
Abstract
Aims/hypothesis
Rotavirus infection in at-risk children correlates with production of serum autoantibodies indicative of type 1 diabetes progression. Oral infection with rhesus monkey rotavirus (RRV) accelerates diabetes onset in mice. This relates to their rotavirus-specific serum antibody titre and local pro-inflammatory cytokine induction without pancreatic infection. Our aim was to further investigate the roles of serum antibodies and viral extra-intestinal spread in diabetes acceleration by rotavirus.
Methods
Rotavirus-specific serum antibody production was detected by ELISA in diabetes-prone mice given either inactivated or low-dose RRV, in relation to their diabetes development. Serum anti-rotavirus antibody titres and infectious virus in lymph nodes were measured in mice given RRV or porcine rotavirus CRW-8. In lymph node cells, rotavirus antigen presence and immune activation were determined by flow cytometry, in conjunction with cytokine mRNA levels.
Results
Acceleration of diabetes by RRV required virus replication, which correlated with antibody presence. CRW-8 induced similar specific total immunoglobulin and IgA titres to those induced by RRV, but did not accelerate diabetes. RRV alone elicited specific serum IgG antibodies with a T helper (Th)1 bias, spread to regional lymph nodes and activated antigen-presenting cells at these sites. RRV increased Th1-specific cytokine expression in pancreatic lymph nodes. Diabetes onset was more rapid in the RRV-infected mice with the greater Th1 bias.
Conclusions/interpretation
Acceleration of murine diabetes by rotavirus is virus strain-specific and associated with virus spread to regional lymph nodes, activation of antigen-presenting cells at these sites and induction of a Th1-dominated antibody and cytokine response.
Similar content being viewed by others

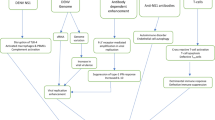

Introduction
The preclinical phase of type 1 diabetes is marked by immune cell infiltration into pancreatic islets, termed insulitis, and has been extensively studied in NOD/Lt (NOD) mice, which develop overt diabetes from 12 weeks of age [1, 2]. Priming of diabetogenic T cells within the pancreatic lymph nodes (PLN) is essential for the development of diabetes in the NOD mouse [3, 4]. Gut-associated lymph nodes also contain diabetogenic T cells, suggesting that T cell activation also occurs at these sites [5]. Gut-derived and islet antigens artificially delivered via the gut are preferentially presented to T cells in the PLN, further implicating the gut–PLN nexus in diabetes development [6].
Although viruses, particularly gastrointestinal pathogens, are considered to play key roles in modulating the development of diabetes in humans, a direct causal relationship is yet to be defined [7, 8]. Viruses delay or accelerate the onset of diabetes in animal models, not necessarily through pancreatic infection. Some viruses may cause beta cell death indirectly by altering immune cell function outside the pancreas [7, 9].
The gastrointestinal pathogen rotavirus is associated with exacerbations in islet autoantibodies in children genetically at risk of type 1 diabetes [10, 11]. Rotavirus infection in at-risk children also relates to increased bovine insulin antibodies, possibly due to enhanced intestinal permeability and dietary insulin uptake [12]. Infection of insulitic older adult NOD mice with rhesus monkey rotavirus (RRV) accelerates the onset of diabetes. This is preceded by elevated Tnf mRNA in islets [13]. As in humans, NOD mouse autoimmune responses target the islet-specific glucose 6-phosphatase catalytic subunit-related protein (IGRP). Transgenic NOD8.3 TCR (NOD8.3) mice, with >90% of their CD8+ T cells specific for a diabetogenic IGRP epitope, provide a model of rapid diabetes development [14, 15]. RRV infection of adult NOD8.3 mice increases islet Tnf mRNA levels and MHC I expression on islet beta cells, and accelerates diabetes onset. Infection of adult NOD and NOD8.3 mice is asymptomatic and aviraemic, and direct pancreatic infection is not involved in acceleration of diabetes by RRV [13]. In contrast, RRV infection of infant NOD mice induces diarrhoea, viraemia and pancreatic infection and delays diabetes onset [16].
Acceleration of murine diabetes by RRV correlates with the specific serum antibody titre, implicating the systemic immune response [13]. Cytokines produced by CD4+ T lymphocyte subsets (T helper, Th) 1 and Th2 differentially regulate the immune response, with beta cell destruction in NOD mice linked to a Th1-dominated response [17, 18]. As murine Th1 and Th2 cells specifically induce antigen-specific B cells to secrete IgG2a and IgG1, respectively, the relative levels of these IgG subclasses in serum represents the dominance of Th1 vs Th2 responses [18]. RRV infection in adult BALB/c mice induces a Th1 response by Peyer’s patch cells and a predominance of RRV-specific IgG2a [19]. We propose that induction of a Th1 response in NOD mice may be a significant factor in diabetes acceleration by rotavirus.
Childhood rotavirus gastroenteritis is associated with the presence of the rotavirus genome in the mesenteric lymph nodes (MLN) [20]. Similarly, MLN in infected infants of diabetes-resistant mice harbour infectious RRV. The cells in the MLN involved are unidentified, although mouse rotavirus replicates in dendritic cells (DC), and possibly B cells and macrophages [21, 22]. The spread of rotavirus to the MLN and PLN in adult mice has not been reported. The importance of the MLN for immune responses to rotavirus, PLN and gut for diabetogenic T cell activation and systemic immune response for RRV-mediated diabetes acceleration show the necessity for this analysis.
The main aim of this study was to obtain a more complete understanding of the involvement of rotavirus-specific serum antibody responses in the acceleration of diabetes and to analyse the Th1 response through measurement of specific serum IgG isotypes and measurement of cytokines in PLN and islets. As efficient rotavirus spread to the MLN and PLN, and activation of immune cells at these sites, would be expected to elicit a strong systemic immune response, these also were analysed. We compared the impact of infection by RRV or the related porcine rotavirus CRW-8 on these variables in diabetes-prone mice, in relation to their diabetes development.
Methods
Mice and rotaviruses
NOD8.3 and NOD mice were obtained, bred and housed in micro-isolator cages under specific pathogen-free conditions as described previously [13]. ‘Principles of laboratory animal care’ (NIH publication No. 85–23) and the ‘Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (2004)’ were followed. All procedures were conducted in accordance with protocols approved by the Animal Ethics Committee of The University of Melbourne. RRV and CRW-8 rotaviruses were amplified, purified and infectious titre was determined in fluorescent cell-forming units (f.f.u.) as previously described [23]. Psoralen/UV inactivation of RRV was performed and verified as before [24, 25].
Mouse inoculation with rotaviruses and diabetes monitoring
Mice were orally inoculated by gavage with virus diluent (mock) or 2 × 106 f.f.u. (unless otherwise indicated) of rotavirus, as before [13, 16]. The inactivated RRV (I-RRV) dose was equivalent to 2 × 106 f.f.u. Mice were inoculated at an age when RRV infection accelerates diabetes onset (5 weeks for NOD8.3 mice; 12 and 15 weeks, respectively, for female and male NOD mice). Approximately equal numbers of male and female NOD8.3 mice were used. Diarrhoea was not detected in any mouse during the first 14 days after inoculation. Mice were monitored weekly for diabetes, as before [16].
Rotavirus-specific antibody assays
Endpoint antibody titres (total immunoglobulin: IgG, IgM and IgA combined) in sera collected at 21 days post inoculation were determined by ELISA using homologous rotavirus antigen and mock-infected-cell control antigen, as before [16]. Rotavirus capture antigens were standardised by chequerboard titration using monoclonal antibody RVA directed to rotavirus capsid protein, to ensure that titres were comparable between virus strains [26]. Rotavirus-specific IgA, IgG1 and IgG2a was measured as above, with horseradish peroxidase-conjugated goat anti-IgA, rat anti-IgG1 and anti-IgG2a (Invitrogen, Carlsbad, CA, USA ) as detector antibodies, respectively. MOPC21 (IgG1) and UPC10 (IgG2a) antibodies were included as isotype specificity controls.
Detection of rotavirus antigen and infectious rotavirus
Stools (days 1–4 post infection), MLN and PLN were processed as 10% homogenates, as before [16]. MLN were digested for 30 min at room temperature with 1 g/l collagenase A and 40 mg/l DNase 1 diluted in RPMI medium supplemented with 2 mmol/l l-glutamine, 5 × 104 U/l penicillin and 50 g/l streptomycin. Tissues were passed through 70 μm mesh and cells dissociated for 5 min in supplemented RPMI containing 5 mmol/l EDTA (pH 7.2) and 5% fetal calf serum. Staining with anti-CD3–phycoerythrin (PE) (BD Biosciences, San Jose, CA, USA) and anti-CD19–FITC (eBioscience, San Diego, CA, USA) allowed FACS sorting into three populations: B cells, T cells and non-T and non-B cells. Infectious rotavirus was detected by ELISA after sample co-culture with MA104 cells for 72 h (unless otherwise indicated), as before [16]. Homogenates also were analysed by ELISA without prior co-culture. A net mean of absorbance at 450 nm minus absorbance at 690 nm (Ab450 − Ab690) with capture antibody ≥2× that of the same sample reacted with negative control capture antibody was considered as positive. Controls included samples from mock-infected mice. Virus titres (f.f.u.) in ELISA-positive samples were determined as before [16].
Detection of intracellular rotavirus and cellular activation
NOD8.3 mouse PLN cells isolated as above were antibody-stained at 4°C using anti-CD4–Pacific Blue, anti-CD8α–peridinin chlorophyll protein complex, anti-CD11c–allophycocyanin and anti-CD11b–biotin (BD Biosciences). Biotin was detected with streptavidin–PE. Stained cells were permeabilised, blocked with 10% heat-inactivated rotavirus antibody-negative sheep serum and stained for intracellular rotavirus antigen with RVA antibody [26] and sheep anti-mouse–FITC (Chemicon, Temecula, CA, USA), in an adaptation of an earlier protocol [27]. For analysis of T cell proportions, cells from PLN and MLN were stained with anti-CD3–AlexaFluor700, anti-CD4 and anti-CD8α.
Cells from the MLN of NOD mice isolated as above were enriched for DC using iso-osmotic Nycodenz medium (density 1.077 g/cm3) as previously described [28]. Surface staining with antibodies was conducted as above using anti-CD3, anti-CD11c–PECy7, anti-RT1B–FITC (MHC II) and anti-MHC I–biotin (BD Biosciences). Biotin was detected with allophycocyanin AlexaFluor750-labelled streptavidin (Invitrogen). Intracellular staining was performed with the BD Cytofix/Cytoperm kit according to the manufacturer’s instructions. Non-specific binding was blocked with 10% heat-inactivated, rotavirus antibody-negative goat serum and intracellular rotavirus was detected with rabbit anti-rotavirus polyclonal antibodies and goat anti-rabbit–Pacific Blue (Invitrogen). Activation of cells in the MLN segregated by the presence or absence of detectable rotavirus was determined using anti-MHC I, anti-MHC II, anti-CD80–PE and anti-CD86–allophycocyanin. Analyses were performed in an LSRII flow cytometer with FACS DIVA version 6.1.1 (BD Biosciences) and FlowJo version 8.7.3 software (Tree star, Ashland, OR, USA)
Cytokine mRNA assay
Total RNA was extracted and reverse transcribed as before [13]. Real-time PCR was performed using Assay-on-demand kits (Applied Biosystems, Foster City, CA, USA), with β-actin as the reference gene.
Statistical analysis
Tests (t, Mann–Whitney and Fisher’s) were used as appropriate. Log10-transformed antibody titres were analysed. Diabetes curves were evaluated by Kaplan–Meier life-table analysis.
Results
RRV accelerated diabetes only in the presence of a specific antibody response
Diabetes acceleration by RRV in seroconverting NOD8.3 and NOD mice correlates with their serum anti-rotavirus antibody level [13]. Here, the relation of diabetes acceleration to anti-rotavirus antibody production was examined. NOD8.3 mice, used because of their earlier diabetes onset, were given either I-RRV or low-dose (2 × 104 f.f.u.) RRV or they were mock inoculated. Mock inoculation produced negative serum anti-rotavirus antibody titres of <1:50, and diabetes curves similar to those in naive mice and our previous experiments [13].
RRV inoculation (2 × 106 f.f.u.) produces virus excretion in 45% of NOD8.3 mice [13]. In contrast, mice given I-RRV or low-dose RRV did not excrete infectious stool rotavirus. RRV antigen was excreted by one mouse given I-RRV on day 2 post inoculation. Serum anti-rotavirus antibodies (total immunoglobulin) were undetectable on days 14 and 56 post inoculation with I-RRV, so rotavirus replication was necessary for a serum antibody response. Significantly fewer mice given low-dose RRV (7/21) than 2 × 106 f.f.u. of RRV (37/37) developed a serum antibody response (p < 0.0001). The geometric mean antibody titre in responding mice (1:300, range 1:50 to 1:6,400) was reduced over mice given the higher RRV dose (1:1,300, range 1:50 to 1:25,600; p = 0.037). Thus, RRV dose influenced the seroconversion rate and antibody titre. I-RRV-inoculated mice developed diabetes at a similar rate to controls (p > 0.05, Fig. 1). Mice given low-dose RRV that seroconverted showed an increased diabetes incidence from 6 to 12 weeks of age (p = 0.02; Fig. 1) whereas diabetes development was unaffected in non-responding mice (p > 0.05). These seroconverting mice progressed more slowly to diabetes than those given 2 × 106 f.f.u. of RRV [13], further confirming the role of antibody titre in diabetes acceleration. Overall, the presence of an anti-RRV antibody response strongly correlated with accelerated diabetes onset.

RRV acceleration of diabetes onset depended on replicative ability and was associated with a specific serum antibody response. NOD8.3 mice given I-RRV (triangles, n = 20) or 2 × 104 f.f.u. of RRV were monitored for diabetes in parallel with mice given virus diluent (mock; circles, n = 76). Data for mice given 2 × 104 f.f.u. of RRV are stratified by the presence (black squares, n = 7) or absence (white squares, n = 14) of a serum antibody response to RRV
CRW-8 infection did not accelerate diabetes onset
Porcine rotavirus CRW-8 is serotypically identical to the Ohio State University (OSU) rotavirus strain, which replicates and causes disease in mice [29, 30]. The effect of CRW-8 on diabetes development is unknown. These experiments were conducted in NOD mice, as the skewed immune response of NOD8.3 mice renders them less useful for further mechanistic studies. CRW-8 induced a specific serum antibody response in 90% (68/77) of NOD mice, with a geometric mean titre of 1:2,700 (range 1:100 to 1:25,600). Compared with RRV [13], 10% fewer NOD mice responded to CRW-8, although their specific antibody titres were similar (p = 0.13). Diabetes curves for seroconverting male and female NOD mice given CRW-8 were similar to those of mock-inoculated mice (p > 0.05; Fig. 2a). No correlation between antibody titre and diabetes onset was observed in CRW-8-infected NOD mice (Fig. 2b, p > 0.05).

CRW-8 infection did not modulate diabetes development in NOD mice. (a) Mice given CRW-8 (female mice, black triangles, n = 31; male mice, black circles, n = 38) or diluent (female mice, white triangles, n = 29; male mice, white circles, n = 44) were monitored for diabetes. Only mice producing a serum anti-CRW-8 antibody response (90%) are included. (b) Lack of correlation (p > 0.05) between anti-CRW-8 antibody titre and age at diabetes onset for the female (black triangles) and male (black circles) mice given CRW-8 that are shown in (a)
RRV but not CRW-8 predominantly induced a Th1-biased specific serum antibody response in NOD mice, which was associated with earlier diabetes onset
As shown above and by Graham et al [13], although RRV accelerated diabetes in NOD mice but CRW-8 did not, these viruses induced serum antibody responses of similar titre in ≥90% of mice. These responses were further investigated by assay of rotavirus-specific serum immunoglobulin classes at 21 days after infection. Total immunoglobulin, IgA, IgG1 and IgG2a was detected in all RRV-infected mice (16/16). In contrast, IgG1 and/or IgG2a was undetectable in 62% (13/21) of mice given CRW-8. RRV- and CRW-8-infected mice showing titres in all these immunoglobulin classes did not differ in total immunoglobulin or IgA titres (Fig. 3a; p > 0.05). However, RRV induced higher IgG1 and IgG2a titres than CRW-8 (Fig. 3b; p = 0.0003 and p < 0.0001, respectively). In addition, RRV-infected mice showed greater titres of specific IgG2a than IgG1 (p < 0.0001), whereas these titres were comparable in CRW-8-infected mice (p = 0.095). This indicated that RRV but not CRW-8 infection favoured the development of an IgG2a antibody response and showed a Th1 bias.

Rotavirus-specific serum antibody titres in relation to virus strain and age of NOD mouse at diabetes onset. (a, b) Total immunoglobulin (black bars) and IgA titres (white bars) (a) and IgG1 (white bars) and IgG2a titres (black bars) (b) are shown as the mean ± SEM in sera collected at day 21 post infection from mice given RRV or CRW-8. Analysis is restricted to sera with IgG subclass endpoint titres to RRV (n = 16; 12 female mice) and CRW-8 (n = 8) of ≥1:25. All mock-infected mice showed total immunoglobulin, IgA, IgG1 and IgG2a titres at day 21 post infection of <1:25. (c) IgG1 (white circles) and IgG2a antibody titres (black circles) in relation to age at diabetes onset of female mice given RRV (n = 12). Bar, mean; **p < 0.01; ***p < 0.001; ns, not significant (p ≥ 0.095). A trend towards increased IgG2a titre in mice that developed diabetes by 20 weeks of age compared with mice that developed diabetes after 20 weeks of age in (c) was observed (p = 0.056)
The relation between IgG titres and mouse age at diabetes onset was examined in female mice given RRV (Fig. 3c). Male mice were excluded due to their low diabetes rate. Female mice that developed diabetes by 20 weeks of age showed significantly higher IgG2a than IgG1 titres (p = 0.0017). However, IgG1 and IgG2a titres in mice developing diabetes after 20 weeks were indistinguishable (p = 0.29). Specific IgG1 titres did not differ between mice that developed diabetes up to or after 20 weeks of age (p = 0.60). However, IgG2a titres in mice developing diabetes by 20 weeks tended to be higher than those in mice with later diabetes onset (p = 0.056). No difference in IgA titres between these groups was observed (p > 0.05; data not shown). Thus, development of a higher level of specific IgG2a than IgG1 was associated with RRV acceleration of diabetes in NOD mice.
RRV infection upregulated pro-inflammatory cytokines
Islet Tnf mRNA is increased on days 7 to 14 in our NOD model [13]. To confirm that RRV infection induces a Th1-biased cytokine response, mRNA levels of several cytokines were determined. In islets, Tnf expression was increased on day 7 (Fig. 4, p = 0.023), Ifnγ (also known as Ifng) was undetectable and Il-1β (also known as Il1b) unaltered (Table 1, p > 0.05). In the PLN, Il-1β and Il-4 (also known as Il4) were unaffected by RRV infection (Table 1, p > 0.05), whereas Tnf and Ifnγ were upregulated (Fig. 4) on days 3 (p = 0.0006 and p = 0.0060, respectively) and 7 post inoculation (p = 0.0014 and p = 0.016, respectively). Thus, pro-inflammatory cytokines were induced in the PLN whereas Il-4 expression was unaltered.

Elevated Th1-related cytokine levels in the islets and PLN of RRV-infected NOD mice. Levels of Tnf mRNA in islets (white bars) and PLN (black bars), and Ifnγ mRNA in PLN (hatched bars) from groups of three to five mock- or RRV-infected non-diabetic mice at days 3 and 7 after inoculation are shown. Ifnγ mRNA levels in islets were below the limit of detection of the assay. Data (mean ± SEM) for RRV-infected mice are presented relative to the mean mRNA level for mock-infected mice at the same time, which was standardised to 1.0 (dotted line). *p < 0.05; **p < 0.01; ***p < 0.001, cytokine mRNA levels in RRV-infected mice vs mock-infected mice
Infectious RRV but not CRW-8 spread to lymph nodes
Infectious RRV was found in 41% (12/29) of MLN from NOD mice given RRV on days 1–4 post infection only, peaking at day 3; RRV antigen was detected in 28% (13/47) of MLN on days 2–14 after inoculation (Fig. 5a). Fewer PLN than MLN contained RRV, with 15% (2/13) of PLN positive for infectious virus on days 1 and 3 and 19% (5/26) showing RRV antigen on day 3 and days 5–10 (Fig. 5b). With one exception, mice with infectious RRV or antigen in the PLN were similarly positive in the MLN. RRV titres in these MLN and PLN were comparable and ranged from 45 to 1 × 103 f.f.u./organ. In contrast, infectious virus was not found in these sites in the first 5 days after CRW-8 infection, and only 8% (2/25) and 4% (1/25) of these MLN and PLN contained CRW-8 antigen, respectively (Fig. 5c, d). RRV- and CRW-8-infected mice showed similar low rotavirus antigen levels in these MLN and PLN (data not shown).

Proportions of lymph nodes in mice given RRV or CRW-8 that contained infectious rotavirus (circles) and rotavirus antigen (crosses). NOD mouse MLN (a, c) and PLN (b, d) were analysed. RRV-infected mice studied (a, b) comprised five mice per day on days 1, 7, 10 and 14 post infection, and eight mice per day on days 2–5. Five CRW-8-infected mice were analysed per day on days 1–5 (c, d). Nodes from RRV-infected NOD8.3 mice were analysed daily (four mice per day) for 5 days (e, f)
Infectious RRV was detected in 2/8 MLN of NOD8.3 mice on days 2 and 4 post infection at titres/organ of 2.7 × 103 and 32 f.f.u., respectively (Fig. 5e). Infectious RRV was found in 1/4 PLN on day 4 at a titre of 3 × 102 f.f.u. (Fig. 5f). RRV antigen was present in 25% (3/16) of MLN on days 2, 3 and 5 (Fig. 5e), and 3/8 PLN on days 1 and 2 (Fig. 5f). RRV was observed in both PLN and MLN in 1/4 RRV-positive NOD8.3 mice.
Characterisation of RRV-infected cells in lymph nodes
Cells sorted from MLN of female NOD mice on day 3 after RRV inoculation were co-cultured with MA104 cells. Infectious RRV was detected in the original unsorted population and non-T and non-B cells, but not B or T cells (Table 2). Prolonged co-culture was necessary to detect RRV in sorted cells.
As an alternative, cells from the MLN pooled from five NOD mice and enriched for DC were analysed. In three independent experiments, cells in the positive cytometry gate from mock- and RRV-infected mice numbered 50 and 120, 477 and 744, and 380 and 1,800, respectively, so 70–1,420 cells contained RRV antigen. Consistent with sorted cell data, rotavirus antigen was detected in CD3−, not CD3+, cells at a maximum frequency of 1.75%. All rotavirus-positive cells expressed MHC I and II, confirming their identity as specialised antigen-presenting cells (APC). Many of these cells were likely to be DC, as a mean of 28.5% expressed CD11c. The remainder probably comprised macrophages and other DC subsets. Although a B cell marker was not included in this analysis, infectious RRV was not detected in B cells (Table 2). No CRW-8 antigen was detected in cells from the MLN, as cell numbers in the positive gate of mock- and CRW-8-infected mice were similar in each of three experiments.
RRV antigen was detected in 0.63% of PLN cells from 1/4 NOD8.3 mice on day 2 post inoculation (Fig. 6), but not on days 1, 3 and 4. Cells in the positive gate numbered 1,133 and 7,763 for the mock- and RRV-infected mouse, respectively. Most RRV antigen (83%) was associated with CD4−CD8− cells, comprising 2.31% of this population. RRV-positive CD4−CD8− cells showed heterogeneous expression of CD11c and CD11b. The remaining 17% of RRV-positive cells also showed variable CD4, CD8, CD11c and CD11b expression. No RRV antigen was detected in PLN cells from CRW-8-inoculated mice. The CD8+:CD4+ T cell ratio was increased in MLN of RRV-infected mice (Fig. 7; p = 0.043), with PLN cells showing a similar trend (p = 0.062), indicating that RRV antigen in lymph nodes is associated with expansion of autoreactive T cells in NOD8.3 mice. Overall, RRV was associated with a minor cell population in the PLN and MLN. Many of these RRV-containing cells in MLN of NOD mice were APC and included DC.

PLN cells of NOD8.3 mice contained RRV. PLN collected on days 1–4 after RRV dosage (n = 4 per day) were analysed. The percentage of PLN leucocytes stained by anti-rotavirus antibody in the matched control mouse (a, day 2) and the single positive mouse (b, day 2) is shown

The ratio of CD8+ to CD4+ T cells in MLN of NOD8.3 mice was increased by RRV infection. The relative proportions of CD8+ T cells to CD4+ T cells in MLN and PLN at day 7 after mock- (white circles) or RRV-inoculation (black circles) are shown. Groups of three mice were analysed. Bar, mean; *p < 0.05. A trend towards an increased CD8+ to CD4+ T cell ratio in PLN was observed (p = 0.062)
RRV but not CRW-8 infection induced activation of APC
As fewer NOD mouse PLN than MLN contained RRV (Fig. 5a, b), RRV-positive APC in PLN probably were too rare for detection and this was not attempted. APC (CD3−MHCII+) in MLN enriched for DC pooled from five mock-, RRV- or CRW8-infected NOD mice at day 3 post infection were analysed for rotavirus antigen and activation markers in three replicate experiments (Fig. 8). RRV-positive APC exhibited a trend towards increased MHC I levels over controls (p = 0.053; Fig. 8a), and expressed significantly more MHC II than RRV-negative APC from RRV-infected mice, and mock APC (p = 0.028 and 0.034, respectively; Fig. 8a). MHC I was elevated on RRV-negative APC (p = 0.044). RRV-positive APC showed a possible trend towards increased CD80 and CD86 expression over controls (Fig. 8b). RRV-positive cells expressing CD11c (mainly DC) also showed elevated MHC I (p = 0.042; Fig. 8c). RRV-positive and RRV-negative cells lacking CD11c showed elevated MHC I (p = 0.0015 and p = 0.041, respectively) and MHC II also was increased on RRV-positive, CD11c− cells (p = 0.046; Fig. 8c). RRV inoculation did not alter the frequency, size or granularity of CD11c+ or CD11c− APC. In contrast, neither CD11c+ nor CD11c− APC in the MLN of CRW-8-infected mice expressed increased levels of these markers (data not shown). In summary, APC (including DC) in the MLN of NOD mice showed signs of activation following infection with RRV but not with CRW-8.

Activation marker expression by APC in the MLN of RRV-infected and mock-infected NOD mice in relation to RRV antigen presence. Levels of MHC I and II (a, c) and CD80 and CD86 (b) on APC (CD3−MHCII+) are shown. Cells from mock-infected (Mock) mice (white circles), cells lacking detectable RRV antigen that were obtained from RRV-infected mice (RRV−; white triangles) and cells containing RRV antigen that were obtained from RRV-infected mice (RRV+; black triangles) are shown. (c) These three populations of APC are further stratified by CD11c expression. Bar, mean; *p < 0.05; **p < 0.01. A trend towards increased MHC I expression on RRV+ cells in (a) was observed (p = 0.053)
Discussion
These findings identify important relationships between the presence and nature of the rotavirus-specific antibody response, the extent of rotavirus infection and the ability of rotavirus to accelerate diabetes in mice. Acceleration of diabetes occurs only in the presence of a specific serum antibody response. In NOD mice, acceleration of diabetes by RRV correlated with spread of virus to the MLN and PLN, association of the virus with APC in MLN, activation of APC in the MLN, increased pro-inflammatory cytokine expression in the PLN and islets and the generation of a Th1-biased serum antibody response. Infectious CRW-8 rotavirus, which was unable to accelerate diabetes, did not spread to the MLN or PLN, affect APC in the MLN or induce Th1-skewed antibodies.
RRV acceleration of diabetes and RRV-specific serum antibody responses in NOD8.3 mice each depended on virus dose and required virus replication, providing further evidence that diabetes acceleration relates to antibody responses. Although childhood rotavirus infection frequently induces specific intestinal IgA without detectable serum antibody [31], the childhood diabetes studies identified rotavirus infections by seroconversion [10, 11]. Our mouse data indicate that these infections would be more likely to modulate childhood diabetes.
The Th1 bias of the immune response elicited, and capacity for spread to regional lymph nodes, correlated with rotavirus-accelerated diabetes. While RRV and CRW-8 infection elicited equivalent levels of total immunoglobulin and IgA, RRV-infected mice developed higher IgG1 and IgG2a responses. This may reflect differences in titres of other IgG subclasses and/or IgM. RRV but not CRW-8 infection in NOD mice produced a bias towards rotavirus-specific IgG2a antibodies over IgG1, indicating a Th1-dominated systemic immune response. Furthermore, the RRV-infected mice with earlier diabetes onset showed an elevated IgG2a but not IgG1 response. PLN and islets also exhibited this pro-inflammatory response. Children with rotavirus diarrhoea also show a strong Th1 bias in their rotavirus-specific serum antibody responses, with IgG1 and IgG3 predominating [32–35], and virus in the MLN [20]. Specific human IgG1 and IgG3 both result from Th1 responses, and human IgG1 is considered the equivalent of mouse IgG2a [18]. Our findings imply that rotavirus-specific IgG1 and/or IgG3 serum antibody responses in at-risk children may mark infections with diabetes modulation potential.
These Th1-biased antibodies likely act as markers of the overall systemic immune response, rather than direct participants in diabetes development. Active involvement of islet autoantibodies in human diabetes has not been identified. However, a higher frequency and strength of serum IgG3 responses to insulin and GAD65 is associated with diabetes progression [36]. It is noteworthy that both rotavirus and enterovirus infections are linked to childhood autoantibody exacerbations [10, 37], as viruses are the most effective inducers of a Th1-biased response. In addition to Th1-related antibodies, we found that RRV-infected NOD mice also exhibited a Th1 bias in PLN cytokine responses. We propose that rotavirus infections capable of inducing Th1-biased antibodies and virus spread to lymph nodes may be associated with production of pro-inflammatory cytokines in PLN and islets. Therefore, screening for rotavirus-specific serum IgG antibody subclasses may assist in identification of childhood rotavirus infections that might accelerate diabetes progression.
Although RRV does not spread to the pancreas in adult diabetes-prone mice [13], we detected infectious RRV in the MLN and PLN. This is the first report of rotavirus infection in the PLN. The non-T and non-B cell population in the MLN contained infectious RRV. APC in the NOD mouse MLN and NOD8.3 mouse PLN harbour RRV antigen. This lymph node spread is likely to play a role in the acceleration of diabetes by RRV, being absent from CRW-8-infected mice. Our data indicate that DC and/or macrophages in the PLN and MLN of diabetes-prone mice harbour RRV. Although their rarity precludes further experimental marker delineation, their variable CD4, CD8 and CD11b expression suggests that RRV associates with several cell subsets. A similar pattern of infectious rotavirus association with multiple subsets of APC occurs in diabetes-resistant mouse strains and human mononuclear cells [22, 38, 39]. The uptake of infectious rotavirus by CD3−CD19− cells indicates that some APC may support virus replication.
Whether or not they contained detectable RRV, APC from the MLN, including CD11c+ DC, expressed increased MHC. Some RRV-negative APC might contain RRV at levels undetectable in our assay, explaining their activation. However, cytokines produced by RRV-positive APC in the MLN also may induce bystander activation of other APC. In cultures of RRV-exposed human DC, infected cells upregulate co-stimulatory factors whereas uninfected cells secrete IFNα [38, 40]. Thus, cytokine profiles of activated RRV-positive and RRV-negative APC from NOD mice may differ. Crucially, the presence of RRV in the PLN was associated with the production of pro-inflammatory cytokines, which likely involves DC and T cells. The interactions of RRV with APC may lead to bystander B and T cell activation. Additionally, the elevated expression of MHC suggests that the presentation of antigen to T cells is enhanced, so the possibility of T cell molecular mimicry remains [10, 41]. CD4+ and CD8+ T cells are critical for diabetes development, while B cells contribute to T cell priming [42–45]. RRV infection increases the number of B cell in PLN and islets and the number of CD8+ T cells in islets [13], supporting the proposed role of RRV interactions with APC in lymphocyte activation. Overall, these interactions in the MLN are likely to play an important role in the activation of autoreactive T and B cells during diabetes development, involving cytokine production and/or antigen presentation.
Abbreviations
- APC:
-
Antigen-presenting cells
- DC:
-
Dendritic cells
- f.f.u.:
-
Fluorescent cell-forming units
- IGRP:
-
Islet-specific glucose 6-phosphatase catalytic subunit-related protein
- I-RRV:
-
Inactivated RRV
- MLN:
-
Mesenteric lymph nodes
- NOD8.3:
-
NOD8.3 TCR-transgenic
- NOD:
-
NOD/Lt
- PE:
-
Phycoerythrin
- PLN:
-
Pancreatic lymph nodes
- RRV:
-
Rhesus monkey rotavirus
- Th:
-
T helper
References
Daneman D (2006) Type 1 diabetes. Lancet 367:847–858
Solomon M, Sarvetnick N (2004) The pathogenesis of diabetes in the NOD mouse. Adv Immunol 84:239–264
Gagnerault MC, Luan JJ, Lotton C, Lepault F (2002) Pancreatic lymph nodes are required for priming of β cell reactive T cells in NOD mice. J Exp Med 196:369–377
Hoglund P, Mintern J, Waltzinger C, Heath W, Benoist C, Mathis D (1999) Initiation of autoimmune diabetes by developmentally regulated presentation of islet cell antigens in the pancreatic lymph nodes. J Exp Med 189:331–339
Jaakkola I, Jalkanen S, Hanninen A (2003) Diabetogenic T cells are primed both in pancreatic and gut-associated lymph nodes in NOD mice. Eur J Immunol 33:3255–3264
Turley SJ, Lee JW, Dutton-Swain N, Mathis D, Benoist C (2005) Endocrine self and gut non-self intersect in the pancreatic lymph nodes. Proc Natl Acad Sci USA 102:17729–17733
Tracy S, Drescher KM, Jackson JD, Kim K, Kono K (2010) Enteroviruses, type 1 diabetes and hygiene: a complex relationship. Rev Med Virol 20:106–116
Van Belle TL, Coppieters KT, von Herrath MG (2011) Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev 91:79–118
Lehuen A, Diana J, Zaccone P, Cooke A (2010) Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol 10:501–513
Honeyman MC, Coulson BS, Stone NL et al (2000) Association between rotavirus infection and pancreatic islet autoimmunity in children at risk of developing type 1 diabetes. Diabetes 49:1319–1324
Lempainen J, Tauriainen S, Vaarala O et al (2012) Interaction of enterovirus infection and cow’s milk-based formula nutrition in type 1 diabetes-associated autoimmunity. Diabetes Metabol Res Rev 28:177–185
Makela M, Vaarala O, Hermann R et al (2006) Enteral virus infections in early childhood and an enhanced type 1 diabetes-associated antibody response to dietary insulin. J Autoimmun 27:54–61
Graham KL, Sanders N, Tan Y, Allison J, Kay TWH, Coulson BS (2008) Rotavirus infection accelerates type 1 diabetes in mice with established insulitis. J Virol 82:6139–6149
Verdaguer J, Yoon JW, Anderson B et al (1996) Acceleration of spontaneous diabetes in TCR-β-transgenic nonobese diabetic mice by β-cell cytotoxic CD8+ T cells expressing identical endogenous TCR-α chains. J Immunol 157:4726–4735
Lieberman SM, Evans AM, Han B et al (2003) Identification of the β cell antigen targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes. Proc Natl Acad Sci USA 100:8384–8388
Graham KL, O’Donnell JA, Tan Y et al (2007) Rotavirus infection of infant and young adult nonobese diabetic mice involves extraintestinal spread and delays diabetes onset. J Virol 81:6446–6458
Rabinovitch A (1998) An update on cytokines in the pathogenesis of insulin-dependent diabetes mellitus. Diabetes Metabol Rev 14:129–151
Abbas AK, Murphy KM, Sher A (1996) Functional diversity of helper T lymphocytes. Nature 383:787–793
VanCott JL, Franco MA, Greenberg HB et al (2000) Protective immunity to rotavirus shedding in the absence of interleukin-6: Th1 cells and immunoglobulin A develop normally. J Virol 74:5250–5256
Lappalainen S, Ylitalo S, Arola A, Halkosalo A, Rasanen S, Vesikari T (2012) Simultaneous presence of human herpesvirus 6 and adenovirus infections in intestinal intussusception of young children. Acta Paediatr 101:663–670
Mossel EC, Ramig RF (2003) A lymphatic mechanism of rotavirus extraintestinal spread in the neonatal mouse. J Virol 77:12352–12356
Fenaux M, Cuadras MA, Feng N, Jaimes M, Greenberg HB (2006) Extraintestinal spread and replication of a homologous EC rotavirus strain and a heterologous rhesus rotavirus in BALB/c mice. J Virol 80:5219–5232
Holloway G, Coulson BS (2006) Rotavirus activates JNK and p38 signaling pathways in intestinal cells, leading to AP-1-driven transcriptional responses and enhanced virus replication. J Virol 80:10624–10633
Groene WS, Shaw RD (1992) Psoralen preparation of antigenically intact noninfectious rotavirus particles. J Virol Meth 38:93–102
Halasz P, Holloway G, Turner SJ, Coulson BS (2008) Rotavirus replication in intestinal cells differentially regulates integrin expression by a phosphatidylinositol 3-kinase-dependent pathway, resulting in increased cell adhesion and virus yield. J Virol 82:148–160
Coulson BS, Unicomb LE, Pitson GA, Bishop RF (1987) Simple and specific enzyme immunoassay using monoclonal antibodies for serotyping human rotaviruses. J Clin Microbiol 25:509–515
Webster NL, Kedzierska K, Azzam R et al (2006) Phagocytosis stimulates mobilization and shedding of intracellular CD16A in human monocytes and macrophages: inhibition by HIV-1 infection. J Leukoc Biol 79:294–302
Vremec D, Shortman K (2008) The isolation and identification of murine dendritic cell populations from lymphoid tissues and their production in culture. Meth Mol Biol 415:163–178
Ciarlet M, Isa P, Conner ME, Liprandi F (2001) Antigenic and molecular analyses reveal that the equine rotavirus strain H-1 is closely related to porcine, but not equine, rotaviruses: interspecies transmission from pigs to horses? Virus Genes 22:5–20
Zhang M, Zeng CQ, Dong Y et al (1998) Mutations in rotavirus nonstructural glycoprotein NSP4 are associated with altered virus virulence. J Virol 72:3666–3672
Coulson BS, Grimwood K, Masendycz PJ et al (1990) Comparison of rotavirus IgA coproconversion with other indices of rotavirus infection in a longitudinal study in childhood. J Clin Microbiol 28:1367–1374
Istrate C, Hinkula J, Hammarstrom L, Svensson L (2008) Individuals with selective IgA deficiency resolve rotavirus disease and develop higher antibody titers (IgG, IgG1) than IgA competent individuals. J Med Virol 80:531–535
Xu J, Dennehy P, Keyserling H et al (2005) Serum antibody responses in children with rotavirus diarrhea can serve as proxy for protection. Clin Diagn Lab Immunol 12:273–279
Azim T, Zaki MH, Podder G et al (2003) Rotavirus-specific subclass antibody and cytokine responses in Bangladeshi children with rotavirus diarrhoea. J Med Virol 69:286–295
Grauballe PC, Hornsleth A, Hjelt K, Krasilnikoff PA (1986) Detection by ELISA of immunoglobulin G subclass-specific antibody responses in rotavirus infections in children. J Med Virol 18:277–281
Ronkainen MS, Hoppu S, Korhonen S et al (2006) Early epitope- and isotype-specific humoral immune responses to GAD65 in young children with genetic susceptibility to type 1 diabetes. Eur J Endocrinol 155:633–642
Lonnrot M, Korpela K, Knip M et al (2000) Enterovirus infection as a risk factor for beta-cell autoimmunity in a prospectively observed birth cohort: the Finnish Diabetes Prediction and Prevention Study. Diabetes 49:1314–1318
Deal EM, Jaimes MC, Crawford SE, Estes MK, Greenberg HB (2010) Rotavirus structural proteins and dsRNA are required for the human primary plasmacytoid dendritic cell IFNα response. PLoS Pathog 6:e1000931
Narvaez CF, Angel J, Franco MA (2005) Interaction of rotavirus with human myeloid dendritic cells. J Virol 79:14526–14535
Mesa MC, Rodriguez LS, Franco MA, Angel J (2007) Interaction of rotavirus with human peripheral blood mononuclear cells: plasmacytoid dendritic cells play a role in stimulating memory rotavirus specific T cells in vitro. Virology 366:174–184
Honeyman MC, Stone NL, Falk BA, Nepom G, Harrison LC (2010) Evidence for molecular mimicry between human T cell epitopes in rotavirus and pancreatic islet autoantigens. J Immunol 184:2204–2210
Phillips JM, Parish NM, Raine T et al (2009) Type 1 diabetes development requires both CD4+ and CD8+ T cells and can be reversed by non-depleting antibodies targeting both T cell populations. Rev Diabet Stud 6:97–103
Chatenoud L, Thervet E, Primo J, Bach JF (1994) Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc Natl Acad Sci USA 91:123–127
Marino E, Batten M, Groom J et al (2008) Marginal-zone B cells of nonobese diabetic mice expand with diabetes onset, invade the pancreatic lymph nodes, and present autoantigen to diabetogenic T cells. Diabetes 57:395–404
Marino E, Villanueva J, Walters S, Liuwantara D, Mackay F, Grey ST (2009) CD4(+)CD25(+) T cells control autoimmunity in the absence of B cells. Diabetes 58:1568–1577
Acknowledgements
We are grateful to P. Santamaria (Department of Microbiology, University of Calgary, Canada) for provision of the NOD8.3 mouse, D. Godfrey and S. Turner (Department of Microbiology and Immunology, The University of Melbourne, Australia) for helpful suggestions and S. Bedoui and A. Turner (Department of Microbiology and Immunology, The University of Melbourne, Australia) for their expert assistance with the studies on APC. F. E. Fleming and N. Sanders (Department of Microbiology and Immunology, The University of Melbourne, Australia) provided excellent technical assistance. We thank D. Taylor, R. Hall and E. Rooney (Department of Microbiology and Immunology, The University of Melbourne, Australia) for mouse husbandry.
Funding
This work was supported by Project Grants (299862 and 509008) and Senior Research Fellowships (350253 and 628319) to B. S. Coulson from the National Health and Medical Research Council of Australia.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
BSC conceived the study, designed many of the experiments and interpreted these data. JAP, NLW, KLG, GH and CZ conceived and designed some studies, and acquired, analysed and interpreted the data. JAP and BSC drafted the article. BSC, NLW, KLG, GH and CZ revised it critically for important intellectual content. All authors approved the final version.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pane, J.A., Webster, N.L., Graham, K.L. et al. Rotavirus acceleration of murine type 1 diabetes is associated with a T helper 1-dependent specific serum antibody response and virus effects in regional lymph nodes. Diabetologia 56, 573–582 (2013). https://doi.org/10.1007/s00125-012-2798-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2798-4