimulated due to hyperinsulinaemia, which may contribute to development of the metabolic syndrome and other complications. In view of the selective nature of insulin resistance, it seems evident that treatments designed to enhance insulin secretion, independently of other adaptations, may be counterproductive.
Abbreviations
- AS160:
-
Akt substrate of 160 kDa
- CT:
-
Computed tomography
- DEXA:
-
Dual energy X-ray absorptiometry
- FABP4:
-
Fatty acid-binding protein 4
- FFM:
-
Fat-free mass
- FGF21:
-
Fibroblast growth factor 21
- FOXO:
-
Forkhead box O
- GIR:
-
Glucose infusion rate
- GSK3:
-
Glycogen synthase kinase 3
- HOMA-IR:
-
HOMA of insulin resistance
- HU:
-
Hounsfield units
- IFG:
-
Impaired fasting glucose
- IGT:
-
Impaired glucose tolerance
- IR group:
-
Insulin resistant group
- IS group:
-
Insulin sensitive group
- Obresistant :
-
Overweight/obese non-diabetic insulin-resistant individuals
- Obsensitive :
-
Overweight/obese non-diabetic insulin-sensitive individuals
- PDX1:
-
Pancreatic and duodenal homeobox 1
- PRAS40:
-
Proline-rich Akt substrate of 40 kDa
References
Samuel VT, Petersen KF, Shulman GI (2010) Lipid-induced insulin resistance: unravelling the mechanism. Lancet 375:2267–2277
Taniguchi CM, Emanuelli B, Kahn CR (2006) Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 7:85–96
Petersen KF, Shulman GI (2006) Etiology of insulin resistance. Am J Med 119:S10–S16
Li S, Brown MS, Goldstein JL (2010) Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A 107:3441–3446
Biddinger SB, Hernandez-Ono A, Rask-Madsen C et al (2008) Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab 7:125–134
Shulman GI, Rothman DL, Jue T, Stein P, DeFronzo RA, Shulman RG (1990) Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N Engl J Med 322:223–228
Rothman DL, Shulman RG, Shulman GI (1992) 31P nuclear magnetic resonance measurements of muscle glucose-6-phosphate. Evidence for reduced insulin-dependent muscle glucose transport or phosphorylation activity in non-insulin-dependent diabetes mellitus. J Clin Invest 89:1069–1075
Thorburn AW, Gumbiner B, Bulacan F, Wallace P, Henry RR (1990) Intracellular glucose oxidation and glycogen synthase activity are reduced in non-insulin-dependent (type II) diabetes independent of impaired glucose uptake. J Clin Invest 85:522–529
Hojlund K, Birk JB, Klein DK et al (2009) Dysregulation of glycogen synthase COOH- and NH2-terminal phosphorylation by insulin in obesity and type 2 diabetes mellitus. J Clin Endocrinol Metab 94:4547–4556
Brozinick JT Jr, Roberts BR, Dohm GL (2003) Defective signaling through Akt-2 and −3 but not Akt-1 in insulin-resistant human skeletal muscle: potential role in insulin resistance. Diabetes 52:935–941
Cozzone D, Frojdo S, Disse E et al (2008) Isoform-specific defects of insulin stimulation of Akt/protein kinase B (PKB) in skeletal muscle cells from type 2 diabetic patients. Diabetologia 51:512–521
Meyer MM, Levin K, Grimmsmann T, Beck-Nielsen H, Klein HH (2002) Insulin signalling in skeletal muscle of subjects with or without type II-diabetes and first degree relatives of patients with the disease. Diabetologia 45:813–822
Krook A, Roth RA, Jiang XJ, Zierath JR, Wallberg-Henriksson H (1998) Insulin-stimulated Akt kinase activity is reduced in skeletal muscle from NIDDM subjects. Diabetes 47:1281–1286
Hojlund K, Glintborg D, Andersen NR et al (2008) Impaired insulin-stimulated phosphorylation of Akt and AS160 in skeletal muscle of women with polycystic ovary syndrome is reversed by pioglitazone treatment. Diabetes 57:357–366
Vind BF, Pehmoller C, Treebak JT et al (2011) Impaired insulin-induced site-specific phosphorylation of TBC1 domain family, member 4 (TBC1D4) in skeletal muscle of type 2 diabetes patients is restored by endurance exercise-training. Diabetologia 54:157–167
Bandyopadhyay GK, Yu JG, Ofrecio J, Olefsky JM (2005) Increased p85/55/50 expression and decreased phosphotidylinositol 3-kinase activity in insulin-resistant human skeletal muscle. Diabetes 54:2351–2359
Karlsson HK, Zierath JR, Kane S, Krook A, Lienhard GE, Wallberg-Henriksson H (2005) Insulin-stimulated phosphorylation of the Akt substrate AS160 is impaired in skeletal muscle of type 2 diabetic subjects. Diabetes 54:1692–1697
Friedrichsen M, Poulsen P, Richter EA et al (2010) Differential aetiology and impact of phosphoinositide 3-kinase (PI3K) and Akt signalling in skeletal muscle on in vivo insulin action. Diabetologia 53:1998–2007
Rowland AF, Fazakerley DJ, James DE (2011) Mapping insulin/GLUT4 circuitry. Traffic 12:672–681
Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH (2007) Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 9:316–323
Kovacina KS, Park GY, Bae SS et al (2003) Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J Biol Chem 278:10189–10194
Sano H, Kane S, Sano E et al (2003) Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem 278:14599–14602
Ramm G, Larance M, Guilhaus M, James DE (2006) A role for 14-3-3 in insulin-stimulated GLUT4 translocation through its interaction with the RabGAP AS160. J Biol Chem 281:29174–29180
Brunet A, Bonni A, Zigmond MJ et al (1999) Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 96:857–868
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378:785–789
Fabbrini E, Magkos F, Mohammed BS et al (2009) Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc Natl Acad Sci U S A 106:15430–15435
Stefan N, Kantartzis K, Machann J et al (2008) Identification and characterization of metabolically benign obesity in humans. Arch Intern Med 168:1609–1616
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC (1985) Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28:412–419
Greenfield JR, Samaras K, Hayward CS, Chisholm DJ, Campbell LV (2005) Beneficial postprandial effect of a small amount of alcohol on diabetes and cardiovascular risk factors: modification by insulin resistance. J Clin Endocrinol Metab 90:661–672
Xu A, Wang Y, Xu JY et al (2006) Adipocyte fatty acid-binding protein is a plasma biomarker closely associated with obesity and metabolic syndrome. Clin Chem 52:405–413
Milner KL, van der Poorten D, Xu A et al (2009) Adipocyte fatty acid binding protein levels relate to inflammation and fibrosis in nonalcoholic fatty liver disease. Hepatology 49:1926–1934
Yu H, Xia F, Lam KS et al (2011) Circadian rhythm of circulating fibroblast growth factor 21 is related to diurnal changes in fatty acids in humans. Clin Chem 57:691–700
R_Development_Core_Team (2011) R: a language and environment for statistical computing. R Foundation for Statistical Computing Vienna, Austria (http://www.R-project.org/)
Yang YH, Dudoit S, Luu P et al (2002) Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res 30:e15
Smyth GK (2004) Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article3
van der Vos KE, Coffer PJ (2011) The extending network of FOXO transcriptional target genes. Antioxid Redox Signal 14:579–592
Kolterman OG, Gray RS, Griffin J et al (1981) Receptor and postreceptor defects contribute to the insulin resistance in noninsulin-dependent diabetes mellitus. J Clin Invest 68:957–969
Milner KL, van der Poorten D, Trenell M et al (2010) Chronic hepatitis C is associated with peripheral rather than hepatic insulin resistance. Gastroenterology 138(932–941):e931–e933
Alessi DR, Andjelkovic M, Caudwell B et al (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15:6541–6551
Kim YB, Nikoulina SE, Ciaraldi TP, Henry RR, Kahn BB (1999) Normal insulin-dependent activation of Akt/protein kinase B, with diminished activation of phosphoinositide 3-kinase, in muscle in type 2 diabetes. J Clin Invest 104:733–741
Plomgaard P, Bouzakri K, Krogh-Madsen R, Mittendorfer B, Zierath JR, Pedersen BK (2005) Tumor necrosis factor-alpha induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes 54:2939–2945
Chen S, Wasserman DH, MacKintosh C, Sakamoto K (2011) Mice with AS160/TBC1D4-Thr649Ala knockin mutation are glucose intolerant with reduced insulin sensitivity and altered GLUT4 trafficking. Cell Metab 13:68–79
Cho H, Mu J, Kim JK et al (2001) Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 292:1728–1731
Friedman JE, Caro JF, Pories WJ, Azevedo JL Jr, Dohm GL (1994) Glucose metabolism in incubated human muscle: effect of obesity and non-insulin-dependent diabetes mellitus. Metabolism 43:1047–1054
Jensen J, Tantiwong P, Stuenaes JT et al (2012) Effect of acute exercise on glycogen synthase in muscle from obese and diabetic subjects. Am J Physiol Endocrinol Metab 303:E82–E89
Bouskila M, Hunter RW, Ibrahim AF et al (2010) Allosteric regulation of glycogen synthase controls glycogen synthesis in muscle. Cell Metab 12:456–466
Gonzalez E, Flier E, Molle D, Accili D, McGraw TE (2011) Hyperinsulinemia leads to uncoupled insulin regulation of the GLUT4 glucose transporter and the FoxO1 transcription factor. Proc Natl Acad Sci U S A 108:10162–10167
Tan SX, Ng Y, Meoli CC et al (2012) Amplification and demultiplexing in insulin-regulated Akt protein kinase pathway in adipocytes. J Biol Chem 287:6128–6138
Xie Q, Chen J, Yuan Z (2012) Post-translational regulation of FOXO. Acta Biochim Biophys Sin (Shanghai) 44:897–901
Gogg S, Smith U, Jansson PA (2009) Increased MAPK activation and impaired insulin signaling in subcutaneous microvascular endothelial cells in type 2 diabetes: the role of endothelin-1. Diabetes 58:2238–2245
Kubota H, Noguchi R, Toyoshima Y et al (2012) Temporal coding of insulin action through multiplexing of the AKT pathway. Mol Cell 46:820–832
Acknowledgements
We would like to thank A. Douglas and O. Wong (Clinical Research Facility, Garvan Institute of Medical Research, Sydney), study nurses, for their assistance in performing these studies. We also acknowledge J. Freund, P. Speight and M. Lee (Department of Radiology, St Vincent’s Hospital, Sydney) for analysis of CT and DEXA scans.
Funding
The study was funded by a Diabetes Australia Research Trust Millennium Grant and, in part, by a National Health and Medical Research Council (NHMRC) Program Grant. K. T. Tonks is funded by an NHMRC Postgraduate Research Scholarship. A. Xu is supported by a Collaborative Research Fund (HKU4/CRF10) from the Research Council of Hong Kong. D. E. James is an NHMRC Senior Principal Research Fellow. J. R. Greenfield is the recipient of an NHMRC Neil Hamilton Fairley Fellowship and the Don Chisholm Fellowship (funds from Garvan Research Foundation, including support from GlaxoSmithKline, Australia, Diabetes Australia Research Trust, the Commonwealth Department of Health and Ageing). No funding bodies had any role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Duality of interest
J. R, Junutula, Z. Modrusan and G. Kolumam are employees of Genentech Inc., South San Francisco, California 94080, USA.
Contribution statement
KTT, DJC, DEJ and JRG designed the studies. KTT performed the clinical studies. KTT, YN, AM, DS-B, TJI, AX, EP, JYHY, JRJ, ZM and GK performed laboratory analysis of the data collected. KTT, ACFC and JS analysed the data. KTT, YN, SM, ACFC, DS-B, JS, DJC, DEJ and JRG interpreted the data. KTT, DJC, DEJ and JRG wrote the first draft of the manuscript. All authors edited and approved the final manuscript. JRG and DEJ are the guarantors of this work, had full access to all the data, and take full responsibility for the integrity of data and the accuracy of data analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
D. E. James and J. R. Greenfield contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 88.3 kb)
ESM Fig. 1
Correlation between phosphorylated Akt at 145 minutes after insulin exposure, and insulin sensitivity measured by hyperinsulinaemie-euglycaemic clamp (GIR/FFM). (a) pAkt-Ser474, R=0.29, p=0.0127 (b) pAkt-Thr309, R=0.53, p<0.0001 (PDF 44 kb)
ESM Table 1
(PDF 63 kb)
Rights and permissions
About this article
Cite this article
Tonks, K.T., Ng, Y., Miller, S. et al. Impaired Akt phosphorylation in insulin-resistant human muscle is accompanied by selective and heterogeneous downstream defects. Diabetologia 56, 875–885 (2013). https://doi.org/10.1007/s00125-012-2811-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2811-y