
Abstract
Aims/hypothesis
The renin–angiotensin system (RAS) potentially has a role in the development of end-organ damage, and tissue RAS activation has been suggested as a risk factor for diabetic retinopathy. We have recently shown significant involvement of (pro)renin receptor ([P]RR) in retinal inflammation in a rodent model of early diabetes. In this study we aim to elucidate the (P)RR-associated pathogenesis of fibrovascular proliferation, a late-stage angiogenic complication in human diabetic retinopathy.
Methods
Vitreous fluids from 23 eyes of patients with proliferative diabetic retinopathy (PDR) and 16 eyes of controls with non-diabetic, idiopathic macular diseases (macular hole and epiretinal membrane) were collected. Protein levels of soluble (P)RR were measured by ELISA, and immunofluorescence was performed to assess the localisation of (P)RR and related molecules in fibrovascular tissues from PDR eyes.
Results
(P)RR immunoreactivity was detected in neovascular endothelial cells, colocalised with prorenin, phosphorylated extracellular signal-regulated kinase (ERK) and vascular endothelial growth factor (VEGF). Prorenin application to human retinal microvascular endothelial cells significantly upregulated mRNA expression of VEGF, especially the VEGF165 isoform, which was abolished by (P)RR or ERK signalling blockade. Proteases known to cleave (P)RR, including furin, were positive in endothelial cells in fibrovascular tissues. Protein levels of soluble (P)RR in vitreous fluids were higher in PDR eyes than in non-diabetic control eyes, and correlated significantly with vitreous prorenin and VEGF levels and the vascular density of fibrovascular tissues.
Conclusions/interpretation
Our data using human samples provide the first evidence that (P)RR is associated with angiogenic activity in PDR.
Similar content being viewed by others

Introduction
Diabetic retinopathy is a leading cause of severe vision loss and blindness when it progresses to the stage of proliferative diabetic retinopathy (PDR), which is characterised by fibrovascular proliferation. Fibrovascular tissue is formed by the extension of retinal angiogenesis into the vitreous cavity, and formation of fibrovascular tissue results in severe complications, such as vitreous haemorrhage and tractional retinal detachment. A number of studies have shown that vascular endothelial growth factor (VEGF) is a major angiogenic factor in PDR [1–3]. We have also revealed a significant contribution by VEGF165 to angiogenic activity in PDR, showing that fibrovascular tissues coexpressing VEGF receptor (VEGFR)-2 and neuropilin (NRP)1, the specific receptor for VEGF165, are highly vascularised [4–6].
The renin–angiotensin system (RAS), originally regarded as an important controller of systemic blood pressure (circulatory RAS), plays a pivotal role in pathological vascular conditions including inflammation and angiogenesis (tissue RAS). Several types of organ damage are known to result from activation of tissue RAS. Clinical trials have found that angiotensin II type 1 receptor (AT1R) blockade resulted in beneficial effects on the incidence and progression of diabetic retinopathy [7, 8]. We have previously revealed, using animal models of diabetes, the molecular mechanisms by which tissue RAS promotes retinal inflammation and angiogenesis [9–12], while RAS components proved to be present in the retina [11, 13, 14]. These multiple reports suggest that tissue RAS activation in the retina is a significant risk factor in the development of diabetic retinopathy.
Prorenin is known to interact with (pro)renin receptor ([P]RR) to exert renin activity through conformational change of the prorenin molecule (non-proteolytic activation of prorenin causing tissue RAS) instead of the conventional proteolysis of the prorenin prosegment basically achieved by processing enzymes (proteolytic activation of prorenin causing circulatory RAS). Prorenin binding to (P)RR activates RAS-independent signal transduction via mitogen-activated protein kinases (MAPKs) including extracellular signal-regulated kinase (ERK) 1/2 and p38, both of which contribute to the pathogenesis of organ damage [15, 16]. The dual activation of tissue RAS and RAS-independent signalling pathways, referred to as the receptor-associated prorenin system (RAPS), has been shown to be involved in the molecular pathogenesis of retinal vascular disorders such as inflammation and angiogenesis [14, 17–21].
In this study, we investigated localisation and expression of (P)RR in fibrovascular tissues and vitreous fluids from patients with PDR and evaluated the molecular mechanisms in vitro to confirm ideas about (P)RR from previous animal studies [14, 17–21]. We herein report the first evidence of a close association between (P)RR and angiogenic activity in human PDR.
Methods
Human surgical samples
Vitreous fluids were collected from 23 eyes of 23 patients (13 male and 10 female, mean ± SEM age 60.3 ± 1.6 years) with PDR, who had undergone pars plana vitrectomy for prolonged vitreous haemorrhage and tractional retinal detachment involving the macula. Control vitreous samples were obtained from 16 eyes of 16 patients (seven male and nine female, mean ± SEM age 64.0 ± 1.1 years) with non-diabetic, idiopathic macular diseases including epiretinal membrane and macular hole. Eight fibrovascular tissue samples were surgically removed from the PDR eyes that provided vitreous fluids, and were used for immunohistochemistry. An additional three fibrovascular tissue samples were processed for gene expression analyses. From all the patients, vitreous fluids were collected without dilution at the start of the pars plana vitrectomy. In addition, from the patients with PDR, fibrovascular tissue samples were obtained after complete shaving of the vitreous gel. After sonication, vitreous samples were centrifuged at 15,000 g for 20 min at 4°C, and then supernatant fractions of the samples were stored at −80°C until use in ELISA. The fibrovascular tissue samples were embedded in paraffin after fixation in 4% paraformaldehyde. This study was conducted in accordance with the tenets of the Declaration of Helsinki after approval from the institutional review board of Hokkaido University Hospital. Written consent was obtained from all patients after they had received an explanation of the purpose and procedures of the study.
Plasmid construction and antibody generation
Human (P)RR cDNA (GenBank No. NM_005765; also known as ATP6AP2) was subcloned into pGEX4T-2 vector (GE Healthcare, Piscataway, NJ, USA), and glutathione S-transferase (GST) fusion (P)RR protein was expressed in Escherichia coli strain Rosetta-gami 2 (DE3) (Novagen, Madison, WI, USA). GST fusion proteins were purified by binding to glutathione–Sepharose (GE Healthcare). All deletion mutants from the (P)RR expression construct were generated by PCR and sequence-verified before use. A mouse monoclonal antibody against (P)RR (Ms3-24) was raised against GST fusion full-length (P)RR protein and purified using a protein G column (ITM, Nagano, Japan).
Immunoblot analysis of GST and (P)RR
Protein extracts from mouse tissues and transfected E. coli extracts were solubilised in 2 × SDS sample buffer by heating to 100°C for 5 min and separated by 10% SDS-PAGE. Proteins were transferred to polyvinylidene fluoride membrane by electroblotting, and immunoblot analyses were performed using Ms3-24 and antibodies against GST (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and ß-actin (Cell Signaling Technology, Danvers, MA, USA), as previously described [22].
Immunofluorescence microscopy for (P)RR, VEGF and related molecules
Paraffin sections of mouse retinas and fibrovascular tissues were deparaffinised and hydrated by exposure to xylene and graded alcohols followed by water. As a pretreatment, microwave-based antigen retrieval was performed in 10 mmol/l citrate buffer (pH 6). Sections were probed with the following primary antibodies: Ms3-24; goat polyclonal antibody against (P)RR (R&D systems, Minneapolis, MN, USA); rabbit polyclonal antibodies against phosphorylated (p-)ERK1/2 (Cell Signaling Technology), α-smooth muscle actin (α-SMA), CD31, CD34, VEGF (Abcam, Cambridge, MA, USA), furin (Enzo Life Sciences, Farmingdale, NY, USA) and ADAM (a disintegrin and metalloproteinase) 19 (Novus Biologicals, Littleton, CO, USA); and mouse monoclonal antibody against prorenin (Abcam). The purchased (P)RR antibody crossreacts with both human and mouse (P)RR. The prorenin antibody used in this study detects the prosegment region of prorenin, thus not recognising renin. The secondary antibodies used for fluorescence detection were Alexa Fluor 488 and 546 (Life Technologies, Carlsbad, CA, USA). Sections were visualised under a FluoView FV10i-DOC (Olympus, Tokyo, Japan).
Cell culture and chemicals
hTERT-RPE, ARPE-19 and Y79 cells were purchased from American Type Culture Collection. Primary human retinal microvascular endothelial cells (HRMECs), CS-C medium, a growth medium optimised for HRMECs, and culture boost were purchased from Cell Systems (Kirkland, WA, USA). HRMECs were cultured following the manufacturer's instructions. To cover the handle region of the prorenin molecule, which is the binding site of (P)RR [23], we synthesised decoy peptides NH2-RIFLKRMPSI-COOH as a human (P)RR blocker (PRRB), and purified them by high-pressure liquid chromatography on a C-18 reverse-phase column. After being serum deprived, cells were pretreated with 1 μmol/l PRRB for 1 h or 10 μmol/l MAPK/ERK kinase (MEK) inhibitor U0126 (Promega, Madison, WI, USA) for 15 min. Prorenin was then added at a final concentration of 10 nmol/l, and the cells were incubated for 3 h and processed for analyses to detect the mRNA expression of VEGF isoforms and their receptors.
RT-PCR and real-time quantitative (q)PCR analysis of (P)RR, VEGF and related molecules
Total RNA was isolated from tissues and cells using TRIzol (Life Technologies). Reverse transcription was performed with GoScrip Reverse Transcriptase (Promega) and oligo dT(20) primers, essentially as described [24]. Real-time qPCR was performed using the GoTaq qPCR Master Mix (Promega), THUNDERBIRD Probe qPCR Mix (TOYOBO, Tokyo, Japan) and StepOne plus System (Life Technologies). All primers are listed in electronic supplementary material (ESM) Table 1. The correct amplification of a specific product was confirmed by the dissociation temperature of the product and agarose gel electrophoresis [24, 25]. Taqman gene expression assays for hypoxanthine phosphoribosyltransferase 1 (HPRT1) (Life Technologies) were performed. All experimental samples were normalised using human HPRT1 as an internal control.
ELISA for VEGF and soluble form of (P)RR (s[P]RR) in vitreous fluids
Goat polyclonal antibodies against (P)RR and Ms3-24 were used in a double-antibody sandwich ELISA to detect a soluble form of (P)RR in the vitreous fluids. The polyclonal antibody against (P)RR was fixed to 96-well plates. Vitreous samples were immobilised on the plates, followed by washing with PBS containing 0.05% Tween 20 and the addition of Ms3-24. After incubation and washing, signals were visualised using tetramethylbenzidine detection reagent per standard protocol [26]. A standard curve was plotted from the measurements made with the standard solution (50–50,000 pg/ml), and the concentration of s(P)RR in each sample was determined from this curve. The protein levels of prorenin and VEGF were measured in vitreous samples by ELISA using kits for human prorenin (Innovative Research, Novi, MI, USA) and VEGF (R&D Systems) per the manufacturers' instructions. Detection ranges of prorenin and VEGF are 10–10,000 and 15.6–2,000 pg/ml, respectively.
Calculation for activated prorenin concentration in vitreous fluids
Activated prorenin corresponds to prorenin bound to s(P)RR, and the dissociation constant (K D) for the binding of prorenin to s(P)RR was calculated as in a previous report [27]: K D (4.0 nmol/l) = (prorenin) × (s[P]RR)/(activated prorenin). Based on the K D, we determined the activated prorenin concentration in the vitreous fluids.
Morphometric analysis
The degree of angiogenesis was evaluated by morphometric analysis of the tissue sections immunostained for CD34, as described previously [4]. Briefly, the entire tissue in each section was photographed to measure the whole area of the tissue, and the number of CD34-immunoreactive vessels with a distinct lumen was counted in a masked fashion. The number of vessels per mm2 was calculated as the vascular density (vessels/mm2) of the section.
Statistical analysis
All results ware expressed as mean ± SEM. The values were analysed by Student's t test and Spearman rank correlation. Differences between the means were considered significant when the p values were <0.05.
Results
Validation of mouse monoclonal antibody against human (P)RR
To understand the biological function of (P)RR, a mouse monoclonal antibody (Ms3-24) was raised against recombinant (P)RR fusion GST protein (GST-fl[P]RR). (P)RR is a single transmembrane protein consisting of 350 amino acids [15]; the gene that encodes it is also named ATP6AP2 because the C-terminal (P)RR fragment is identical with ATP6AP2 [28]. fl(P)RR encodes the full-length (P)RR protein without its signal peptide sequence. To map the epitope of antibody Ms3-24, we generated four (P)RR cDNA deletion constructs (Fig. 1a). s(P)RR, a solubilised form of (P)RR (residues 17-274), lacks the transmembrane domain at the C-terminus, and each of the two additional N-terminal constructs contains a progressive C-terminal deletion ([P]RR-225, residues 17-225; [P]RR-124, residues 17-124). The last fragment, ATP6AP2, encodes the C-terminal region after the furin cleavage site (residues 279-350). All five GST-(P)RR fusion proteins were found in E. coli and purified and examined by immunoblot analysis. Antibody Ms3-24 detected fl(P)RR, s(P)RR, (P)RR-125 and (P)RR-225, but not ATP6AP2 and GST proteins (Fig. 1b). Epitope mapping with different length (P)RR proteins demonstrated that antibody Ms3-24 has an epitope at the N-terminus, and can detect both fl(P)RR and s(P)RR proteins.

Validation of mouse monoclonal antibody against human (P)RR. (a) Schematic representation of (P)RR constructs used. SP, signal peptide; TM, transmembrane domain. The Grey box (position from 275 to 278) indicates the furin cleavage site. (b) Immunoblot analysis of recombinant (P)RR fusion GST protein. The GST fusion proteins were detected using antibody against GST or (P)RR (Ms3-24). (c) Immunoblot analysis of mouse tissue. (d–f, f′, f″) Localisation of (P)RR in the mouse retina. Double labelling of Ms3-24 (green), (P)RR (red) and DAPI (blue). (g) No primary antibodies. OS, outer segment; IS, inner segment; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. Scale bar 30 μm
Next, we checked the specificity of this antibody through immunoblot and immunolocalisation experiments using mouse tissue. Immunoblot analysis showed the predicted protein species of molecular mass ∼35 kDa in mouse retina, brain and kidney (Fig. 1c). The overall immunofluorescence signals of antibody Ms3-24 (Fig. 1d, f) colocalised with a characterised commercial (P)RR antibody (Fig. 1e, f). Immunolocalisation of (P)RR in mouse retina showed staining in the retinal vessels within the ganglion cell layer (Fig. 1d–f, f″) and in Müller cells within the inner nuclear layer (Fig. 1d–f), which is consistent with previous reports [14, 17, 29]. Antibody Ms3-24 also detected a signal in the photoreceptor layer (Fig. 1d–f, f′), consistent with a gene expression profiling report that showed (P)RR production in flow-sorted mouse rod photoreceptors [30]. There was no signal detected in control experiments where Ms3-24 and (P)RR antibodies were omitted (Fig. 1g).
Endothelial localisation of (P)RR in human PDR fibrovascular tissues
(P)RR localisation to retinal vessels was previously demonstrated in rodents [14, 17, 19, 29]; however, no studies on human ocular tissue have been reported. First, we confirmed mRNA expression of (P)RR and RAS components (prorenin [REN], angiotensinogen [AGT], angiotensin-converting enzyme [ACE], AT1R and angiotensin II type 2 receptor [AT2R]) in fibrovascular tissue samples surgically excised from human PDR eyes and several human retinal cell lines including HRMECs (primary cell line), hTERT-RPE, ARPE-19 and Y79 (established cell lines). RT-PCR analyses showed that (P)RR and RAS components were expressed in fibrovascular tissues, HRMECs and other retinal cell lines (Fig. 2a).

Gene expression and localisation of (P)RR in human PDR fibrovascular tissues. (a) Gene expression analysis of (P)RR and RAS components in fibrovascular tissues and human retinal cell lines. RT-PCR products of (P)RR and RAS components (REN, AGT, ACE, AT1R [also known as AGTR1] and AT2R [also known as AGTR2]) from three fibrovascular tissues (FVT 1–3) and primary (HRMEC) and established (hTERT-RPE ARPE-19, and Y79) human retinal cell lines. HPRT1 was used as an internal control. (b–d, d′) Double labelling in fibrovascular tissues of Ms3-24 (green), CD31 (red) and DAPI (blue). (e–g, g′) Double labelling in fibrovascular tissues of (P)RR (green), α-SMA (red) and DAPI (blue). Scale bar 30 μm
Next, using antibody Ms3-24, we carried out immunofluorescence analysis on fibrovascular tissue samples. Double-staining experiments demonstrated colocalisation of Ms3-24 signal with CD31, a vascular endothelial cell marker (Fig. 2b–d, d′), but not with α-SMA, a vascular smooth muscle cell or pericyte marker (Fig. 2e–g, g′), showing that (P)RR protein was selectively produced in vascular endothelial cells, but not in pericytes.
(P)RR downstream signalling of p-ERK and VEGF in human PDR fibrovascular tissues
Interaction of prorenin and (P)RR has been reported to stimulate the phosphorylation of ERK and the production of VEGF in vivo and in vitro [18, 19]. To examine the production and tissue localisation of the prorenin–(P)RR downstream molecules, human fibrovascular tissues were immunostained with antibodies against prorenin, (P)RR, p-ERK1/2 and VEGF. Immunofluorescence analysis of the fibrovascular tissue samples showed colocalisation of prorenin (Fig. 3a–c), p-ERK1/2 (Fig. 3d–f) and VEGF (Fig. 3g–i) in (P)RR-positive vascular endothelial cells (Fig. 3b, d and g), suggesting the robust activation of prorenin-induced (P)RR signal transduction in the pathogenesis of fibrovascular proliferation in human PDR.

(P)RR downstream signal p-ERK and VEGF in human PDR fibrovascular tissues. Localisation of (P)RR and related proteins in the human fibrovascular tissues. (a–c) Double labelling of prorenin (green), (P)RR (red) and DAPI (blue). (d–f) Double labelling of Ms3-24 (green), p-ERK1/2 (red) and DAPI (blue). (g–i) Double labelling of Ms3-24 (green), VEGF (red) and DAPI (blue). Scale bar 30 μm
(P)RR-mediated upregulation of VEGF in primary human retinal endothelial cells
To confirm an association between prorenin–(P)RR signalling and its downstream VEGF upregulation in vitro, we examined mRNA expression levels of three major VEGF isoforms (VEGF121, VEGF165 and VEGF189) in HRMECs stimulated with prorenin. HRMECs were stimulated with prorenin after being serum deprived, and total RNAs were harvested and processed for real-time qPCR. Interestingly, all three VEGF isoform expression profiles, especially VEGF165, were significantly increased after prorenin treatment (VEGF121, fold change 1.38, p < 0.05; VEGF165, fold change 2.16, p < 0.01; VEGF189, fold change 1.31, p < 0.05). Importantly, pretreatment with PRRB or the MEK inhibitor, U0126, inhibited VEGF expression (PRRB pretreatment [VEGF121, fold change 1.12, p > 0.05; VEGF165, fold change 1.09, p < 0.01; VEGF189, fold change 0.92, p < 0.05], U0126 pretreatment [VEGF121, fold change 1.24, p > 0.05; VEGF165, fold change 1.38, p < 0.05; VEGF189, fold change 0.91, p < 0.05]) (Fig. 4a). VEGF mRNA expression levels were not affected by much lower concentrations of prorenin (1–10 pmol/l) or by U0126 or PRRB alone (data not shown).

(P)RR-mediated upregulation of VEGF in primary human retinal endothelial cells. (a) VEGF mRNA expression in prorenin-treated HRMECs. Real-time PCR amplification of VEGF121, VEGF165 and VEGF189 at 3 h after stimulation with prorenin. (b) mRNA expression of VEGF receptors in prorenin-treated HRMECs. Real-time PCR amplification of VEGFR-1, VEGFR-2 and NRP1 at 3 h after administration of prorenin. qPCR data were normalised to HPRT1 expression with control HRMECs as a reference. White, black, grey and hatched bars indicate control cells, prorenin-treated cells, prorenin-treated cells with PRRB preincubation and prorenin-treated cells with U0126 preincubation, respectively. *p < 0.05, **p < 0.01
VEGFR-1 (FLT1), VEGFR-2 (KDR) and NRP1 mRNA expression levels were also checked; however, there were no significant differences in expression levels after prorenin treatment (VEGFR-1, fold change 1.02, p > 0.05; VEGFR-2, fold change 1.13, p > 0.05; NRP1, fold change 1.02, p > 0.05) (Fig. 4b). These data suggest ligand-based activation of the VEGF system through the prorenin–(P)RR-ERK signalling axis.
Upregulation of s(P)RR levels in human PDR vitreous fluids
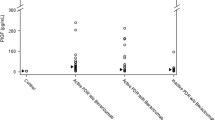
(P)RR also exists in a solubilised form, s(P)RR, in the plasma after cleavage by proteases such as the proprotein convertase, furin, and ADAM19 [31, 32]. To investigate whether s(P)RR levels are changed in PDR eyes, we performed ELISA experiments to measure s(P)RR in the vitreous fluids aspirated from PDR and non-diabetic control eyes. s(P)RR protein was detectable in all the PDR and control vitreous samples, but s(P)RR levels were significantly (p < 0.01) increased in the vitreous fluids of PDR eyes (3.88 ± 0.43 ng/ml) compared with those of control eyes (0.56 ± 0.16 ng/ml) (Fig. 5a).

Upregulation of s(P)RR protein levels in human PDR vitreous fluids. (a) Protein levels of s(P)RR in PDR and control vitreous fluids. Black symbols indicate individual samples, and bar shows the average. ERM, epiretinal membrane; MH, macular hole. **p < 0.01. (b–d) Double labelling of Ms3-24 (green), furin (red) and DAPI (blue) in the human fibrovascular tissues. (e–g) Double labelling of Ms3-24 (green), ADAM19 (red) and DAPI (blue) in the human fibrovascular tissues. Scale bar 30 μm
Next, we carried out immunofluorescence analysis of furin (Fig. 5b–d) and ADAM19 (Fig. 5e–g), both of which are known to cleave (P)RR, in the fibrovascular tissue samples surgically excised from human PDR eyes. Both signals were observed in (P)RR-positive vascular endothelial cells in the fibrovascular tissues, confirming the validity of increased levels of s(P)RR in the vitreous fluids of PDR eyes but not of control eyes lacking fibrovascular tissues.
Upregulation of prorenin and activated prorenin levels in human PDR vitreous fluids
Protein levels of prorenin in the vitreous fluids of PDR have been reported to be increased compared with controls [33]. In accordance with previous data, prorenin protein levels were significantly (p < 0.01) higher in the vitreous fluids of PDR patients (184.09 ± 26.02 pg/ml) than in those of controls (65.16 ± 12.85 pg/ml) (Fig. 6a), and increased prorenin levels correlated significantly (p < 0.05, r 2 = 0.242) with s(P)RR levels (Fig. 6b). Based on the K D for the binding of prorenin to s(P)RR [27], we determined the vitreous levels of activated prorenin. Importantly, activated prorenin levels in PDR samples (157.45 fmol/l) were significantly (p < 0.01) elevated compared with controls (7.78 fmol/l) (Fig. 6c).

Upregulation of prorenin and activated prorenin levels in human PDR vitreous fluids. (a) Protein levels of prorenin in PDR and control vitreous fluids. Black symbols indicate individual samples, and bar shows the average. ERM, epiretinal membrane; MH, macular hole. **p < 0.01. (b) Correlation between s(P)RR and prorenin in PDR vitreous fluids. (c) Protein levels of activated prorenin in PDR and control vitreous fluids. **p < 0.01
Correlation of s(P)RR with VEGF and the vascular density of fibrovascular tissues
Intraocular levels of VEGF, a key molecule responsible for angiogenesis, have been shown to be increased in PDR eyes [1, 2, 34]. To investigate the relationship between s(P)RR and VEGF, we checked VEGF protein levels in the PDR vitreous samples. Interestingly, VEGF protein levels in PDR eyes, which were significantly (p < 0.01) increased compared with controls (536.20 ± 115.94 vs 66.39 ± 32.22 pg/ml) (Fig. 7a), correlated significantly with those of s(P)RR (p < 0.01, r 2 = 0.546) (Fig. 7b) and activated prorenin (p < 0.01, r 2 = 0.243) (Fig. 7c).

Correlation of s(P)RR with VEGF and the vascular density of fibrovascular tissues. (a) Protein levels of VEGF in PDR and control vitreous fluids. Black symbols indicate individual samples, and bar shows the average. ERM, epiretinal membrane; MH, macular hole. **p < 0.01. (b) Correlation between s(P)RR and VEGF in PDR vitreous fluids. (c) Correlation between activated prorenin and VEGF in PDR vitreous fluids. (d) Correlation between s(P)RR in the vitreous fluids and the vascular density of fibrovascular tissues obtained from PDR eyes
Moreover, the vascular density of fibrovascular tissues, which represents angiogenic activity in PDR [4], was evaluated by CD34 immunolabelling in eight surgical samples from the patients who donated vitreous fluids. Importantly, there was a significant (p < 0.05, r 2 = 0.520) correlation between s(P)RR protein levels in the vitreous fluids and the vascular density of fibrovascular tissues, both of which were derived from the same PDR eyes (Fig. 7d).
Discussion
This study reveals, for the first time to our knowledge, several important findings concerning the relationship of (P)RR to angiogenic activity in human PDR. First, (P)RR and RAS component genes were expressed in surgically excised fibrovascular tissues and human retinal cell lines (Fig. 2). (P)RR protein was immunopositive in neovascular endothelial cells in the tissues (Fig. 2), and colocalised with its ligand, prorenin, and downstream molecules, p-ERK and VEGF (Fig. 3). Second, stimulation of primary human retinal vascular endothelial cell culture with prorenin triggered the upregulation of VEGF, especially VEGF165, which was mediated via (P)RR and its downstream ERK signalling (Fig. 4). Finally, s(P)RR, a truncated form of (P)RR, was increased in vitreous fluids of human PDR (Fig. 5), and this correlated with vitreous prorenin (Fig. 6) and VEGF levels and the vascular density of fibrovascular tissues (Fig. 7). These novel findings on human samples, in concert with our previous data using rodent models [17, 18], further support the pathogenic role of (P)RR in the development of PDR, which is characterised by VEGF-driven angiogenesis.
Induction of diabetes in a mouse model led to a significant increase in retinal mRNA expression of REN and production of VEGF protein, and administration of PRRB reduced diabetes-induced retinal expression of VEGF and pathological angiogenesis in ischaemic retinopathy [17, 19]. In the present study, prorenin application to HRMECs elevated mRNA expression of VEGF isoforms, especially VEGF165, which was abolished by pretreatment with PRRB or the MEK inhibitor, U0126 (Fig. 4a). This is compatible with the results of animal studies showing that the interaction of prorenin and (P)RR induces expression of VEGF capable of promoting inflammation-related angiogenesis in the eye [18]. Angiotensin II induced the in vitro expression of VEGF and VEGFR-2 mRNA through AT1R on cultured bovine retinal vascular cells, enhancing VEGF-induced angiogenic activity [35]. In contrast, in prorenin-treated HRMECs (Fig. 4b), VEGFR-2 mRNA expression did not change from the physiological baseline, suggesting that the marked increase in VEGF165 mRNA expression (Fig. 4a) was not induced by AT1R signalling pathways mediated by RAS activation despite the constitutive expression of RAS components (Fig. 2a), but by RAS-independent intracellular signal transduction via (P)RR bound to prorenin.
In accordance with these in vitro findings, immunofluorescence studies in human fibrovascular tissues showed that (P)RR was colocalised with prorenin, p-ERK1/2 and VEGF in vascular endothelial cells (Figs 2 and 3). Binding of prorenin to (P)RR has been reported to initiate the ERK1/2 signalling pathway and increase the protein level of VEGF [15, 18]. Taking these findings together, it is possible that prorenin in human fibrovascular tissues triggers VEGF (VEGF165) expression through the (P)RR–ERK axis leading to enhanced inflammatory response and angiogenic activity, both of which mediate the pathogenesis of diabetic retinopathy [4, 5, 36].
To quantitatively examine the relationship between (P)RR and angiogenic activity in PDR, we performed ELISAs to check s(P)RR protein concentration in the vitreous fluids. Protein levels of s(P)RR were significantly increased in the vitreous fluids of PDR patients (Fig. 5a), and this correlated with increased levels of both prorenin (Fig. 6a, b) and VEGF (Fig. 7a, b) and the degree of angiogenesis in PDR (Fig. 7d). fl(P)RR has been shown to localise in the cellular membrane and endoplasmic reticulum [37]. (P)RR has a predilection for the trans-Golgi network, where it undergoes cleavage by proteases such as the proprotein convertase, furin, and ADAM19 to generate an N-terminal soluble fragment of (P)RR (28 kDa) and the transmembrane–cytoplasmic domain [31, 32]. s(P)RR is secreted into the plasma, and the remaining 8 kDa C-terminal residue represents ATP6AP2. The latter corresponds to the V-ATPase segment of the (P)RR and is retrieved in lysosomes, whereas non-cleaved (escaped protease action) fl(P)RR is transported to the plasma membrane [31]. The Ms3-24 monoclonal antibody we generated in this study recognises both fl(P)RR and s(P)RR but not ATP6AP2 (Fig. 1), indicating that (P)RR immunoreactivity in neovascular endothelial cells (Figs 2b, e, 3d, g, 5b, e) basically originated from fl(P)RR and definitely not from V-ATPase. The fl(P)RR protein in fibrovascular tissues was colocalised with the (P)RR-shedding proteases, furin (Fig. 5b–d) and ADAM19 (Fig. 5e–g), and the vitreous s(P)RR protein levels were significantly elevated in PDR eyes but not control eyes lacking fibrovascular tissues (Fig. 5a). These data suggest that neovascular endothelial cells in fibrovascular tissues were the major source of s(P)RR in the vitreous fluids of PDR. Given that (P)RR is related to Wnt/β-catenin signalling pathways [38] and acts as the V-ATPase-associated protein that is essential for cell survival [39], (P)RR is speculated to play a physiological role in the eye, which is currently under investigation in our laboratory.
The vitreous levels of RAS components, including prorenin, angiotensinogen and angiotensin II, have been reported to increase in PDR eyes [33, 34, 40, 41]. We confirmed the increased levels of prorenin (Fig. 6a), as well as s(P)RR (Fig. 5a). Interestingly, although it lacks intracellular signalling, s(P)RR has recently been shown to have the ability to bind to prorenin and enhance the catalytic activity of renin, leading to generation of angiotensin I from angiotensinogen in vitro [27]. Importantly, we revealed markedly increased activated prorenin (Fig. 6c) in PDR vitreous fluids and its positive correlation with VEGF (Fig. 7c). Activated prorenin (Fig. 6c) together with angiotensinogen [34, 41] is thought to trigger tissue RAS activation causing angiotensin II generation [34, 41] in PDR. Therefore s(P)RR accumulation in vitreous fluids, in concert with fl(P)RR in fibrovascular tissues, is likely to enhance tissue RAS activation in PDR. Indeed, we previously showed endothelial localisation of AT1R, the pathogenic receptor for tissue RAS, in surgically excised human PDR fibrovascular tissues [10], which was further confirmed by our gene expression analysis (Fig. 2a). We have previously reported significant involvement of the AT1R signalling pathway in inflammation-related angiogenesis in the eye, causing enhanced production of VEGF, intercellular adhesion molecule-1 and monocyte chemotactic protein-1 [11, 12], all of which proved to be responsible for the pathogenesis of PDR. It is reasonable therefore to think that both vitreous s(P)RR (Figs 5a, 6b, 7b, d) and endothelial fl(P)RR (Figs 2b, e, 3d, g, 5b, e) bind to increased prorenin (Fig. 6a) and cooperatively facilitate the dual action of RAPS—i.e. tissue RAS activation (due to fl[P]RR and s[P]RR) and RAS-independent signal transduction (due to fl[P]RR only)—resulting in VEGF-induced angiogenic activity in PDR. The present data support the validity of (P)RR as a molecular target for the treatment of PDR.
Abbreviations
- ADAM:
-
A disintegrin and metalloproteinase
- AT1R:
-
Angiotensin II type 1 receptor
- ERK:
-
Extracellular signal-regulated kinase
- GST:
-
Glutathione S-transferase
- HRMEC:
-
Human retinal microvascular endothelial cell
- MAPK:
-
Mitogen-activated protein kinase
- MEK:
-
MAPK/ERK kinase
- NRP:
-
Neuropilin
- PDR:
-
Proliferative diabetic retinopathy
- (P)RR:
-
(Pro)renin receptor
- PRRB:
-
(Pro)renin receptor blocker
- qPCR:
-
Quantitative PCR
- RAPS:
-
Receptor-associated prorenin system
- RAS:
-
Renin–angiotensin system
- α-SMA:
-
α-Smooth muscle actin
- s(P)RR:
-
Soluble form of (P)RR
- VEGF:
-
Vascular endothelial growth factor
- VEGFR:
-
VEGF receptor
References
Adamis AP, Miller JW, Bernal MT et al (1994) Increased vascular endothelial growth factor levels in the vitreous of eyes with proliferative diabetic retinopathy. Am J Ophthalmol 118:445–450
Malecaze F, Clamens S, Simorre-Pinatel V et al (1994) Detection of vascular endothelial growth factor messenger RNA and vascular endothelial growth factor-like activity in proliferative diabetic retinopathy. Arch Ophthalmol 112:1476–1482
Aiello LP, Avery RL, Arrigg PG et al (1994) Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med 331:1480–1487
Ishida S, Shinoda K, Kawashima S, Oguchi Y, Okada Y, Ikeda E (2000) Coexpression of VEGF receptors VEGF-R2 and neuropilin-1 in proliferative diabetic retinopathy. Invest Ophthalmol Vis Sci 41:1649–1656
Ishida S, Usui T, Yamashiro K et al (2003) VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J Exp Med 198:483–489
Usui T, Ishida S, Yamashiro K et al (2004) VEGF164(165) as the pathological isoform: differential leukocyte and endothelial responses through VEGFR1 and VEGFR2. Invest Ophthalmol Vis Sci 45:368–374
Chaturvedi N, Sjolie AK, Stephenson JM et al (1998) Effect of lisinopril on progression of retinopathy in normotensive people with type 1 diabetes. The EUCLID Study Group. EURODIAB Controlled Trial of Lisinopril in Insulin-Dependent Diabetes Mellitus. Lancet 351:28–31
Chaturvedi N, Porta M, Klein R et al (2008) Effect of candesartan on prevention (DIRECT-Prevent 1) and progression (DIRECT-Protect 1) of retinopathy in type 1 diabetes: randomised, placebo-controlled trials. Lancet 372:1394–1402
Nagai N, Oike Y, Noda K et al (2005) Suppression of ocular inflammation in endotoxin-induced uveitis by blocking the angiotensin II type 1 receptor. Invest Ophthalmol Vis Sci 46:2925–2931
Nagai N, Noda K, Urano T et al (2005) Selective suppression of pathologic, but not physiologic, retinal neovascularization by blocking the angiotensin II type 1 receptor. Invest Ophthalmol Vis Sci 46:1078–1084
Nagai N, Izumi-Nagai K, Oike Y et al (2007) Suppression of diabetes-induced retinal inflammation by blocking the angiotensin II type 1 receptor or its downstream nuclear factor-kappaB pathway. Invest Ophthalmol Vis Sci 48:4342–4350
Nagai N, Oike Y, Izumi-Nagai K et al (2006) Angiotensin II type 1 receptor-mediated inflammation is required for choroidal neovascularization. Arterioscler Thromb Vasc Biol 26:2252–2259
Fletcher EL, Phipps JA, Ward MM, Vessey KA, Wilkinson-Berka JL (2010) The renin-angiotensin system in retinal health and disease: its influence on neurons, glia and the vasculature. Prog Retin Eye Res 29:284–311
Satofuka S, Ichihara A, Nagai N et al (2006) Suppression of ocular inflammation in endotoxin-induced uveitis by inhibiting nonproteolytic activation of prorenin. Invest Ophthalmol Vis Sci 47:2686–2692
Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer JD (2002) Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest 109:1417–1427
Kaneshiro Y, Ichihara A, Takemitsu T et al (2006) Increased expression of cyclooxygenase-2 in the renal cortex of human prorenin receptor gene-transgenic rats. Kidney Int 70:641–646
Satofuka S, Ichihara A, Nagai N et al (2007) Role of nonproteolytically activated prorenin in pathologic, but not physiologic, retinal neovascularization. Invest Ophthalmol Vis Sci 48:422–429
Satofuka S, Ichihara A, Nagai N et al (2008) (Pro)renin receptor promotes choroidal neovascularization by activating its signal transduction and tissue renin-angiotensin system. Am J Pathol 173:1911–1918
Satofuka S, Ichihara A, Nagai N et al (2009) (Pro)renin receptor-mediated signal transduction and tissue renin-angiotensin system contribute to diabetes-induced retinal inflammation. Diabetes 58:1625–1633
Satofuka S, Ichihara A, Nagai N, Tsubota K, Itoh H, Ishida S (2008) Pathologic roles of prorenin and (pro)renin receptor in the eye. Front Biosci 13:3884–3895
Satofuka S, Kanda A, Ishida S (2012) Receptor-associated prorenin system in the pathogenesis of retinal diseases. Front Biosci 4:1449–1460
Kanda A, Friedman JS, Nishiguchi KM, Swaroop A (2007) Retinopathy mutations in the bZIP protein NRL alter phosphorylation and transcriptional activity. Hum Mutat 28:589–598
Suzuki F, Hayakawa M, Nakagawa T et al (2003) Human prorenin has “gate and handle” regions for its non-proteolytic activation. J Biol Chem 278:22217–22222
Kanda A, Stambolian D, Chen W, Curcio CA, Abecasis GR, Swaroop A (2010) Age-related macular degeneration-associated variants at chromosome 10q26 do not significantly alter ARMS2 and HTRA1 transcript levels in the human retina. Mol Vis 16:1317–1323
Wang XF, Cui JZ, Prasad SS, Matsubara JA (2005) Altered gene expression of angiogenic factors induced by calcium-mediated dissociation of retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 46:1508–1515
Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (1989) Current protocols in molecular biology. Wiley, New York
Biswas KB, Nabi AN, Arai Y et al (2010) Qualitative and quantitative analyses of (pro)renin receptor in the medium of cultured human umbilical vein endothelial cells. Hypertens Res 34:735–739
Ludwig J, Kerscher S, Brandt U et al (1998) Identification and characterization of a novel 9.2-kDa membrane sector-associated protein of vacuolar proton-ATPase from chromaffin granules. J Biol Chem 273:10939–10947
Wilkinson-Berka JL, Heine R, Tan G et al (2010) RILLKKMPSV influences the vasculature, neurons and glia, and (pro)renin receptor expression in the retina. Hypertension 55:1454–1460
Parapuram SK, Cojocaru RI, Chang JR et al (2010) Distinct signature of altered homeostasis in aging rod photoreceptors: implications for retinal diseases. PLoS One 5:e13885
Cousin C, Bracquart D, Contrepas A, Corvol P, Muller L, Nguyen G (2009) Soluble form of the (pro)renin receptor generated by intracellular cleavage by furin is secreted in plasma. Hypertension 53:1077–1082
Yoshikawa A, Aizaki Y, Kusano K et al (2011) The (pro)renin receptor is cleaved by ADAM19 in the Golgi leading to its secretion into extracellular space. Hypertens Res 34:599–605
Danser AH, van den Dorpel MA, Deinum J et al (1989) Renin, prorenin, and immunoreactive renin in vitreous fluid from eyes with and without diabetic retinopathy. J Clin Endocrinol Metab 68:160–167
Funatsu H, Yamashita H, Nakanishi Y, Hori S (2002) Angiotensin II and vascular endothelial growth factor in the vitreous fluid of patients with proliferative diabetic retinopathy. Br J Ophthalmol 86:311–315
Otani A, Takagi H, Oh H et al (2000) Angiotensin II-stimulated vascular endothelial growth factor expression in bovine retinal pericytes. Invest Ophthalmol Vis Sci 41:1192–1199
Ishida S, Usui T, Yamashiro K et al (2003) VEGF164 is proinflammatory in the diabetic retina. Invest Ophthalmol Vis Sci 44:2155–2162
Schefe JH, Menk M, Reinemund J et al (2006) A novel signal transduction cascade involving direct physical interaction of the renin/prorenin receptor with the transcription factor promyelocytic zinc finger protein. Circ Res 99:1355–1366
Cruciat CM, Ohkawara B, Acebron SP et al (2010) Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science 327:459–463
Kinouchi K, Ichihara A, Sano M et al (2010) The (pro)renin receptor/ATP6AP2 is essential for vacuolar H+-ATPase assembly in murine cardiomyocytes. Circ Res 107:30–34
Gao BB, Clermont A, Rook S et al (2007) Extracellular carbonic anhydrase mediates hemorrhagic retinal and cerebral vascular permeability through prekallikrein activation. Nat Med 13:181–188
Gao BB, Chen X, Timothy N, Aiello LP, Feener EP (2008) Characterization of the vitreous proteome in diabetes without diabetic retinopathy and diabetes with proliferative diabetic retinopathy. J Proteome Res 7:2516–2525
Acknowledgements
We thank J. Friedman (University of Colorado Denver) for reviewing the manuscript, and I. Hirose and S. Namba (Hokkaido University) for technical assistance.
Funding
This work was supported in part by the Matching Program for Innovations in Future Drug Discovery, Medical Care, Tokyo Biochemical Research Foundation, Takeda Science Foundation, Mishima Saiichi Memorial Ophthalmic Research Japan Foundation, and a grant-in-aid from the Ministry of Education, Science and Culture of Japan (to A. Kanda, No. 24791823).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
AK and SI were responsible for the conception and design; AK, KN and WS performed analysis and interpretation of data described in the study; AK drafted the article; and KN, WS and SI contributed to discussion and reviewed the article. All authors approved the final version submitted for publication.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
Primer sequences used in RT-PCR and real-time qPCR (PDF 18 kb)
Rights and permissions
About this article
Cite this article
Kanda, A., Noda, K., Saito, W. et al. (Pro)renin receptor is associated with angiogenic activity in proliferative diabetic retinopathy. Diabetologia 55, 3104–3113 (2012). https://doi.org/10.1007/s00125-012-2702-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2702-2