
Abstract
Adipocyte fatty acid binding protein (AFABP, also known as aP2 and FABP4) has recently been introduced as a novel fat-derived circulating protein. AFABP serum concentrations are positively correlated with markers of the metabolic syndrome and vascular disease in various cross-sectional and interventional studies. Furthermore, a small set of prospective studies indicates that high AFABP serum levels at baseline predict the risk for metabolic and vascular morbidity and mortality. Studies in Afabp (also known as Fabp4) knockout mice and AFABP inhibitor-treated animals suggest that total AFABP promotes insulin resistance, hypertriacylglycerolaemia and atherosclerosis by ligand/ligand delivery, as well as ligand-independent mechanisms. In contrast, the pathophysiological significance of circulating AFABP and the mechanisms leading to its release remain to be established. The current review summarises recent findings on the regulation and potential role of AFABP in metabolic and vascular disease.
Similar content being viewed by others


Introduction
Adipocytes produce and secrete a variety of bioactive peptides, termed adipokines, which influence body weight, insulin sensitivity, lipid metabolism and vascular function. Adipocyte fatty acid binding protein (AFABP), also known as fatty acid binding protein-4 and aP2, has recently been suggested as a third adipokine, in addition to leptin [1] and adiponectin [2], that is preferentially produced in and released from adipocytes. AFABP accounts for up to 6% of total cytosolic protein in cultured differentiated fat cells [3]. In addition to production in adipocytes, AFABP is also produced in significant amounts in macrophages [4] and endothelial cells [5].
This review summarises findings on the production, structure and pathophysiological significance of total AFABP. Recent data on regulation and the potential role of circulating AFABP in metabolic and vascular disease are presented and discussed.
Production and structure of AFABP
The gene coding for AFABP has been identified from a cDNA library constructed from poly(A)+ RNA isolated from differentiated 3T3–L1 adipocytes [6]. The gene, AFABP (also known as FABP4), encodes a polypeptide with 132 amino acids and a molecular mass of 14.6 kDa (GenBank accession number NM_024406). Afabp mRNA and protein production are significantly induced during adipocyte differentiation [7]. AFABP has frequently been used as an adipocyte differentiation marker [8–10], based on these observations. The DNA-binding protein CCAAT/enhancer-binding protein interacts with the AFABP promoter and elevates expression of the AFABP gene [11]. Furthermore, cyclic adenosine monophosphate induces AFABP gene expression during adipocyte differentiation by relieving the inhibitory effect of a negative regulatory element in the AFABP promoter [10]. Moreover, insulin-sensitising and pro-adipogenic peroxisome proliferator-activated receptor (PPAR)γ agonists induce AFABP production and secretion [12, 13]. In addition, AFABP mRNA and protein production are elevated in subcutaneous adipose tissue compared with the visceral depot in lean and obese humans [14].
AFABP serves as a lipid-binding chaperone for long-chain NEFA [15]. Structure–function studies show that AFABP consists of a beta barrel formed by ten antiparallel beta strands defining an interior water-filled binding cavity that is used as the NEFA-binding site [16, 17]. One end of the beta barrel is closed by protein side chains, while the other end is partially restricted by a helix-turn-helix motif that serves as the portal for NEFA binding and dissociation. AFABP transfers NEFA to membranes via collisional interaction, and lysine residues in the helical cap domain are vital for this collisional transfer process [18]. Furthermore, AFABP increases the hydrolytic activity of hormone-sensitive lipase (HSL) via a specific protein–protein interaction [19]. Work using mutational analysis coupled with fluorescence resonance energy transfer suggests that interaction between AFABP and HSL depends on AFABP-bound NEFA and HSL phosphorylation [20]. In accordance with these findings, a small molecular inhibitor blocking NEFA binding to AFABP also antagonises physical interaction with HSL [21]. Furthermore, four amino acids on the helical domain of AFABP (D17, D18, K21 and R30), termed the charge quartet, are responsible for HSL interaction (Fig. 1) [22]. Interestingly, the adipokine is also an intercompartmental shuttle from the cytosol to the nucleus for PPARγ agonists, including thiazolidinediones and linoleic acid [23, 24]. The primary AFABP sequence does not harbour a readily identifiable nuclear localisation signal or nuclear export signal [25]. However, both signals are found in the three-dimensional structure of the protein bound to troglitazone and linoleic acid [24, 25]. In agreement with these findings, AFABP selectively relocates to the nucleus in response to troglitazone and enhances the transcriptional activity of PPARγ [26]. Elegant X-ray crystal structure analyses have revealed that troglitazone and linoleic acid bind in the internal pocket of AFABP within a plane that is parallel to and just below the helix-turn-helix motif, revealing a stabilised AFABP conformation [24]. In contrast, AFABP bound to oleate and stearate, two ligands that do not activate PPARγ, does not lead to a stable nuclear localisation signal [24].

Structure of murine AFABP showing the positions of the charge quartet D17, D18, K21 and R30 (labelled red) on helix 1 and helix 2 with bound linoleic acid (labelled blue); from the Protein Data Bank (PDB ID: 2Q9S [24]). The structure was generated in PyMOL 1.4.1
Taken together, published evidence suggests that AFABP is produced in fat cells, regulates transport of NEFA and PPARγ agonists via a ligand/ligand delivery mechanism, and interacts with proteins influencing lipid metabolism and insulin sensitivity, including HSL and PPARγ.
The role of AFABP in body-weight control, glucose metabolism, lipid metabolism and beta cell function
Animal studies on AFABP function are summarised in Table 1. Afabp knockout (Afabp −/−) mice develop high-fat diet-induced obesity (DIO) to a greater extent than wild-type controls [27]. Unlike wild-type mice, however, they do not develop insulin resistance or diabetes [27]. Interestingly, epidermal fatty acid binding protein (EFABP) is upregulated in Afabp −/− mice, potentially compensating for the loss of AFABP [27–29]. Lack of insulin resistance in DIO Afabp −/− mice is correlated with diminished obesity-induced TNF-α production in adipose tissue [27]. Mice with double Afabp and Efabp (also known as Fabp5 and Mal1) knockout (Afabp −/−–Efabp −/−) develop a comparable phenotype with significant protection from diet-induced and obesity-associated insulin resistance and type 2 diabetes mellitus [30]. Afabp −/− animals with additional leptin deficiency (ob/ob–Afabp −/− mice) also become more obese than ob/ob controls [31]. Paradoxically, AFABP deficiency again improves insulin sensitivity and lipid variables in ob/ob–Afabp −/− mice compared with control animals, despite extreme obesity [31]. Loss of AFABP significantly influences two major metabolic processes: decreased adipose NEFA efflux and preferential use of glucose relative to fatty acids [32]. These results are in accordance with findings in triple knockout ob/ob–Afabp −/−–Efabp −/− animals [33]. Adiposity detected in both epididymal and subcutaneous fat pads is increased in these animals compared with ob/ob control mice [33]. However, ob/ob–Afabp −/−–Efabp −/− animals demonstrate increased insulin responsiveness relative to controls, despite severe obesity [33].
The influence of AFABP deficiency on beta cell function has been assessed in various studies. Thus, the lipolytic response to β-adrenergic stimulation is significantly reduced in ob/ob–Afabp −/− animals [31]. These findings are in agreement with results from two independent studies obtained in Afabp −/− mice [28, 34]. Reduced β-adrenoceptor-stimulated lipolysis translates into significantly reduced lipolysis-associated insulin secretion from beta cells in both ob/ob–Afabp −/− mice [31] and Afabp −/− mice [34]. However, loss of AFABP does not generally impair beta cell function. Thus, glucose-stimulated insulin secretion is significantly improved in ob/ob–Afabp −/− mice compared with controls [31]. Furthermore, other insulin secretagogues such as arginine and glibenclamide (known as glyburide in the USA and Canada) are as effective in stimulating an insulin secretory response in Afabp −/− mice as in controls [34]. No apparent morphological differences in the structure or size of the pancreatic islets are detectable between ob/ob–Afabp −/− animals and ob/ob controls [31].
Taking these findings in AFABP-deficient animals into consideration, AFABP does not contribute to weight gain but promotes insulin resistance and hypertriacylglycerolaemia. Furthermore, AFABP deficiency increases glucose-stimulated insulin secretion from beta cells, whereas it decreases lipolysis-induced insulin secretion. Mechanistically, stimulation of TNF-α production in adipose tissue and induction of lipolysis by AFABP have been suggested as mediators of these effects.
Studies on AFABP function have not only been performed with genetically altered animal models, but also with specific inhibitors. Thus, the small-molecule and orally available synthetic AFABP inhibitor BMS309403 significantly improves insulin sensitivity in ob/ob mice [35]. These findings are in accordance with results in Afabp −/− animals [27, 29, 31, 32]. Mechanistically, macrophage infiltration in adipose tissue is less evident in BMS309403- compared with vehicle-treated animals [35]. Reduced macrophage infiltration in AFABP inhibitor-treated mice is accompanied by diminished abundance of proinflammatory adipokines in adipose tissue, including monocyte chemoattractant protein (MCP)-1, IL-1β, IL-6 and TNF-α [35]. Concerning beta cell function, islet and pancreas morphology are not affected by the AFABP inhibitor [35]. BMS309403 has also been studied in DIO mice [36]. In agreement with findings in ob/ob mice [35], the AFABP inhibitor reduces plasma triacylglycerol (TG) levels [36]. However, insulin and glucose tolerance are not improved by AFABP inhibition in DIO mice [36], in contrast to findings in ob/ob animals [35]. In agreement with these findings, knockdown of AFABP mediated by RNA interference technology does not significantly influence glucose homeostasis in DIO mice, but these mice significantly increase in body weight compared with vehicle-treated controls [37]. Interestingly, no compensatory increase in adipocyte Efabp expression is detectable in AFABP knockdown mice [37] in contrast to previous studies in Afabp −/− animals [27–29].
Taking the findings in AFABP inhibitor-treated animals into consideration, AFABP promotes hypertriacylglycerolaemia similar to findings in Afabp −/− mice. Whereas AFABP inhibition improves insulin sensitivity in genetically obese mice, this effect is not seen in DIO animals. These studies indicate that the influence of AFABP on insulin sensitivity might depend on the extent of AFABP loss and the mode of body weight gain (DIO vs genetically determined obesity).
The role of AFABP in the pathogenesis of atherosclerosis
Afabp–ApoE double-knockout (Afabp −/−–ApoE −/−) animals show protection from atherosclerosis in the absence of significant differences in serum lipids and insulin sensitivity compared with control ApoE −/− animals (Table 1) [38]. In more detail, quantitative analysis of the proximal aorta reveals that the mean atherosclerotic lesion area in Afabp −/−–ApoE −/− male mice is reduced by 66% compared with control mice [38]. Interestingly, ApoE −/− mice with AFABP deficiency specifically restricted to macrophages show a comparable reduction in atherosclerotic lesion area compared with animals with whole-body AFABP deficiency [38]. In accordance with these results, Afabp −/−–ApoE −/− mice have reduced atherosclerotic lesion formation after 14 weeks of Western diet [39]. In a second paper, Boord et al investigated Afabp–Efabp–ApoE triple knockout (Afabp −/−–Efabp −/−–ApoE −/−) mice, with similar results [40]. Interestingly, lipid metabolism in both sexes and survival in males were also improved in Afabp −/−–Efabp −/−–ApoE −/−mice. In accordance with these findings, the AFABP inhibitor BMS309403 markedly reduces atherosclerotic lesion area in the en face aorta by more than 50% in ApoE −/− mice [35]. Similar results have been obtained in an independent study using the same AFABP inhibitor [41].
One study in human participants supports the hypothesis that AFABP might also contribute to human atherosclerotic disease. Thus, a functionally significant genetic variation (T-87C polymorphism) at the AFABP locus in humans not only results in reduced adipose tissue AFABP expression, but is also associated with lower serum TG levels as well as a lower risk for coronary heart disease and type 2 diabetes mellitus [42]. However, the relationship between AFABP genetic variants and type 2 diabetes mellitus has not been confirmed recently (e.g. in the Women’s Health Initiative observational study) [43]. Here, none of the AFABP single-nucleotide polymorphisms analysed showed a significant association with type 2 diabetes mellitus risk.
The studies summarised above convincingly demonstrate that AFABP promotes atherosclerotic disease. The impact of AFABP on atherosclerosis appears to be related exclusively to its action in macrophages [38]. Thus, AFABP-deficient macrophages display reduced inhibitor of κB kinase and nuclear factor κB activity, resulting in decreased levels of the proinflammatory cytokines TNF-α, IL-6 and MCP-1 [44]. In agreement with these findings, treatment with the AFABP inhibitor BMS309403 significantly decreases TNF-α, IL-6 and MCP-1 production in THP-1 macrophages [35]. In addition, AFABP-deficient mice are resistant to several inflammatory disorders, including allergic airway inflammation [45] and experimental autoimmune encephalomyelitis/multiple sclerosis [46]. Several proinflammatory stimuli induce AFABP production, including phorbol myristate acetate, oxidised (ox)LDL and toll-like receptor (TLR) agonists [4, 35, 47, 48]. Among these, oxLDL upregulates AFABP during the transformation of macrophages into foam cells [4]. This AFABP elevation involves activation of PPARγ by components of oxLDL that function as PPARγ ligands [4]. Lipopolysaccharides (LPS) are potent TLR4 agonists that induce AFABP production in macrophages [47]. LPS stimulates AFABP transcription through Jun N-terminal kinase (JNK), which in turn induces c-Jun recruitment to a highly conserved activator protein-1 recognition site within the proximal region of the AFABP promoter [49]. In addition, LPS-induced JNK phosphorylation, activator protein-1 stimulation and production of proinflammatory cytokines are significantly attenuated by pharmacological or genetic AFABP suppression in macrophages [49]. Besides TLR4-agonistic LPS, the TLR2 ligand zymosan and the TLR3 ligand polyinosine:polycytidylic acid significantly upregulate AFABP in murine macrophages [47]. Furthermore, saturated fatty acids including palmitate induce AFABP production in macrophages [50]. Most interestingly, AFABP is an obligatory mediator coupling toxic lipids (i.e. saturated fatty acids) to endoplasmic reticulum (ER) stress in macrophages in vitro and in vivo [50]. The impact of AFABP on macrophage ER stress is mediated by upregulation of key lipogenic enzymes by the liver X receptor [50]. It is interesting to note from a clinical standpoint that mitigation of ER stress with a chemical chaperone not only prevents macrophage AFABP production, but also leads to marked protection against macrophage ER stress, cell death and atherosclerosis [50].
Upregulation of AFABP in foam cells regulates cholesterol and TG accumulation in macrophages. Thus, adenovirus- or lentivirus-mediated overproduction of AFABP enhances the accumulation of cholesteryl esters in macrophage foam cells [51]. In accordance with these results, human THP-1 macrophages treated with an AFABP inhibitor exhibit 44% reduced cholesteryl ester accumulation and significantly increased cholesterol efflux relative to vehicle-treated cells [35]. Moreover, upregulation of AFABP gene expression by advanced glycation end-products has also been shown to induce cholesterol and TG accumulation in THP-1 macrophages in vitro in an independent study [52].
It is interesting to note in a clinical context that upregulation of AFABP is also found in vivo in macrophage/foam cells from human atherosclerotic plaques [4]. Furthermore, independent studies suggest that increased AFABP synthesis in atherosclerotic plaques is associated with disease severity [53–56]. Various studies agree that AFABP synthesis is localised in macrophages in atherosclerotic plaques [54–56]. Moreover, atorvastatin, which has antiatherogenic properties, potently reduces Afabp mRNA and AFABP protein level in oxLDL-treated human macrophages [57].
AFABP is also detected in endothelial cells as well as in adipocytes [3] and macrophages [4]. Afabp mRNA expression and AFABP protein synthesis are significantly induced in cultured endothelial cells by vascular endothelial growth factor (VEGF). Interestingly, knockdown of AFABP has been reported to dramatically reduce proliferation of endothelial cells both under baseline conditions and in response to VEGF in vitro [5]. The same group has identified AFABP as a mediator of angiogenesis [58]. Thus, AFABP-deficient human umbilical vein endothelial cells demonstrate a markedly increased susceptibility to apoptosis, as well as decreased migration and capillary network formation [58]. Furthermore, aortic rings from Afabp −/− mice show decreased angiogenic sprouting, which is reversible upon Afabp re-expression [58].
Taken together, these studies indicate that AFABP is upregulated by proatherogenic stimuli and induces atherosclerosis by (1) promoting cholesterol and TG accumulation in and inhibiting cholesterol efflux from macrophages; and (2) inducing proinflammatory and proatherogenic cytokines in macrophages. Furthermore, AFABP might have a role in the proliferation of endothelial cells, as well as in angiogenesis.
Circulating AFABP—a potential novel adipokine
The first description of AFABP as a circulating protein was published in 2006 [59]. Since this publication, several cross-sectional, interventional and prospective studies have investigated the regulation and predictive value of circulating AFABP in humans. Furthermore, potential mechanisms leading to the release of AFABP, as well as potential effects of circulating AFABP, have been elucidated recently and are summarised below.
Potential mechanisms by which AFABP reaches the circulation
While AFABP has been established as a circulating protein, the mechanisms by which the adipokine reaches the circulation are unclear. So far, there is no solid evidence that AFABP is, in fact, secreted or that any regulated release of AFABP from adipocytes or macrophages occurs. Thus, there is no secretory signal sequence in the primary and three-dimensional structure of AFABP [60, 61]. However, proteins lacking exocytosis-targeting signal sequences can cross the plasma membrane by different, unconventional, secretion mechanisms [62]. As an example, fibroblast growth factor-1 is exported through an unconventional release pathway involving the formation of a specific multiprotein complex [63]. Furthermore, murine adipocytes release various proteins lacking a secretory signal sequence by microvesicles and, most interestingly, AFABP is detected in microvesicles [64]. These findings are supported by an independent study that convincingly demonstrated that the microvesicle fraction prepared by ultracentrifugation of human adipocyte supernatant fractions contains AFABP [65]. Taking these findings into consideration, AFABP might get into the circulation at least in part by adipocyte-derived microvesicles. Nevertheless, it is also possible that AFABP reaches the circulation from lysis of large adipocytes. In agreement with this hypothesis, most studies on circulating AFABP have shown a positive correlation with measures of adiposity, including BMI and percentage body fat. However, whereas both β-tubulin and AFABP are present in the cell lysate of cultured 3T3–L1 adipocytes, only AFABP is present in the culture medium, suggesting the secretion of AFABP from cultured adipocytes [59]. Clearly, more work is needed to define the mechanisms by which AFABP reaches the circulation.
Potential effects of circulating AFABP
Only one study to date has elucidated the potential role of circulating AFABP in vitro [65]. In an elegant study, Lamounier-Zepter et al identified AFABP as a cardiodepressant factor. Recombinant AFABP in a concentration of 100 nmol/l decreased shortening amplitude by more than 50% and intracellular systolic peak Ca2+ by more than 20% in isolated adult rat cardiomyocytes. The N-terminal amino acids 1–20 of AFABP were the most effective cardiodepressive structure, showing approximately 40% suppression of rat cardiomyocyte contraction. Interestingly, adipocyte-secreted microvesicles contained up to 8% of the total amount of AFABP detected in the supernatant fraction, and the microvesicle fraction from human adipocytes had a low but significant cardiodepressant effects on rat cardiomyocytes (−28% decrease in shortening amplitude). These findings suggest that ligand-independent mechanisms might also contribute to actions of AFABP in addition to ligand/ligand delivery mechanisms. However, these potential ligand-independent mechanisms need to be confirmed and further characterised in additional studies. Furthermore, future experiments should aim to determine whether AFABP exists in its apo- and/or holo-form in human serum.
Circulating AFABP, the metabolic syndrome and atherosclerosis—cross-sectional studies
Several cross-sectional studies have demonstrated that AFABP is independently and positively associated with markers of the metabolic syndrome and, most notably, obesity-related variables (Table 2). Thus, in the first description of circulating AFABP, Xu et al observed a significant upregulation of AFABP levels in obesity [59]. An increase in AFABP serum levels in patients with the metabolic syndrome compared with controls has also been described in various independent studies [12, 66, 67]. Furthermore, circulating AFABP has been reported to be elevated in patients with familial combined hyperlipidaemia [68] and in those with non-alcoholic fatty liver disease, a disorder regarded as the hepatic manifestation of the metabolic syndrome [69]. In agreement with these findings, serum AFABP values were positively and independently correlated with markers of the metabolic syndrome, including waist circumference, blood pressure, dyslipidaemia, fasting insulin and HOMA of insulin resistance, in the initial study [59]. A similar association of circulating AFABP with facets of the metabolic syndrome has also been shown in other studies [12, 66, 67, 70–73].
The first description of the association between circulating AFABP and atherosclerosis was published in 2007 [74]. This study reported that circulating AFABP levels are significantly higher in women with atherosclerotic plaques compared with those without plaques. Furthermore, AFABP concentrations significantly increase with the number of stenotic coronary arteries [75]. In accordance with these findings, three independent studies have reported that serum AFABP is significantly elevated in patients with coronary artery disease (CAD) [76–78]. Very recently, a small case–control study detected significantly elevated serum AFABP levels in patients with cerebrovascular disease, whereas no difference was found in patients with CAD [79]. Similarly, AFABP plasma concentrations are increased in patients with carotid atherosclerosis compared with sex- and age-matched healthy controls [54]. Furthermore, the adipokine positively correlates with vascular inflammation measured by 18F-fluorodeoxyglucose positron emission tomography [80].
Taken together, these studies provide evidence that circulating AFABP is increased in patients with the metabolic syndrome and atherosclerosis, and might be a marker for these disease states. Whether AFABP directly contributes to these disorders needs to be determined in future studies.
Interventional studies on circulating AFABP
Several interventional studies have evaluated the influence of weight reduction and statin use on circulating AFABP (Table 3). All [81–83] except one [84] published studies have demonstrated that the serum AFABP level is reduced following successful weight reduction through lifestyle or surgical measures. Furthermore, atorvastatin treatment over 3 months has been found to significantly decrease circulating AFABP levels in statin-naive participants with atherosclerosis [85], in agreement with in vitro findings [57].
In summary, interventional studies suggest that circulating AFABP decreases after weight loss and statin therapy, and might contribute to the beneficial effects of these interventions.
Prospective studies on circulating AFABP
Elevated concentrations of circulating AFABP independently predict the risk of developing a metabolic syndrome [86]. In agreement with these findings, baseline serum AFABP concentrations are significantly higher in children who develop a metabolic syndrome compared with controls [87]. High baseline AFABP serum levels are also a biomarker predicting the development of type 2 diabetes mellitus independent of obesity, insulin resistance and glycaemic indices [88]. Furthermore, circulating AFABP predicts prognosis in patients with end-stage renal disease, a high-risk population for atherosclerosis-associated morbidity and mortality [89]. Moreover, serum AFABP is independently and positively associated with stroke and 3-month mortality in ischaemic stroke patients [90]. Similar findings have been described in two independent studies [54, 56]. Very recently, it was shown that circulating AFABP concentrations are independently associated with cardiovascular morbidity and mortality in patients with CAD [91].
Taken together, data from prospective studies (Table 4) suggest that high baseline circulating AFABP concentrations predict the risk of developing metabolic and vascular disease states independent of established risk markers. Whether AFABP directly contributes to these disease states needs to be determined in future studies.
Summary and concluding remarks
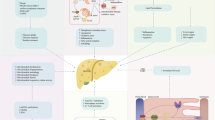
A model of AFABP action in metabolic and vascular disease is shown in Fig. 2.

A schematic model of AFABP in metabolic and vascular disease; IKK, inhibitor of κB kinase; NFκB, nuclear factor κB
Convincing evidence has been presented that AFABP is an adipocyte- and macrophage-produced protein that promotes insulin resistance, hypertriacylglycerolaemia and atherosclerosis by ligand/ligand delivery, as well as ligand-independent, mechanisms (Table 1). Results from animal experiments suggest that AFABP production in adipocytes leads to insulin resistance, NEFA release and proinflammatory gene expression. Furthermore, AFABP synthesis in macrophages is crucial for its effects on atherosclerosis. Thus, AFABP overproduction leads to foam cell development by increasing cholesterol and TG accumulation, as well as by inducing expression of proinflammatory genes. Furthermore, macrophage infiltration into adipose tissue is stimulated by AFABP. These data indicate that AFABP might be an interesting therapeutic target for metabolic and vascular disease.
Since 2006, data have accumulated that AFABP is also a circulating protein, the levels of which are independently and positively associated with various facets of the metabolic syndrome and vascular disease (Table 2). Furthermore, interventional studies suggest that circulating AFABP decreases after weight loss and statin therapy (Table 3). In addition, a small set of prospective studies indicates that high AFABP serum levels at baseline predict the risk for metabolic and vascular morbidity, as well as mortality (Table 4).
Despite these recent advances in our understanding of AFABP physiology, various important questions remain unanswered, particularly concerning the role of AFABP as an adipokine. These include: (1) Is AFABP secreted from adipocytes and, if yes, what are the mechanisms? (2) Is circulating AFABP only a marker of metabolic and vascular disease or does it directly influence these conditions? (3) In the latter case, does AFABP mediate its effects via ligand-dependent and/or independent mechanisms? Furthermore, what are the target tissues of circulating AFABP?
These questions need to be addressed in future studies and are critical to determining the role of circulating AFABP in metabolic and vascular disease in more detail.
Abbreviations
- AFABP:
-
Adipocyte fatty acid binding protein
- CAD:
-
Coronary artery disease
- DIO:
-
Diet-induced obesity
- EFABP:
-
Epidermal-type fatty acid binding protein
- ER:
-
Endoplasmic reticulum
- HSL:
-
Hormone-sensitive lipase
- JNK:
-
Jun N-terminal kinase
- LPS:
-
Lipopolysaccharides
- MCP:
-
Monocyte chemoattractant protein
- oxLDL:
-
Oxidised LDL
- PPAR:
-
Peroxisome proliferator-activated receptor
- TG:
-
Triacylglycerol
- TLR:
-
Toll-like receptor
- VEGF:
-
Vascular endothelial growth factor
References
Ahima RS, Flier JS (2000) Leptin. Annu Rev Physiol 62:413–437
Fruebis J, Tsao TS, Javorschi S et al (2001) Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci USA 98:2005–2010
Baxa CA, Sha RS, Buelt MK et al (1989) Human adipocyte lipid-binding protein—purification of the protein and cloning of its complementary-DNA. Biochemistry 28:8683–8690
Fu YC, Luo NL, Lopes-Virella MF, Garvey WT (2002) The adipocyte lipid binding protein (ALBP/aP2) gene facilitates foam cell formation in human THP-1 macrophages. Atherosclerosis 165:259–269
Elmasri H, Karaaslan C, Teper Y et al (2009) Fatty acid binding protein 4 is a target of VEGF and a regulator of cell proliferation in endothelial cells. FASEB J 23:3865–3873
Bernlohr DA, Angus CW, Lane MD, Bolanowski MA, Kelly TJ (1984) Expression of specific mRNAs during adipose differentiation: identification of an mRNA encoding a homologue of myelin P2 protein. Proc Natl Acad Sci USA 81:5468–5472
Bernlohr DA, Bolanowski MA, Kelly TJ, Lane MD (1985) Evidence for an increase in transcription of specific mRNAs during differentiation of 3t3-l1 preadipocytes. J Biol Chem 260:5563–5567
Bernlohr DA, Doering TL, Kelly TJ, Lane MD (1985) Tissue specific expression of p422 protein, a putative lipid carrier, in mouse adipocytes. Biochem Biophys Res Commun 132:850–855
Smith PJ, Wise LS, Berkowitz R, Wan C, Rubin CS (1988) Insulin-like growth factor-I is an essential regulator of the differentiation of 3T3-L1 adipocytes. J Biol Chem 263:9402–9408
Yang VW, Christy RJ, Cook JS, Kelly TJ, Lane MD (1989) Mechanism of regulation of the 422(Ap2) gene by CAMP during preadipocyte differentiation. Proc Natl Acad Sci USA 86:3629–3633
Christy RJ, Yang VW, Ntambi JM et al (1989) Differentiation-induced gene expression in 3T3-L1 preadipocytes: CCAAT/enhancer binding protein interacts with and activates the promoters of two adipocyte-specific genes. Genes Dev 3:1323–1335
Cabré A, Lázaro I, Girona J et al (2007) Fatty acid binding protein 4 is increased in metabolic syndrome and with thiazolidinedione treatment in diabetic patients. Atherosclerosis 195:e150–e158
Kletzien RF, Foellmi LA, Harris PKW, Wyse BM, Clarke SD (1992) Adipocyte fatty acid-binding protein: regulation of gene expression in vivo and in vitro by an insulin-sensitizing agent. Mol Pharmacol 42:558–562
Fisher RM, Eriksson P, Hoffstedt J et al (2001) Fatty acid binding protein expression in different adipose tissue depots from lean and obese individuals. Diabetologia 44:1268–1273
Coe NR, Bernlohr DA (1998) Physiological properties and functions of intracellular fatty acid-binding proteins. Biochim Biophys Acta 1391:287–306
Lalonde JM, Bernlohr DA, Banaszak LJ (1994) X-ray crystallographic structures of adipocyte lipid-binding protein complexed with palmitate and hexadecanesulfonic acid—properties of cavity binding-sites. Biochemistry 33:4885–4895
Xu ZH, Bernlohr DA, Banaszak LJ (1993) The adipocyte lipid-binding protein at 1.6-a resolution—crystal-structures of the apoprotein and with bound saturated and unsaturated fatty-acids. J Biol Chem 268:7874–7884
Liou HL, Storch J (2001) Role of surface lysine residues of adipocyte fatty acid-binding protein in fatty acid transfer to phospholipid vesicles. Biochemistry 40:6475–6485
Shen WJ, Liang Y, Hong R et al (2001) Characterization of the functional interaction of adipocyte lipid-binding protein with hormone-sensitive lipase. J Biol Chem 276:49443–49448
Smith AJ, Thompson BR, Sanders MA, Bernlohr DA (2007) Interaction of the adipocyte fatty acid-binding protein with the hormone-sensitive lipase—regulation by fatty acids and phosphorylation. J Biol Chem 282:32424–32432
Hertzel AV, Hellberg K, Reynolds JM et al (2009) Identification and characterization of a small molecule inhibitor of fatty acid binding proteins. J Med Chem 52:6024–6031
Smith AJ, Sanders MA, Juhlmann BE, Hertzel AV, Bernlohr DA (2008) Mapping of the hormone-sensitive lipase binding site on the adipocyte fatty acid-binding protein (AFABP) identification of the charge quartet on the AFABP/aP2 helix-turn-helix domain. J Biol Chem 283:33536–33543
Adida A, Spener F (2006) Adipocyte-type fatty acid-binding protein as inter-compartmental shuttle for peroxisome proliferator activated receptor γ agonists in cultured cell. Biochim Biophys Acta 1761:172–181
Gillilan RE, Ayers SD, Noy N (2007) Structural basis for activation of fatty acid-binding protein 4. J Mol Biol 372:1246–1260
Ayers SD, Nedrow KL, Gillilan RE, Noy N (2007) Continuous nucleocytoplasmic shuttling underlies transcriptional activation of PPAR gamma by FABP4. Biochemistry 46:6744–6752
Tan N-S, Shaw NS, Vinckenbosch N, Liu P, Yasmin R et al (2002) Selective cooperation between fatty acid binding proteins and peroxisome proliferator-activated receptors in regulating transcription. Mol Cell Biol 22:5114–5127
Hotamisligil GS, Johnson RS, Distel RJ et al (1996) Uncoupling of obesity from insulin resistance through a targeted mutation in aP2, the adipocyte fatty acid binding protein. Science 274:1377–1379
Coe NR, Simpson MA, Bernlohr DA (1999) Targeted disruption of the adipocyte lipid-binding protein (aP2 protein) gene impairs fat cell lipolysis and increases cellular fatty acid levels. J Lipid Res 40:967–972
Shaughnessy S, Smith ER, Kodukula S, Storch J, Fried SK (2000) Adipocyte metabolism in adipocyte fatty acid binding protein knockout (aP2(−/−)) mice after short-term high-fat feeding—functional compensation by the keritinocyte fatty acid binding protein. Diabetes 49:904–911
Maeda K, Cao H, Kono K, Gorgun CZ, Furuhashi M et al (2005) Adipocyte/macrophage fatty acid binding proteins control integrated metabolic responses in obesity and diabetes. Cell Metab 1:107–119
Uysal KT, Scheja L, Wiesbrock SM, Bonner-Weir S, Hotamisligil GS (2000) Improved glucose and lipid metabolism in genetically obese mice lacking aP2. Endocrinology 141:3388–3396
Baar RA, Dingfelder CS, Smith LA et al (2005) Investigation of in vivo fatty acid metabolism in AFABP/aP2(−/−) mice. Am J Physiol Endocrinol Metab 288:E187–E193
Cao H, Maeda K, Gorgun CZ et al (2006) Regulation of metabolic responses by adipocyte/macrophage fatty acid-binding proteins in leptin-deficient mice. Diabetes 55:1915–1922
Scheja L, Makowski L, Uysal KT et al (1999) Altered insulin secretion associated with reduced lipolytic efficiency in aP2−/− mice. Diabetes 48:1987–1994
Furuhashi M, Tuncman G, Gorgun CZ et al (2007) Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2. Nature 447:959–965
Lan H, Cheng CC, Kowalski TJ et al (2011) Small-molecule inhibitors of FABP4/5 ameliorate dyslipidaemia but not insulin resistance in mice with diet-induced obesity. J Lipid Res 52:646–656
Yang R, Castriota G, Chen Y et al (2011) RNAi-mediated germline knockdown of FABP4 increases body weight but does not improve the deranged nutrient metabolism of diet-induced obese mice. Int J Obes (Lond) 35:217–225
Makowski L, Boord JB, Maeda K et al (2001) Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nat Med 7:699–705
Boord JB, Maeda K, Makowski L et al (2002) Adipocyte fatty acid-binding protein, aP2, alters late atherosclerotic lesion formation in severe hypercholesterolemia. Arterioscler Thromb Vasc Biol 22:1686–1691
Boord JB, Maeda K, Makowski L et al (2004) Combined adipocyte-macrophage fatty acid-binding protein deficiency improves metabolism, atherosclerosis, and survival in apolipoprotein E-deficient mice. Circulation 110:1492–1498
Lee MYK, Li HY, Xiao Y et al (2011) Chronic administration of BMS309403 improves endothelial function in apolipoprotein E-deficient mice and in cultured human endothelial cells. Br J Pharmacol 162:1564–1576
Tuncman G, Erbay E, Hom X et al (2006) A genetic variant at the fatty acid-binding protein aP2 locus reduces the risk for hypertriglyceridemia, type 2 diabetes, and cardiovascular disease. Proc Natl Acad Sci USA 103:6970–6975
Chan KHK, Song YQ, Hsu YH et al (2010) Common genetic variants in fatty acid-binding protein-4 (FABP4) and clinical diabetes risk in the Women’s Health Initiative observational study. Obesity 18:1812–1820
Makowski L, Brittingham KC, Reynolds JM, Suttles J, Hotamisligil GS (2005) The fatty acid-binding protein, aP2, coordinates macrophage cholesterol trafficking and inflammatory activity. Macrophage expression of aP2 impacts peroxisome proliferator-activated receptor gamma and IkappaB kinase activities. J Biol Chem 280:12888–12895
Shum BO, Mackay CR, Gorgun CZ et al (2006) The adipocyte fatty acid-binding protein aP2 is required in allergic airway inflammation. J Clin Invest 116:2183–2192
Reynolds JM, Liu Q, Brittingham KC et al (2007) Deficiency of fatty acid-binding proteins in mice confers protection from development of experimental autoimmune encephalomyelitis. J Immunol 179:313–321
Kazemi MR, McDonald CM, Shigenaga JK, Grunfeld C, Feingold KR (2005) Adipocyte fatty acid-binding protein expression and lipid accumulation are increased during activation of murine macrophages by toll-like receptor agonists. Arterioscler Thromb Vasc Biol 25:1220–1224
Shi H, Kokoeva MV, Inouye K et al (2006) TLR4 links innate immunity and fatty acid–induced insulin resistance. J Clin Invest 116:3015–3025
Hui X, Li H, Zhou Z et al (2010) Adipocyte fatty acid-binding protein modulates inflammatory responses in macrophages through a positive feedback loop involving c-Jun NH2-terminal kinases and activator protein-1. J Biol Chem 285:10273–10280
Erbay E, Babaev VR, Mayers JR et al (2009) Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat Med 15:1383–1391
Fu YC, Luo LH, Luo NL, Garvey WT (2006) Lipid metabolism mediated by adipocyte lipid binding protein (ALBP/aP2) gene expression in human THP-1 macrophages. Atherosclerosis 188:102–111
Wang XQ, Yang K, He YS, Lu L, Shen WF (2011) Receptor mediated elevation in FABP4 levels by advanced glycation end products induces cholesterol and triacylglycerol accumulation in THP-1 Macrophages. Lipids 46:479–486
Saksi J, Nuotio K, Sonninen R et al (2011) Gene expression differences between stroke-associated and asymptomatic carotid plaques. J Mol Med 89:1015–1026
Holm S, Ueland T, Dahl TB et al (2011) Fatty acid binding protein 4 is associated with carotid atherosclerosis and outcome in patients with acute ischemic stroke. PLoS One 6:e28785
Agardh HE, Folkersen L, Ekstrand J et al (2011) Expression of fatty acid-binding protein 4/aP2 is correlated with plaque instability in carotid atherosclerosis. J Int Med 269:200–210
Peeters W, de Kleijn DPV, Vink A et al (2011) Adipocyte fatty acid binding protein in atherosclerotic plaques is associated with local vulnerability and is predictive for the occurrence of adverse cardiovascular events. Eur Heart J 32:1758–1768
Llaverias G, Noé V, Peñuelas S et al (2004) Atorvastatin reduces CD68, FABP4, and HBP expression in oxLDL-treated human macrophages. Biochem Biophys Res Commun 318:265–274
Elmasri H, Ghelfi E, Yu C et al (2012) Endothelial cell-fatty acid binding protein 4 promotes angiogenesis: role of stem cell factor/c-kit pathway. Angiogenesis 15:457–468
Xu A, Wang Y, Xu JY et al (2006) Adipocyte fatty acid-binding protein is a plasma biomarker closely associated with obesity and metabolic syndrome. Clin Chem 52:405–413
Veerkamp JH, Maatman RG (1995) Cytoplasmic fatty acid-binding proteins: their structure and genes. Prog Lipid Res 34:17–52
Reese-Wagoner A, Thompson J, Banaszak L (1999) Structural properties of the adipocyte lipid binding protein. Biochim Biophys Acta 1441:106–116
Radisky DC, Stallings-Mann M, Hirai Y, Bissell MJ (2009) Single proteins might have dual but related functions in intracellular and extracellular microenvironments. Nat Rev Mol Cell Biol 10:228–234
Mohan SK, Rani SG, Yu C (2010) The heterohexameric complex structure, a component in the non-classical pathway for fibroblast growth factor 1 (FGF1) secretion. J Biol Chem 285:15464–15475
Aoki N, Jin-no S, Nakagawa Y et al (2007) Identification and characterization of microvesicles secreted by 3T3-L1 adipocytes: redox- and hormone-dependent induction of milk fat globule-epidermal growth factor 8-associated microvesicles. Endocrinology 148:3850–3862
Lamounier-Zepter V, Look C, Alvarez J et al (2009) Adipocyte fatty acid-binding protein suppresses cardiomyocyte contraction: a new link between obesity and heart disease. Circ Res 105:326–334
Stejskal D, Karpisek M (2006) Adipocyte fatty acid binding protein in a Caucasian population: a new marker of metabolic syndrome? Eur J Clin Invest 36:621–625
Coll B, Cabre A, Alonso-Villaverde C et al (2008) The fatty acid binding protein-4 (FABP4) is a strong biomarker of metabolic syndrome and lipodystrophy in HIV-infected patients. Atherosclerosis 199:147–153
Cabre A, Lazaro I, Cofan M et al (2010) FABP4 plasma levels are increased in familial combined hyperlipidemia. J Lipid Res 51:1173–1178
Milner KL, van der Poorten D, Xu A et al (2009) Adipocyte fatty acid binding protein levels relate to inflammation and fibrosis in nonalcoholic fatty liver disease. Hepatology 49:1926–1934
Yun KE, Kim SM, Choi KM, Park HS (2009) Association between adipocyte fatty acid-binding protein levels and childhood obesity in Korean children. Metabolism 58:798–802
Tonjes A, Kralisch S, Lossner U et al (2012) Metabolic and genetic predictors of circulating adipocyte fatty acid-binding protein. Int J Obes 36:766–773
Cabre A, Lazaro I, Girona J et al (2008) Plasma fatty acid binding protein 4 is associated with atherogenic dyslipidemia in diabetes. J Lipid Res 49:1746–1751
Hsu BG, Chen YC, Lee RP et al (2010) Fasting serum level of fatty-acid-binding protein 4 positively correlates with metabolic syndrome in patients with coronary artery disease. Circ J 74:327–331
Yeung DC, Xu A, Cheung CW et al (2007) Serum adipocyte fatty acid-binding protein levels were independently associated with carotid atherosclerosis. Arterioscler Thromb Vasc Biol 27:1796–1802
Rhee EJ, Lee WY, Park CY et al (2009) The association of serum adipocyte fatty acid-binding protein with coronary artery disease in Korean adults. Eur J Endocrinol 160:165–172
Doi M, Miyoshi T, Hirohata S et al (2011) Association of increased plasma adipocyte fatty acid-binding protein with coronary artery disease in non-elderly men. Cardiovasc Diabetol 10:44
Miyoshi T, Onoue G, Hirohata A et al (2010) Serum adipocyte fatty acid-binding protein is independently associated with coronary atherosclerotic burden measured by intravascular ultrasound. Atherosclerosis 211:164–169
Bao YQ, Lu ZG, Zhou M et al (2011) Serum levels of adipocyte fatty acid-binding protein are associated with the severity of coronary artery disease in chinese women. PLoS One 6:e19115
Pala L, Monami M, Ciani S et al (2012) Adipokines as possible new predictors of cardiovascular diseases: a case control study. J Nutr Metab 2012:1–5
Yoo HJ, Kim S, Park MS et al (2011) Serum adipocyte fatty acid-binding protein is associated independently with vascular inflammation: analysis with (18)F-fluorodeoxyglucose positron emission tomography. J Clin Endocrinol Metab 96:E488–E492
Choi KM, Kim TN, Yoo HJ et al (2009) Effect of exercise training on A-FABP, lipocalin-2 and RBP4 levels in obese women. Clin Endocrinol 70:569–574
Simon I, Escote X, Vilarrasa N et al (2009) Adipocyte fatty acid-binding protein as a determinant of insulin sensitivity in morbid-obese women. Obesity 17:1124–1128
Corripio R, Gonzalez-Clemente JM, Perez-Sanchez J et al (2010) Weight loss in prepubertal obese children is associated with a decrease in adipocyte fatty-acid-binding protein without changes in lipocalin-2: a 2-year longitudinal study. Eur J Endocrinol 163:887–893
Stejskal D, Karpisek M, Bronsky J (2008) Serum adipocyte-fatty acid binding protein discriminates patients with permanent and temporary body weight loss. J Clin Lab Ana 22:380–382
Wu Y-W, Kao H-L, Huang C-L et al (2012) The effects of 3-month atorvastatin therapy on arterial inflammation, calcification, abdominal adipose tissue and circulating biomarkers. Eur J Nucl Med Mol Imaging 39:399–407
Xu A, Tso AW, Cheung BM et al (2007) Circulating adipocyte-fatty acid binding protein levels predict the development of the metabolic syndrome: a 5-year prospective study. Circulation 115:1537–1543
Choi KM, Yannakoulia M, Park MS et al (2011) Serum adipocyte fatty acid-binding protein, retinol-binding protein 4, and adiponectin concentrations in relation to the development of the metabolic syndrome in Korean boys: a 3-y prospective cohort study. Am J Clin Nutr 93:19–26
Tso AW, Xu A, Sham PC et al (2007) Serum adipocyte fatty acid binding protein as a new biomarker predicting the development of type 2 diabetes: a 10-year prospective study in a Chinese cohort. Diabetes Care 30:2667–2672
Furuhashi M, Ishimura S, Ota H et al (2011) Serum fatty acid-binding protein 4 is a predictor of cardiovascular events in end-stage renal disease. PLoS One 6:e27356
Tso AWK, Lam TKY, Xu A et al (2011) Serum adipocyte fatty acid-binding protein associated with ischemic stroke and early death. Neurology 76:1968–1975
von Eynatten M, Breitling LP, Roos M et al (2012) Circulating adipocyte fatty acid-binding protein levels and cardiovascular morbidity and mortality in patients with coronary heart disease. Arterioscler Thromb Vasc Biol 32:2327–2335
Acknowledgements
Studies from our group summarised in this review were supported by grants to M. Fasshauer from the Deutsche Forschungsgemeinschaft (DFG, KFO 152 ‘Atherobesity’, FA476/4–1), the Federal Ministry of Education and Research (BMBF), Germany, FKZ: 01EO1001 (IFB Adiposity Diseases, projects K7-3, K7-9, and K7-31), and the Deutsche Hochdruckliga e.V. The authors would like to thank S. Lorenz, J. Richter and J. Schaarschmidt (all Department of Endocrinology and Nephrology, University of Leipzig, Leipzig, Germany) for their help in generating the figures.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
Both authors were responsible for the conception, design and drafting of the manuscript, and approved the final version for publication.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kralisch, S., Fasshauer, M. Adipocyte fatty acid binding protein: a novel adipokine involved in the pathogenesis of metabolic and vascular disease?. Diabetologia 56, 10–21 (2013). https://doi.org/10.1007/s00125-012-2737-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2737-4