
Abstract
Aims/hypothesis
Inflammation in obesity increases the levels of the suppressor of cytokine signalling-3 (SOCS3) protein in adipose tissue, but the physiological importance of this protein in regulating whole-body insulin sensitivity in obesity is not known.
Methods
We generated Socs3 floxed (wild-type, WT) and Socs3 aP2 (also known as Fabp4)-Cre null (Socs3 AKO) mice. Mice were maintained on either a regular chow or a high-fat diet (HFD) for 16 weeks during which time body mass, adiposity, glucose homeostasis and insulin sensitivity were assessed.
Results
The HFD increased SOCS3 levels in adipose tissue of WT but not Socs3 AKO mice. WT and Socs3 AKO mice had similar body mass and adiposity, assessed using computed tomography (CT) imaging, irrespective of diet or sex. On a control chow diet there were no differences in insulin sensitivity or glucose tolerance. When fed a HFD, female but not male Socs3 AKO mice had improved glucose tolerance as well as lower fasting glucose and insulin levels compared with WT littermates. Hyperinsulinaemic–euglycaemic clamps and positron emission tomography (PET) imaging demonstrated that improved insulin sensitivity was due to elevated adipose tissue glucose uptake. Increased insulin-stimulated glucose uptake in adipose tissue was associated with enhanced levels and activating phosphorylation of insulin receptor substrate-1 (IRS1).
Conclusions/interpretation
These data demonstrate that inhibiting SOCS3 production in adipose tissue of female mice is effective for improving whole-body insulin sensitivity in obesity.
Similar content being viewed by others


Introduction
Adipocytes and resident macrophages are a critical source of inflammatory cytokines that cause metabolic abnormalities including the development of insulin resistance. The suppressor of cytokine signalling (SOCS) family of proteins are negative regulators of cytokine signalling that act via their central SH2 domain and conserved C-terminal SOCS box to modulate inflammatory responses [1]. In obese rodents, SOCS3 is upregulated in hypothalamus [2, 3], liver [4], skeletal muscle [5, 6] and adipose tissue [7, 8]. In humans, SOCS3 abundance is also increased in mononuclear cells in response to pro-inflammatory conditions such as obesity [9] and a high-fat diet (HFD) [10], which may have important implications for adipose tissue inflammation. SOCS3 has been shown to be important for the regulation of insulin sensitivity, as it inhibits tyrosine phosphorylation of the insulin receptor [11, 12] and insulin receptor substrate-1 (IRS1) [4, 12] and may also target IRS proteins for proteasomal degradation [13]. Importantly, in-vitro studies indicate that SOCS3-deficient adipocytes derived from mouse embryonic fibroblasts are protected against the development of TNF-α-induced insulin resistance [14].
In addition to its role in regulating insulin sensitivity, SOCS3 has also been shown to be important for regulating sensitivity to leptin. Bjorbaek et al were the first to demonstrate that SOCS3 binding to Y985 of the leptin receptor inhibited leptin activation of the signal transducer and activator of transcription-3 in hypothalamic nuclei [15, 16]. Both Socs3 hypothalamic specific null mice [2] and Socs3 mice with haploinsufficiency [17] have enhanced leptin sensitivity and are resistant to diet-induced obesity. Similarly, the overproduction of SOCS3 in proopiomelanocortin neurons induces glucose intolerance and obesity [18]. In addition female, but not male, mice with a point mutation at Y985 within the leptin receptor are protected against developing diet-induced obesity and insulin resistance [19]. In adipocytes, leptin has been shown to increase the activity of AMP-activated protein kinase (AMPK) resulting in increased phosphorylation of its downstream substrate acetyl-CoA carboxylase (ACC) [20]. This activation of AMPK is associated with the rapid depletion of lipid from adipocytes and results in increases in glycerol but not NEFA, suggesting that leptin reduces adipose tissue mass by increasing the rate of adipose tissue fatty-acid oxidation [21, 22]. Consistent with the potential for SOCS3 to inhibit adipose tissue leptin signalling, recent reports in both rodents [23] and humans [24] have found that AMPK phosphorylation is reduced with obesity.
Evaluating the physiological importance of SOCS3 in vivo has been difficult because Socs3 −/− mice die mid gestation due to placental insufficiency [25]. In adipocytes the transgenic overexpression of Socs3 in adipose tissue by greater than 100-fold reduces adipose tissue but not whole-body insulin sensitivity when mice are fed a standard chow diet [26]. However, when mice were fed an HFD, the overexpression of Socs3 in adipose tissue prevented the development of HFD-induced obesity [26]. This result was attributed to a reduction in adiposity of HFD-fed Socs3 transgenic mice, increased secretion of adiponectin and subsequently elevated levels of mitochondrial enzymes in skeletal muscle [26]. While these studies suggest an important but surprising role for adipose tissue SOCS3 in regulating whole-body energy metabolism, an important caveat is that given the functional redundancy of the SOCS family of proteins and their relatively low abundance in the absence of overt inflammation, this supra-physiological increase in SOCS3 might be expected to overlap with other SOCS-related signalling pathways [25]. For example, SOCS1 shares ∼85% homology with SOCS3 and has also been shown to inhibit insulin signalling [4, 13, 27]. Therefore, it is feasible that the phenotype of these transgenic mice might be a function of inhibiting multiple pathways simultaneously and thus may not be indicative of the physiological role of SOCS3 in adipose tissue following modest increases in production (two- to threefold) as observed with obesity [8, 11].
The generation of Socs3 floxed mice has made it possible to study the tissue-specific effects of Socs3 deletion. In the liver the deletion of Socs3 enhances insulin sensitivity in mice fed a chow diet; however, when fed an HFD, deletion of Socs3 promotes the development of fatty liver disease, systemic inflammation and obesity [28, 29]. In the current study we generated Socs3 Ap2-Cre null mice (Socs3 AKO). We show that Ap2-Cre deletion of Socs3 does not alter body mass or adiposity, and that female, but not male, Socs3 AKO mice were partially protected against the development of obesity-induced glucose intolerance and insulin resistance due to enhanced adipose tissue insulin sensitivity.
Methods
Animal procedures
All procedures were approved by St Vincent’s Hospital and McMaster University Animal Ethics Committee. Mice were generated by crossing Socs3 floxed mice generated on a C57Bl6 background [30] with mice expressing Cre-recombinase under the control of the aP2 (also known as Fabp4) promoter [31] that had been backcrossed onto a C57Bl6 background for seven generations. Socs3 flox/flox Cre −/− (wild-type, WT) and Socs3 flox/flox aP2-Cre +/− (Socs3 AKO) mice were maintained on a 12 h light–dark cycle with lights on at 07:00 hours. For lipopolysaccharide (LPS) experiments female and male WT and Socs3 AKO mice were injected with 2 mg/kg LPS (Invivogen, San Diego, CA, USA) and tissues harvested 2 h later as described [29]. For diet experiments WT and Socs3 AKO littermates starting at 6 weeks of age were maintained on chow, or an HFD (digestible energy of 17 MJ/kg with 46% of energy from fat) for 16 weeks [29]. Body mass was monitored weekly. Energy expenditure and activity levels were measured using a Columbus Instruments Laboratory Animal Monitoring System (Columbus, OH, USA) over 72 h as described [32, 33]. Glucose tolerance tests were performed after 15 weeks of chow or high-fat feeding through intraperitoneal injection of mice with 1 g/kg glucose (Sigma-Aldrich, Oakville, ON, Canada) in saline and were followed by monitoring of tail-blood glucose (Bayer, Leverkusen, Germany) [29, 34]. Euglycaemic–hyperinsulinaemic clamps were conducted in conscious mice as recently described [29, 32, 34]. Briefly, 3 days before application of the clamp two catheters were inserted into the right jugular vein. The clamp was conducted after a 6 h fast (which commenced at the start of the light-cycle), in restrained conscious mice that had been acclimatised to the restrainers for the preceding 3 weeks. At 60 min, a solution containing d-[3-3H]glucose was infused at a constant rate (278 Gbq/h, 0.12 ml/h) for 1 h for determination of basal glucose turnover. At 0 min, insulin diluted in saline was infused at a rate of 10 mU kg−1 min−1 and a 50% dextrose solution was infused at a variable rate to maintain euglycaemia. Once steady state was achieved, glucose specific activity was measured in whole blood after deproteinisation with BaOH and ZnSO4. Hepatic glucose production and glucose disposal rate for the basal and clamp period were calculated using Steele's equation for steady-state conditions.
In vivo glucose uptake and body composition
[18F]Fluorodeoxyglucose (FDG) was synthesised at McMaster University by the nucleophilic substitution method using an FDG synthesising instrument (GE Healthcare, Milwaukee, WI, USA) and a cyclotron (Siemans20-30 gb) [35]. After an 8 h fast mice were injected with insulin (0.5 U/kg) diluted in 0.9% physiological saline and 5 min later received an intravenous administration of FDG (10.8 ± 1.2 MBq/g) [35]. After injection, the mice were maintained under conscious conditions and warmed using a heating pad. At 28 min post insulin injection mice were anaesthetised with isofluorane and at 30 min small-animal positron emission tomography (PET; Philip Mosaic, Andover, MA, USA) and micro-computed tomography (CT) (Gamma Medica-Ideas Xspect System, Northridge, CA, USA) imaging were performed using an acquisition time of 15 min for PET, followed by CT for 5 min [35]. Images were reconstructed using 3D-RAMLA algorithm, with no attenuation correction and no correction for partial-volume effects of the tomograph as previously described [35]. Quantification was performed by region-of-interest analysis using Amide Research Workplace software and FDG tissue uptake calculated using the mean value of standard uptake values (SUV) as described [35]. Analysis of total body fat composition was carried out using Amira software (Visage Imaging; www.amira.com/downloads.html) and the mean value of voxels of segmented adipose were calculated as described [32].
Serum hormones and cytokines analysis
Whole blood was spun at 7,000 g for 5 min at 4°C and the serum removed. Leptin, resistin, TNF-α and IL-6 were measured in serum by BioPlex assay (Millipore, Burlington, ON, Canada), NEFA via colorimetric assay (Wako Chemical, Richmond, VA, USA) and adiponectin (Millipore) via ELISA, all following the manufacturers’ recommendations.
Real-time quantitative PCR
Adipose tissue macrophages were isolated from gonadal adipose tissue following collagenase digestion (Liberase enzyme; Roche, Mannheim, Germany) and isolated using a Mouse CD11b Positive Selection kit (Stemcell Technologies, Vancouver, BC, Canada) as described [32]. Adipose tissue macrophages and gonadal adipose tissue were then lysed in TRIzol-reagent (Invitrogen, Carlsbad, CA, USA) and RNA extracted for real-time quantitative PCR as described [36].
Immunoblotting
Protein lysates were generated from epididymal fat pads homogenised in ice-cold buffer (50 mmol/l HEPES, pH 7.4, 150 mmol/l NaCl, 10 mmol/l NaF, 1 mmol/l sodium pyrophosphate, 0.5 mmol/l EDTA, 250 mmol/l sucrose, 1 mmol/l dithiothreitol, 1% (vol./vol.) Triton X-100, 1 mmol/l Na3VO4 and one Roche protease inhibitor tablet per 50 ml buffer) using an electrical homogeniser [36]. Protein content in lysates was measured by the BCA method (Pierce, Rockford, IL, USA) and immunoblotting was conducted using antibodies (Cell Signaling Technology, Danvers, MA, USA) against total and/or phosphorylated forms of SOCS3, the insulin receptor substrate-1 (Y1122), Akt (T308 and S473), IKK (S176/180) and JNK (T183/Y185) as described [36]. AMPK and ACC antibodies were generated and used as described [37]. Bands were visualised using an enhanced chemiluminescence system (Vilber Fusion-FX1; Eberhardzell Germany) and quantified using Scion image software.
Calculations and statistical analysis
Data are presented as the mean ± SEM and were analysed using Student’s t test or, where appropriate, one-way repeated measures or two-way ANOVA with a Bonferroni post-hoc test (GraphPad PRISM, version 4.0; La Jolla, CA, USA). Differences were considered statistically significant at p < 0.05.
Results
Socs3 AKO mice develop similar degrees of obesity
Two hours after the injection of LPS, Socs3 expression was observed in adipose tissue of WT but not Socs3 AKO mice thus indicating the effective deletion of SOCS3 from adipose tissue of Socs3 AKO mice (see electronic supplementary material [ESM] Fig. 1). Starting at 6 weeks of age, mice were either maintained on a control chow diet or switched to an HFD. When mice were fed a chow diet white adipose tissue Socs3 mRNA expression was at the limits of detection of our real-time quantitative PCR assay (∼35 cycles) and hence we were unable to detect any difference in expression between WT and Socs3 AKO mice. After 16 weeks of HFD feeding there was a ∼fourfold upregulation of Socs3 mRNA expression in adipose tissue of WT but not Socs3 AKO mice (Fig. 1a). SOCS1 shares ∼85% homology with SOCS3 and has also been implicated in the inhibition of insulin sensitivity. However, SOCS1 abundance was not altered by diet or genotype in adipose tissue (data not shown). aP2 has also been shown to be produced in macrophages [38] but in contrast to our findings in adipocytes we found that expression of Socs3 mRNA in adipose tissue macrophages of HFD-fed mice was similar between WT and Socs3 AKO mice (ESM Fig. 2). This finding is consistent with several recent reports demonstrating that the efficiency of aP2-Cre recombination in macrophages is much less efficient than in adipocytes [39–41].

Improved glucose tolerance in female Socs3 AKO mice. (a) Adipose tissue Socs3 mRNA in WT (white bars) and Socs3 AKO mice (black bars) following 16 weeks consumption of a chow or HFD. Body mass of female (b) and male (c) WT (white symbols) and Socs3 AKO mice (black symbols) fed chow (circles) or HFD (triangles, inverted triangles). (d) Glucose tolerance (1 g/kg body mass) and (e) AUC (mmol/l × min) in female WT and Socs3 AKO mice fed a chow or HFD for 16 weeks. AU, arbitrary units. Data are mean ± SEM, n = 6 for real-time quantitative PCR data and n = 14–16 for body mass and GTT data. † p < 0.0001 relative to chow control; *p < 0.05, ***p < 0.0001 relative to WT for same diet
We monitored body mass over 16 weeks in both male and female Socs3 AKO mice relative to WT littermates, fed chow or HFD. As anticipated the HFD led to significant increases in body mass in both male and female mice but there were no differences between genotypes (Fig. 1 b, c). We next assessed glucose tolerance in male and female WT and Socs3 AKO mice. When fed a chow diet there were no differences in glucose tolerance in either female (Fig. 1 d, e) or male mice (ESM Fig. 3 a, b). When fed an HFD, female (Fig. 1 d, e) but not male (ESM Fig. 3 a, b) Socs3 AKO mice had improved glucose tolerance. Given this sexual dimorphism the remainder of the analyses were completed in only female mice.
Socs3 AKO mice have similar adiposity, adipose-tissue cell size
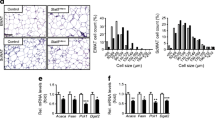
SOCS3 has been proposed to be a negative regulator of leptin signalling in adipocytes [8]; consistent with this idea, recent studies have shown that AMPK activity is reduced in adipose tissue with obesity [23, 24]. We hypothesised that AMPK activity might be elevated in adipose tissue of Socs3 AKO mice and that this might be associated with reductions in adipose-tissue cell size. Using CT imaging we assessed total adiposity and found that there was no difference between genotypes when mice were fed chow or an HFD (Fig. 2a). Similarly, while the HFD increased mean adipocyte cell size compared with chow controls there was no difference between WT and Socs3 AKO mice (Fig. 2b). We found that, consistent with previous reports, AMPK T172 (Fig. 2c) and ACC Ser 79 (Fig. 2d) phosphorylation were dramatically reduced with the HFD but that the reduction in AMPK signalling was comparable between WT and Socs3 AKO mice. These data suggest that adipose tissue Socs3 expression is not a critical regulator of adipose tissue morphology and that AMPK signalling is not maintained in adipose tissue of obese mice despite the absence of Socs3.

Adiposity, adipose tissue cell size and adipose-tissue AMPK activity is not altered in Socs3 AKO mice. (a) Body fat composition assessed by CT analysis (representative image with quantification). (b) Adipose tissue histology (haematoxylin and eosin [H&E] stain) from WT and Socs3 AKO mice fed chow and HFD (representative image with quantification of adipose-tissue cell size). Phosphorylation of AMPK T172 (c) and ACC S79 (d) in adipose tissue from WT and Socs3 AKO mice fed chow or HFD. White bars, WT mice; black bars, Socs3 AKO mice. AU, arbitrary units. Data are means ± SEM, n = 6–8. † p < 0.001 relative to chow control
Socs3 AKO mice do not have altered energy balance or substrate use
The transgenic overexpression of Socs3 in adipose tissue using the aP2-Cre promoter results in mice that have altered rates of energy expenditure and metabolism, an effect which is attributed to altered secretion of adiponectin [26]. We found that, consistent with comparable adiposity between genotypes, serum levels of adiponectin and resistin did not differ between WT and Socs3 AKO mice (Table 1). Surprisingly, leptin levels were lower in Socs3 AKO mice fed a chow diet but were elevated to a similar degree when mice were fed an HFD (Table 1). Serum levels of TNF-α were not altered by either diet or genotype (Table 1). In contrast, serum IL-6 was significantly increased in HFD-fed Socs3 AKO mice (Table 1).
The transgenic overexpression of Socs3 in adipose tissue increases oxygen consumption, activity level and food intake when mice are fed an HFD [26]. However, we found that, consistent with similar adiposity and body mass, energy expenditure, activity level and food intake were similar in WT and Socs3 AKO mice irrespective of diet (Table 2). Socs3 has also been shown to be important in regulating substrate use and in particular has been shown to play a role in regulating fatty acid oxidation and lipogenesis [6, 26, 29]. As anticipated the HFD significantly reduced the respiratory exchange ratio (RER), indicating reduced glucose use, however, there was no difference between genotypes (Table 2). These data indicate that adipose tissue Socs3 does not play a critical role in regulating whole-body energy metabolism.
Female Socs3 AKO mice have improved adipose tissue insulin sensitivity when fed an HFD
Improved glucose tolerance in female HFD-fed Socs3 AKO mice (Fig. 1), was associated with significantly lower fasting blood glucose (Fig. 3a) and serum insulin levels (Fig. 3b). To examine whether improvements in insulin sensitivity were related to changes in hepatic or peripheral insulin sensitivity we conducted hyperinsulinaemic–euglycaemic clamps. We did not clamp chow-fed mice as we did not detect any differences in blood glucose, serum insulin or glucose tolerance. Serum glucose concentrations before and during the clamp did not differ between WT and Socs3 AKO mice (Table 3). We found that Socs3 AKO mice tended to have a slightly higher glucose infusion rate (GINF) (∼10%, p = 0.12, Table 3), which was due to a ∼20% increase in insulin-stimulated glucose disposal rate (IS-GDR) (Table 3). There were no changes in hepatic glucose output or per cent suppression of hepatic glucose production when comparing genotypes (Table 3). To determine whether increased IS-GDR in Socs3 AKO mice involved skeletal muscle or adipose tissue we injected mice with insulin (0.5 U/kg) and measured FDG uptake using PET imaging. We found that FDG uptake was significantly higher in adipose tissue (+56%, p = 0.002) but not in muscle of Socs3 AKO mice relative to WT littermates (Fig. 3c). Brown adipose tissue FDG uptake was also comparable between WT and Socs3 AKO mice (WT, 4.63 ± 0.45; Socs3 AKO, 4.12 ± 0.24 SUV). Taken together these data indicate that improved insulin sensitivity in HFD-fed Socs3 AKO mice was due to enhanced insulin-stimulated glucose disposal into white adipose tissue.

Improved whole-body insulin sensitivity and increased FDG uptake in adipose tissue of Socs3 AKO mice. (a) Reduced fasting glucose and (b) insulin levels in HFD-fed Socs3 AKO mice. (c) Increased insulin-stimulated FDG uptake into adipose tissue but not muscle of Socs3 AKO mice fed an HFD. White bars, WT mice; black bars, Socs3 AKO mice. Data are mean ± SEM, n = 12–16 for blood glucose and insulin, n = 6 for FDG uptake. † p < 0.05 relative to chow control; *p < 0.05 relative to WT for same diet
Socs3 AKO mice have enhanced adipose tissue IRS1 production and phosphorylation
To assess the mechanisms contributing to the increased adipose tissue glucose uptake in HFD-fed Socs3 AKO mice we measured total production and phosphorylation of proximal and distal components of the canonical insulin signalling pathway, 2 min after a bolus of insulin (0.5 U/kg). We found that the HFD significantly reduced IRS1 level in WT mice, but that this inhibitory effect was completely inhibited in Socs3 AKO mice (Fig. 4 a, b). In addition, Socs3 AKO mice were also protected from an HFD-induced reduction in IRS1 Y1222 phosphorylation, as phosphorylation of this site on IRS1 was greater than in HFD-fed WT littermates even when corrected for the higher level of IRS1 (Fig. 4 a, b). Increases in IRS1 production and phosphorylation were associated with enhanced Akt T308 (Fig. 4c) and S473 (Fig. 4d) phosphorylation in HFD-fed Socs3 AKO mice. There was also a tendency for Akt phosphorylation to be increased in Socs3 AKO mice fed a chow diet (Fig. 4 c, d). Consistent with our data demonstrating that skeletal muscle FDG uptake was not altered in Socs3 AKO mice, we found that IRS1 and Akt phosphorylation in muscle were not different between genotypes (data not shown). These data suggest that SOCS3 inhibits adipocyte insulin-stimulated glucose uptake via both reduction in IRS1 production and inhibition of IRS1 tyrosine phosphorylation.

Increased adipose tissue IRS1 content and phosphorylation in HFD-fed Socs3 AKO mice despite normal increases in JNK and IKK phosphorylation. (a, b) Adipose tissue IRS1 production and IRS1 Y1222 phosphorylation, (c) Akt (Thr308) and (d) Akt (Ser473) phosphorylation in adipose tissues of WT and Socs3 AKO mice fed a chow or HFD following a bolus of insulin. Phosphorylation of JNK (e) and IKK (f) in adipose tissue of WT and Socs3 AKO mice fed a chow or HFD. AU, arbitrary units. Data are mean ± SEM, n = 8. † p < 0.05 relative to chow control; *p < 0.05 relative to WT for same diet
In adipose tissue of obese mice the inhibitor κB kinase (IKK) and c-Jun terminal kinase (JNK) inhibit insulin signalling via IRS1 [42, 43]. To examine whether improved adipose tissue insulin sensitivity in Socs3 AKO mice might involve the downregulation of these pathways, we examined activating phosphorylation of IKK and JNK. We found that while as expected the HFD significantly increased adipose tissue JNK (Fig. 4e) and IKK (Fig. 4f) phosphorylation there were no differences between genotypes.
Discussion
The precise regulation of both the magnitude and duration of cytokine signalling is essential for the orchestration of many biological processes. Studies in mouse models of SOCS3 deficiency have shown an important role for this protein in the control of cytokine responses within the central nervous system [2, 17] and the liver [44]. Adipose tissue is the largest source of fuel in the body, and is also an important source of secreted proteins and inflammatory cytokines which are critical for controlling whole-body energy metabolism and insulin sensitivity [45]. In the current study we detected no change in adipose tissue cell size or total adiposity between WT and Socs3 AKO mice when fed either a control chow or HFD. Previous studies from the Unger laboratory have shown that chronically elevated levels of leptin reduce adipose tissue cell size potentially by increasing the phosphorylation of AMPK and ACC in adipocytes and that this effect is blocked when mice become obese; findings which correlate with increases in adipose tissue SOCS3 production [20]. However, we found that even despite higher leptin levels in HFD-fed mice, AMPK and ACC phosphorylation were reduced in both WT and Socs3 AKO mice relative to lean chow-fed controls. These data suggest that SOCS3 is not the only regulator of AMPK activity in adipocytes and that potentially other negative regulators of leptin signalling, such as protein tyrosine phosphatase 1B [46], may also be important in regulating this response.
It was previously shown that transgenic overexpression of Socs3 in adipose tissue increased oxygen consumption and reduced appetite, resulting in reduced body mass when mice were fed an HFD. This surprising effect was attributed to the smaller adipose-tissue cell size of HFD-fed Socs3-aP2 transgenic mice that resulted in increased adiponectin production and muscle mitochondrial biogenesis [26]. In the current study, we did not observe any evidence for a role of aP2 deletion of Socs3 in regulating energy balance as both WT and Socs3 AKO mice had similar activity levels, food intake and energy expenditure and, consistent with this, similar body mass, adiposity and adipose-tissue cell size. Similarly, adiponectin levels and muscle insulin sensitivity were not altered in Socs3 AKO mice. The level of Socs3 overexpression observed in Socs3 aP2 transgenic mice was much higher than observed in obesity, therefore the results may have been due to overlap with Socs1, which was not altered in our model. Taken together these data suggest that it is unlikely that adipose tissue Socs3 is important in regulating whole-body energy balance.
In the current study we demonstrated that under normal chow-fed conditions there is no difference in whole-body glucose tolerance or insulin sensitivity between WT and Socs3 AKO mice. This finding is consistent with the very low levels of SOCS3, which were indistinguishable between WT and Socs3 AKO mice. However, when made obese through chronic high-fat feeding which increased SOCS3 production by approximately fourfold in adipose tissue of WT but not Socs3 AKO mice, female but not male Socs3 AKO mice were partially protected from developing HFD-induced glucose intolerance and insulin resistance. The reason for this sexual dimorphism is not known, but the findings are consistent with previous findings in SOCS3-deficient models [2, 17] or leptin receptor Y985 mutant mice [19] where female, but not male mice, display more robust differences in leptin sensitivity. The findings are also very similar to those previously reported in Tlr4 knockout mice, where only females were partially protected against the development of HFD-induced insulin resistance [47]. As both TLR4 and SOCS3 are important regulators of cytokine signalling these data may suggest that in male mice factors independent of inflammatory signalling are more critical for controlling insulin and leptin sensitivity in obesity. Alternatively, as oestrogen receptor α (ERα) is phosphorylated at Y737 [48] and oestrogen has been shown to upregulate SOCS3 in hepatocytes [49], it is possible that SOCS3 may also be a negative regulator of oestrogen signalling. Hence the removal of this negative regulator in female Socs3 AKO mice may allow oestrogen to more fully activate insulin sensitising pathways [50]. Future studies examining the interaction between SOCS3 and ERα are warranted.
Hyperinsulinaemic–euglycaemic clamps and measurements of glucose uptake using PET revealed that improvements in whole-body insulin sensitivity in Socs3 AKO female mice were due to increased rates of FDG entry into adipose tissue. We found that SOCS3 was critical for mediating HFD-induced suppression of adipose tissue IRS1 production and Y1122 phosphorylation. Importantly, this enhanced IRS production and phosphorylation in Socs3 AKO mice resulted in increased Akt T308 and S473 phosphorylation. These data are consistent with previous in-vitro studies, which found that SOCS3 inhibits TNF-α-induced insulin signalling by downregulating both IRS1 production and tyrosine phosphorylation in adipocytes [14]. Importantly, we found that this improvement in adipose tissue insulin sensitivity occurred despite activation of JNK and IKK, suggesting that the inhibition of SOCS3 is sufficient to improve adipose tissue insulin sensitivity independently of alterations in other aspects of inflammatory signalling.
In conclusion, we demonstrate that the inhibition of SOCS3 in adipose tissue of female mice improves glucose tolerance and whole-body insulin sensitivity due to enhanced adipose tissue insulin sensitivity. Importantly, improvements in insulin sensitivity occurred independently of changes in body mass, energy balance and adiposity/adipose tissue cell size, findings which are very different from those previously reported following the transgenic overexpression of Socs3 in adipose tissue [26]. These findings are also in stark contrast to the obesity-promoting effects of Socs3 deletion in the liver [29]. While these data support an important role for SOCS3 in regulating adipose tissue insulin sensitivity the effects on the IS-GDR were modest. This is consistent with the fact that there was no change in skeletal muscle insulin sensitivity, which quantitatively accounts for a greater percentage of blood glucose disposal than adipose tissue. These data indicate that while SOCS3 is an important negative regulator of adipose tissue insulin signalling, future studies examining the role of SOCS3 in other tissues, such as skeletal muscle, are warranted.
Abbreviations
- ACC:
-
Acetyl-CoA carboxylase
- AMPK:
-
AMP-activated protein kinase
- aP2:
-
Adipocyte protein 2
- CT:
-
Computed tomography
- ERα:
-
Oestrogen receptor alpha
- FABP4:
-
Fatty acid binding protein 4
- FDG:
-
[18F]Fluorodeoxyglucose
- GDR:
-
Glucose disposal rate
- GINF:
-
Glucose infusion rate
- GTT:
-
Glucose tolerance test
- HFD:
-
High-fat diet
- IKK:
-
Inhibitor κB kinase
- IRS1:
-
Insulin receptor substrate-1
- IS-GDR:
-
Insulin-stimulated glucose disposal rate
- JNK:
-
c-Jun terminal kinase
- LPS:
-
Lipopolysaccharide
- PET:
-
Positron emission tomography
- RER:
-
Respiratory exchange ratio
- SOCS:
-
Suppressor of cytokine signalling
- Socs3 AKO:
-
Socs3 Ap2-Cre null
- SUV:
-
Standard uptake value
- WT:
-
Wild-type
References
Wormald S, Hilton DJ (2004) Inhibitors of cytokine signal transduction. J Biol Chem 279:821–824
Mori H, Hanada R, Hanada T et al (2004) Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med 10:739–743
Myers MG Jr (2004) Leptin receptor signaling and the regulation of mammalian physiology. Recent Prog Horm Res 59:287–304
Ueki K, Kondo T, Kahn CR (2004) Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol Cell Biol 24:5434–5446
Steinberg GR, Smith AC, Wormald S, Malenfant P, Collier C, Dyck DJ (2004) Endurance training partially reverses dietary-induced leptin resistance in rodent skeletal muscle. Am J Physiol Endocrinol Metab 286:E57–E63
Steinberg GR, McAinch AJ, Chen MB et al (2006) The suppressor of cytokine signaling 3 inhibits leptin activation of AMP-kinase in cultured skeletal muscle of obese humans. J Clin Endocrinol Metab 91:3592–3597
Emanuelli B, Peraldi P, Filloux C et al (2001) SOCS-3 inhibits insulin signaling and is up-regulated in response to tumor necrosis factor-alpha in the adipose tissue of obese mice. J Biol Chem 276:47944–47949
Wang Z, Zhou YT, Kakuma T et al (2000) Leptin resistance of adipocytes in obesity: role of suppressors of cytokine signaling. Biochem Biophys Res Commun 277:20–26
Ghanim H, Aljada A, Daoud N, Deopurkar R, Chaudhuri A, Dandona P (2007) Role of inflammatory mediators in the suppression of insulin receptor phosphorylation in circulating mononuclear cells of obese subjects. Diabetologia 50:278–285
Ghanim H, Abuaysheh S, Sia CL et al (2009) Increase in plasma endotoxin concentrations and the expression of Toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: implications for insulin resistance. Diabetes Care 32:2281–2287
Emanuelli B, Peraldi P, Filloux C, Sawka-Verhelle D, Hilton D, van Obberghen E (2000) SOCS-3 is an insulin-induced negative regulator of insulin signaling. J Biol Chem 275:15985–15991
Senn JJ, Klover PJ, Nowak IA et al (2003) Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J Biol Chem 278:13740–13746
Rui L, Yuan M, Frantz D, Shoelson S, White MF (2002) SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem 277:42394–42398
Shi H, Tzameli I, Bjorbaek C, Flier JS (2004) Suppressor of cytokine signaling 3 is a physiological regulator of adipocyte insulin signaling. J Biol Chem 279:34733–34740
Bjorbaek C, Lavery HJ, Bates SH et al (2000) SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J Biol Chem 275:40649–40657
Bjorbaek C, El-Haschimi K, Frantz JD, Flier JS (1999) The role of SOCS-3 in leptin signaling and leptin resistance. J Biol Chem 274:30059–30065
Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjorbaek C, Flier JS (2004) Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat Med 10:734–738
Reed AS, Unger EK, Olofsson LE, Piper ML, Myers MG Jr, Xu AW (2010) Functional role of suppressor of cytokine signaling 3 upregulation in hypothalamic leptin resistance and long-term energy homeostasis. Diabetes 59:894–906
Bjornholm M, Munzberg H, Leshan RL et al (2007) Mice lacking inhibitory leptin receptor signals are lean with normal endocrine function. J Clin Invest 117:1354–1360
Wang MY, Orci L, Ravazzola M, Unger RH (2005) Fat storage in adipocytes requires inactivation of leptin's paracrine activity: implications for treatment of human obesity. Proc Natl Acad Sci U S A 102:18011–18016
Wang MY, Lee Y, Unger RH (1999) Novel form of lipolysis induced by leptin. J Biol Chem 274:17541–17544
Orci L, Cook WS, Ravazzola M et al (2004) Rapid transformation of white adipocytes into fat-oxidizing machines. Proc Natl Acad Sci U S A 101:2058–2063
Gaidhu MP, Anthony NM, Patel P, Hawke TJ, Ceddia RB (2010) Dysregulation of lipolysis and lipid metabolism in visceral and subcutaneous adipocytes by high-fat diet: role of ATGL, HSL, and AMPK. Am J Physiol Cell Physiol 298:C961–C971
Gauthier MS, O’Brien EL, Bigornia S et al (2010) Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem Biophys Res Commun 404:382–387
Krebs DL, Hilton DJ (2001) SOCS proteins: negative regulators of cytokine signaling. Stem Cells 19:378–387
Shi H, Cave B, Inouye K, Bjorbaek C, Flier JS (2006) Overexpression of suppressor of cytokine signaling 3 in adipose tissue causes local but not systemic insulin resistance. Diabetes 55:699–707
Jamieson E, Chong MM, Steinberg GR et al (2005) Socs1 deficiency enhances hepatic insulin signaling. J Biol Chem 280:31516–31521
Torisu T, Sato N, Yoshiga D et al (2007) The dual function of hepatic SOCS3 in insulin resistance in vivo. Genes Cells 12:143–154
Sachithanandan N, Fam BC, Fynch S et al (2010) Liver specific SOCS3 deletion in mice enhances hepatic insulin sensitivity and lipogenesis resulting in fatty liver and obesity. Hepatology 52:1632–1642
Kiu H, Greenhalgh CJ, Thaus A et al (2009) Regulation of multiple cytokine signalling pathways by SOCS3 is independent of SOCS2. Growth Factors 27:384–393
Abel ED, Peroni O, Kim JK et al (2001) Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 409:729–733
Galic S, Fullerton MD, Schertzer JD et al (2011) Hematopoietic AMPK beta1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J Clin Invest 121:4903–4915
O’Neill HM, Maarbjerg SJ, Crane JD et al (2011) AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci U S A 108:16092–16097
Dzamko N, Vandenderen BJ, Hevener AL et al (2010) AMPK β1 deletion reduces appetite preventing obesity and hepatic insulin resistance. J Biol Chem 285:115–122
Saab C, Labiris N, Chirakal R, Farncombe T (2007) A database of normal PET and SPECT radiotracer distributions in rats and mice. J Nucl Med Meeting Abstracts 48:292P-b-
Watt MJ, Dzamko N, Thomas WG et al (2006) CNTF reverses obesity-induced insulin resistance by activating skeletal muscle AMPK. Nat Med 12:541–548
Chen Z-P, McConell GK, Michell BJ, Snow RJ, Canny BJ, Kemp BE (2000) AMPK signaling in contracting human skeletal muscle: acetyl-CoA carboxylase and NO synthase phosphorylation. Am J Physiol 279:E1202–E1206
Maeda K, Cao H, Kono K et al (2005) Adipocyte/macrophage fatty acid binding proteins control integrated metabolic responses in obesity and diabetes. Cell Metab 1:107–119
Jiang C, Qu A, Matsubara T et al (2011) Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes 60:2484–2495
Wueest S, Rapold RA, Schumann DM et al (2010) Deletion of Fas in adipocytes relieves adipose tissue inflammation and hepatic manifestations of obesity in mice. J Clin Invest 120:191–202
Kumar A, Lawrence JC Jr, Jung DY et al (2010) Fat cell-specific ablation of rictor in mice impairs insulin-regulated fat cell and whole-body glucose and lipid metabolism. Diabetes 59:1397–1406
Hirosumi J, Tuncman G, Chang L et al (2002) A central role for JNK in obesity and insulin resistance. Nature 420:333–336
Gao Z, Hwang D, Bataille F et al (2002) Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J Biol Chem 277:48115–48121
Croker BA, Krebs DL, Zhang JG et al (2003) SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol 4:540–545
Galic S, Oakhill JS, Steinberg GR (2010) Adipose tissue as an endocrine organ. Mol Cell Endocrinol 316:129–139
Bence KK, Delibegovic M, Xue B et al (2006) Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med 12:917–924
Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS (2006) TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116:3015–3025
Arnold SF, Obourn JD, Jaffe H, Notides AC (1995) Phosphorylation of the human estrogen receptor on tyrosine 537 in vivo and by src family tyrosine kinases in vitro. Mol Endocrinol 9:24–33
Leong GM, Moverare S, Brce J et al (2004) Estrogen up-regulates hepatic expression of suppressors of cytokine signaling-2 and -3 in vivo and in vitro. Endocrinology 145:5525–5531
Ribas V, Nguyen MT, Henstridge DC et al (2010) Impaired oxidative metabolism and inflammation are associated with insulin resistance in ERalpha-deficient mice. Am J Physiol Endocrinol Metab 298:E304–E319
Acknowledgements
We thank W. Alexander (Walter and Eliza Hall Institute of Medical Research, University of Melbourne) and B. Kahn (Beth Israel Deaconess Medical Center, Harvard Medical School) for providing us with Socs3 floxed and aP2-Cre mice, respectively. We are also grateful for the assistance of T. Farncombe and C. Saab from the McMaster Centre for Preclinical and Translational Imaging for completing the CT and PET studies.
Funding
These studies were supported by grants and fellowships from the National Health and Medical Research Council, the Diabetes Australia Research Trust, the Canadian Foundation for Innovation and the Canadian Institutes of Health Research (all G. R. Steinberg). S. B. Jorgensen was supported by a Research fellowship from the Danish Medical Research Council (271-05-0697). M. D. Fullerton is a CIHR Banting Postdoctoral Fellow. G. R. Steinberg is a Canada Research Chair in Metabolism and Obesity.
Duality of interest
The authors declare there is no duality of interest associated with this manuscript.
Contribution statement
RP, MDF, SG, JH, KAH and SBJ researched data. RP, MDF, SG, JH, KAH and SBJ reviewed and edited manuscript. GRS designed experiments and wrote the manuscript. All authors have approved the final version of manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below is the link to the electronic supplementary material.
ESM Fig. 1
(PDF 132 kb)
ESM Fig. 2
(PDF 7 kb)
ESM Fig. 3
(PDF 9 kb)
Rights and permissions
About this article
Cite this article
Palanivel, R., Fullerton, M.D., Galic, S. et al. Reduced Socs3 expression in adipose tissue protects female mice against obesity-induced insulin resistance. Diabetologia 55, 3083–3093 (2012). https://doi.org/10.1007/s00125-012-2665-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2665-3