
Abstract
Aims/hypothesis
Non-invasive diagnostic tools specific for pancreatic beta cells will have a profound impact on our understanding of the pathophysiology of metabolic diseases such as diabetes. The objective of this study was to use molecular imaging probes specifically targeting beta cells on human samples and animal models using state-of-the-art imaging modalities (fluorescence and PET) with preclinical and clinical perspective.
Methods
We generated a monoclonal antibody, 8/9-mAb, targeting transmembrane protein 27 (TMEM27; a surface N-glycoprotein that is highly expressed on beta cells), compared its expression in human and mouse pancreas, and demonstrated beta cell-specific binding in both. In vivo imaging was performed in mice with subcutaneous insulinomas overexpressing the human TMEM27 gene, or transgenic mice with beta cell-specific hTMEM27 expression under the control of rat insulin promoter (RIP-hTMEM27-tg), using fluorescence and radioactively labelled antibody, followed by tissue ex vivo analysis and fluorescence microscopy.
Results
Fluorescently labelled 8/9-mAb showed beta cell-specific staining on human and mouse pancreatic sections. Real-time PCR on islet cDNA indicated about tenfold higher expression of hTMEM27 in RIP-hTMEM27-tg mice than in humans. In vivo fluorescence and PET imaging in nude mice with insulinoma xenografts expressing hTMEM27 showed high 8/9-mAb uptake in tumours after 72 h. Antibody homing was also observed in beta cells of RIP-hTMEM27-tg mice by in vivo fluorescence imaging. Ex vivo analysis of intact pancreas and fluorescence microscopy in beta cells confirmed these findings.
Conclusions/interpretation
hTMEM27 constitutes an attractive target for in vivo visualisation of pancreatic beta cells. Studies in mouse insulinoma models and mice expressing hTMEM27 demonstrate the feasibility of beta cell-targeted in vivo imaging, which is attractive for preclinical investigations and holds potential in clinical diagnostics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetes is a progressive disease ultimately leading to loss of pancreatic beta cells, which produce and secrete insulin to maintain glucose homeostasis. With a chronically increasing demand for insulin in response to an increased metabolic load and peripheral insulin resistance, beta cells adapt by altering both functionality and mass [1–5]. Gradual loss of beta cells and their function causes hyperglycaemia and eventually type 2 diabetes [2, 6–8]. Today, an estimated 300 million people have impaired glucose tolerance, while another 600 million are suffering from fully established diabetes [9]. Non-invasive monitoring of beta cell changes in vivo could serve as an early indicator of diabetes and accelerate the clinical development of novel, possibly preventive, therapies. Molecular imaging could provide specific tools for detecting beta cell mass and even functionality in vivo [10–12]. Beta cells constitute only 1% of the total pancreatic mass, and their scattered organisation in islets of Langerhans within the largely exocrine pancreas makes development and use of cell-specific imaging tools challenging. Moreover, the anatomical location of the pancreas next to large organs involved in drug/label metabolism and excretion makes it difficult to access by imaging-based approaches.
A variety of strategies for imaging beta cells based on MRI, positron emission tomography (PET) and optical imaging are being explored [13]. Probes such as dihydrotetrabenazine, sulfonylurea secretagogues and somatostatin receptor analogues have been evaluated as potential beta cell-detection agents. The ligand dihydrotetrabenazine, targeting the vesicular monoamine transporter 2 expressed on the pancreatic beta cells, is the most promising candidate for imaging pancreatic beta cells in vivo [12, 14]. Alternative methods allow exploitation of physiological properties of pancreatic islet structures with imaging. One example is MRI-based non-targeted superparamagnetic nanobeads, which have been used to visualise peri-islet vascular leakage due to insulitis [15]. Because of its sensitivity and clinical translatability, PET is the modality of choice for non-invasive imaging of molecular and cellular targets [16], although high-energy ionising radiation and the half-life of the radioisotopes may limit achievement of a high signal-to-background ratio [17]. For experimental studies, fluorescence imaging is an attractive alternative to PET, and fluorescent probes can be readily used for histological validation of in vivo imaging results. However, the major limitation of optical imaging is photon scattering by the biological tissue, which renders recovery of geometrical information difficult and limits penetration depth [18]. Therefore, for imaging beta cells in vivo, we examined both optical and PET imaging as tools for research and clinical translation, respectively, using an antibody-based approach.
Given the diffuse distribution of beta cells in the pancreas, it is important to identify a beta cell-specific molecular target with high affinity. Studies on gene expression in beta cells in the pancreas have shown that the transmembrane protein, TMEM27, is selectively expressed on the beta cell surface [19] and in kidney collecting ducts [20].
Here we report on the characterisation of a monoclonal antibody, 8/9-mAb, specific to human TMEM27 (hTMEM27), which, upon suitable labelling, could be used for specific imaging of beta cells in vivo by fluorescence or PET imaging. The specificity of fluorescently labelled 8/9-mAb was evaluated in human tissue sample microarrays and pancreatic sections from transgenic mice expressing hTMEM27 under the control of rat insulin promoter (RIP-hTMEM27-tg). The biodistribution of the antibody and its binding to hTMEM27 in vivo were assessed in a nude mouse subcutaneous insulinoma model expressing high levels of hTMEM27. Finally, target-specific in vivo imaging was used to assess beta cells in RIP-hTMEM27-tg mice.
Methods
Experimental animals
All procedures were in accordance with the Cantonal Veterinary Office in Zurich. C57BL6 mice and BALB/c nude mice were from Charles River Laboratories (Sulzfeld, Germany). RIP-hTMEM27-tg mice were generated in-house (M. Stoffel). All animals used in this study were matched for sex and age.
Vectors and stable cell lines
hTMEM27 is cloned in the pTRE vector (Clontech/Takara Bio, Saint-Germain-en-Laye, France). Clonal selection was used to generate cell lines stably transfected with a pTRE-TMEM27 construct as described by Wang and Iynedjian [21].
Cell culture and transfections
Rat insulinoma INS-1E cells were obtained from C. Wolheim’s laboratory, Geneva University, Switzerland and grown according to standard laboratory protocol [22]. For stable cell lines carrying the pTRE-hTMEM27 construct (INS-1E-hTMEM27), the culture medium also contained the antibiotic, G418 (400 μg/ml; Sigma-Aldrich), to maintain the construct, and doxycycline (20 μg/ml), to induce human TMEM27 gene expression [21].
Tumour generation
The animals were injected subcutaneously in the left thigh with either 3 × 106 cultured INS-1E or INS-1E-hTMEM27 cells in 100 μl saline (154 mmol/l NaCl). The tumour-bearing mice were administered 200 ng/ml doxycycline hydrochloride in drinking water to induce hTMEM27 gene expression [21], with sucrose (1%) added to ameliorate the bitter taste.
Antibody generation and labelling
mAb against hTMEM27 (8/9-mAb) was generated by whole-cell immunisation with repeated injections of living INS-1E-hTMEM27 cells as described by Köhler and Milstein [23], at F. Hoffmann-La Roche Pharmaceuticals, Basel AG. The 8/9-mAb was conjugated to Alexa Fluor (AF) 488 or AF 680 (A200000; Molecular Probes, Eugene, OR, USA). The quality of the conjugate was analysed by immunofluorescence in serial dilutions of the antibodies on INS-1E-hTMEM27 cells.
89Zr–8/9-mAb conjugates were prepared by conjugating 8/9-mAb with p-isothiocyanatobenzyl-desferrioxamine (SCN-Bz-DF; Macrocyclics, Dallas, TX, USA) [24], Df-8/9-mAb. 89Zr was produced by irradiating natural yttrium (89Y) with protons at the 72 MeV accelerator of the Paul Scherrer Institute (Villigen, Switzerland) as described by Hohn et al [25]. Df-8/9-mAb (1.5 mg/ml) was added to 89Zr (100–200 MBq) in 1 mol/l oxalic acid neutralised to pH 7.8 and allowed to react for 30 min at 37°C. The conjugate was then purified by size-exclusion chromatography on a Sepharose 12 column (GE Healthcare Life Sciences, UK) with PBS as running buffer.
Human versus mouse islet mRNA quantification
Human islets were obtained from six independent cadaver donors through the European Consortium of Islet Transplantation (ECIT) supported by the Juvenile Diabetes Research Foundation (JDRF) in the course of 2008–2010. The islets used were frozen 1 week after isolation, after 2 h incubation at 37°C in CMRL (Connaught Medical Research Laboratories) medium (GibcoBRL, CA, USA) upon arrival at our location. Murine islets were isolated from C57Bl6 (five) or RIP-TMEM27 (four) mice using a standard mouse islet-isolation protocol, and were frozen 2 h after isolation. RNA was isolated using Trizol (Invitrogen, Life Technologies, Zug, Switzerland), genomic DNA was digested using the DNA-free kit (Ambion, Life Technologies, Zug, Switzerland), and 400 ng RNA per sample was reverse transcribed using the Superscript III kit (Invitrogen, Life Technologies, Zug, Switzerland). The primer pairs used to quantify total PDX1 and hTMEM27 cDNA were as given in the ESM. Tab All primer pairs were used at an annealing temperature of 60°C, when all pairs amplified single bands from the islet cDNA and showed no PCR product in the non-Superscript-treated control samples. Quantitative PCR was performed on a Stratagene RT-PCR machine.
Near-infrared frequency imaging
Mice bearing subcutaneous tumours were anaesthetised with 2% isoflurane (Attane; Minrad, Buffalo, NY, USA) in oxygen/air mixture (1:4, vol./vol.). Animals were imaged on a prewarmed (37°C) platform in the small-animal imaging system, with maintained anaesthesia. Control (C57BL6) and RIP-hTMEM27-tg mice (n = 3–4) or subcutaneous tumour-bearing nude mice (both INS-1E-hTMEM27 and INS-1E tumours) were administered 2 mg/kg AF 680–8/9-mAb or AF 680–anti-haemagglutinin (HA)-mAb i.v. via the tail vein and imaged in vivo longitudinally for the next 6 days.
Fluorescence molecular tomography
In vivo imaging was performed on a home-made fluorescence molecular tomography (FMT) system [26]. For each animal (n = 3), a region of interest (ROI) containing the tumour was excited by scanning at 671 nm using a continuous-wave laser source across the ROI. The images were recorded with an Andor iKon CCD camera (LOT-Oriel, Moulin-du-Choc, Switzerland). The FMT data were reconstructed using the normalised Born approximation using the slab approximation [27]. For quantification, the mean reconstructed fluorescence intensity was computed over the tumour volume.
Two-dimensional in vivo imaging
This was carried out with a Maestro 500 imaging system (Cambridge Research, Woburn, MA, USA). For AF 680 measurements, a band pass filter from 615 nm to 665 nm and a high-pass filter over 700 nm were used for excitation and emission light, respectively. A series of images were acquired at different wavelengths and then subjected to spectral unmixing as described by Gao et al [28] for unmixing the AF 680 fluorescence pattern from tissue autofluorescence.
Two-dimensional ex vivo imaging
This was also carried out with a Maestro 500 imaging system. Control and RIP-hTMEM27-tg mice were injected with 2 mg/kg AF 680–8/9-mAb or AF 488–8/9-mAb. The animals were killed 72 h (3 days) after antibody administration, and organs (pancreas, liver, kidney and spleen) were excised and imaged. For detection of the AF 488 signal, a band pass filter from 445 nm to 490 nm and a high-pass filter over 515 nm were used for excitation and emission light, respectively, while the filter combination described above was used for AF 680 detection. Spectral unmixing was carried out as previously described [28]. The images were normalised for depth intensity, exposure time and binning differences.
In vivo PET
INS-1E-hTMEM27 (n = 2) and INS-1E (n = 2) tumour-bearing nude mice (19–21 g) were intravenously injected with 1.5 MBq of the radiotracer, (89Zr-8/9-mAb, corresponding to 60 μg 8/9-mAb). PET scanning of the animals was performed at 24 h and 72 h after tracer injection using small-animal GE VISTA eXplore PET/CT tomograph. Animals were anaesthetised and scanned for 60 min. After the PET scan on day 3, the mice were killed, and this was followed by tissue sampling and gamma counting. Radioactivity accumulated in tissue samples was expressed as percentage of the injected dose (%ID)/g tissue. PET data were reconstructed in a single time frame and analysed using PMOD software (PMOD Technologies, Zurich, Switzerland). PET data were quantitatively evaluated by ROI analysis, and the standard uptake value was determined by normalising the activity concentration in the ROI to the injected dose per body weight. Imaged data were visualised by either maximum-intensity projections or horizontal slice series, which were normalised to the injected dose per body weight.
Immunohistochemistry
Cryo-blocks and sections of insulinomas and pancreas were derived from mice that were systemically injected with either prelabelled AF 488–8/9-mAb or AF 488–anti-HA-mAb and were processed as follows. The excised mouse organs were processed to cryo-blocks using cryo-preservant medium (optimal cutting temperature [OCT] medium; Tissue-Tek, Sakura, the Netherlands) into 10 μm tissue sections. Sections were washed (3 × 5 min) with PBS and fixed with 3% paraformaldehyde/2% sucrose in 1× PBS (30 min) at room temperature (RT), followed by washing with 1× PBS (3 × 5 min). Blocking was performed with 10% normal goat serum/BSA/0.3% Triton X-100 in 1× PBS (blocking buffer) (30 min at RT). The sections were incubated with the primary antibody (either unlabelled 8/9-mAb [F. Hoffmann-La Roche] or anti-insulin [Linco; No. 4011-01; St Charles, Missouri, USA]) for 2 h at RT in blocking buffer. Sections were washed with 1× PBS (3 × 5 min) and subsequently incubated with AF 488 goat anti-mouse (Invitrogen Life Technologies, Zug, Switzerland) secondary antibody (dilution 1:400) for 30 min at RT in blocking buffer. The sections were washed with 1× PBS (3 × 5 min) and mounted using ProLong Gold (Molecular Probes) with DAPI. All sections were visualised using a fluorescence microscope (Zeiss Axioplan 2), and images were processed using Zeiss AxioVision (release 4.7) software. Human adult normal tissue microarrays (T8234708; BioChain, Newark, CA, USA; 5 μm thick) were stained with 100 ng/ml Alex Fluor 488–8/9-mAb and insulin following the procedures described above.
Statistical analysis
All quantification was carried out using GraphPad Prism 5 (http://www.softpedia.com/get/Science-CAD/GraphPad-Prism.shtml). Error bars represent SD. p values were calculated using two-way ANOVA; those smaller than 0.05 were considered significant.
Results
TMEM27 as a potential target for pancreatic beta cell visualisation
A series of mAbs was raised against the beta cell surface N-glycoprotein, hTMEM27 [19], and clone 8/9 was identified as the hTMEM27-specific mAb (8/9-mAb) most suitable for imaging applications. This antibody was labelled with either the dye AF 680 (AF 680–8/9-mAb) or a chelating ligand containing zirconium-89 (89Zr-8/9-mAb) for visualisation with fluorescence imaging and PET, respectively. For histological analysis, 8/9-mAb was labelled with the dye AF 488 (AF 488–8/9-mAb).
Human tissue samples were used to evaluate the clinical relevance of hTMEM27 by assessing tissue-specific production and binding specificity of 8/9-mAb to TMEM27 in adult human normal tissue microarrays. US Food and Drug Administration (FDA) standard adult human tissue microarrays containing samples of pancreas, kidney, liver and spleen (T8234708) were stained with AF 488–8/9-mAb. Immunofluorescence analysis of these tissue microarrays showed TMEM27 expression to be restricted to pancreatic and kidney samples, with no TMEM27 immunoreactivity in liver and spleen (Fig. 1a). Importantly, AF 488–8/9-mAb colocalised with insulin, showing that the staining is beta cell specific (Fig. 1b).

TMEM27, a potential target for pancreatic beta cell imaging. (a) Immunofluorescence images of 5 μm FDA standard adult human tissue microarrays containing samples of spleen, liver, pancreas and kidney with AF 488–8/9-mAb, showing uptake in the pancreas and kidney. Scale bar = 20 μm. (b) Immunohistochemistry of 10 μm-thick human pancreatic tissue sections with AF 488–8/9-mAb (identifying hTMEM27) and insulin staining of the same sections. 8/9-mAb and insulin colocalisation images show antibody retention in beta cells. Scale bar = 50 μm. (c) Immunofluorescence of 10 μm pancreatic sections from RIP-hTMEM27-tg and normal C57BL6 (control) mice with 8/9-mAb. Insulin staining, for localising beta cells in an islet, is shown in red and nuclear staining in blue. Scale bar = 20 μm. (d) Real-time PCR of cDNA from murine, human and RIP-TMEM27-tg mouse islets. hTMEM27 levels were determined using primers unique to hTMEM27. Signals were first normalised to real-time PCR signal obtained for PDX1 cDNA (as a beta cell normalisation standard) using primers binding exon flanking sequences identical in human and mouse PDX1 cDNA sequence, and then to the human TMEM27/PDX1 ratio. Murine islets, n = 5, human islets, n = 6, and RIP-TMEM27 islets, n = 4. Error bars represent standard deviations. ***p < 0.001. RU = relative units
Expression analysis of hTMEM27 using the 8/9-mAb antibody in pancreatic sections of C57BL/6 mice (control) compared with RIP-hTMEM27-tg mice [19] showed distinct islet staining (Fig. 1c). Co-staining of AF 488–8/9-mAb and an insulin-specific antibody confirmed the binding to be beta cell specific in RIP-hTMEM27-tg mice (Fig. 1c).
To determine the level of hTMEM27 in the transgenic animals compared with humans and thus evaluate its suitability as a model for future application in human beta cells, real-time PCR specific for hTMEM27 was performed on islet cDNA derived from six different human cadaver donors, four RIP-hTMEM27-tg mice and five C57Bl6-negative control animals, using primer pairs given in ESM Tab. 1. After normalisation to PDX1 cDNA as a beta cell marker, the ratio of hTMEM27 in the transgenic mice to that in human material was about 10:1, while no signal was obtained from the negative control animals (Fig. 1d). Given that the human islets used were compromised compared with the murine samples (see the Methods section), this degree of overexpression was considered close enough to the hTMEM27 expression that might be expected in humans in vivo.
In vivo binding specificity and kinetics of 8/9-mAb in mice bearing subcutaneous insulinomas
To characterise binding properties and distribution kinetics of 8/9-mAb in vivo, we generated a rat beta cell insulinoma cell line stably expressing hTMEM27 under the control of a tetracycline transactivator (INS-1E-hTMEM27). Before in vivo studies, 8/9-mAb binding was assessed in cell cultures using doxycycline-inducible INS-1E-hTMEM27 cells. Because of expression leakage in INS-1E-hTMEM27, the parental INS-1E cells served as controls rather than non-doxycycline-treated cells.
Antibody labelling showed strong uptake of 8/9-mAb by the INS-1E-hTMEM27 cells compared with controls (electronic supplementary material [ESM] Fig. 1). The binding specificity of 8/9-mAb was further confirmed using the same concentration of anti-HA-mAb on INS-1E-hTMEM27 cells, where no specific signal was detected (ESM Fig. 1).
To monitor 8/9-mAb binding in vivo over time, either INS-1E-hTMEM27 or INS-1E cells were subcutaneously injected into nude mice. To induce expression of hTMEM27 in the target cell, all tumour-bearing mice were provided with doxycycline-containing drinking water. After the tumours had reached a visible size (>3 mm), AF 680–8/9-mAb was injected i.v. and fluorescence intensity was measured dynamically during the subsequent 6 days using FMT [27, 29]. Reconstructed FMT images of the tumour-bearing nude mice showed strong fluorescence in the INS-1E-hTMEM27 tumours within 24 h of AF 680–8/9-mAb injection. This signal was retained for the subsequent 3 days (Fig. 2a), and gradually cleared in the next 6 days. In contrast, the control (INS1E) mice injected with AF 680–8/9-mAb displayed significantly less fluorescence intensity during this 6-day observation period (Fig. 2a). Similarly no signal was observed in the INS-1E-hTMEM27 tumour-bearing mice injected with AF 680–anti-HA-mAb (Fig. 2a).

8/9-mAb targets hTMEM27 in vivo using near-infrared frequency FMT. (a) FMT of a beta cell subcutaneous tumour in a nude mouse, 3 days after AF 680–8/9-mAb injection. Uptake of AF 680–8/9-mAb in vivo was observed in INS-1E-hTMEM27 tumours, whereas INS-1E tumours and AF 680–anti-HA-mAb in INS-1E-hTMEM27 tumours show no specific uptake. Below each animal is the reconstructed three-dimensional view of the tumours, with the z axis perpendicular to the tumour surface. (b) 8/9-mAb pharmacokinetics, in vivo. Quantification of antibody uptake in tumours (fluorescence intensity measured by FMT), over a 6-day period after injection. There was a significant difference (p = 0.0001) in antibody uptake by hTMEM27-expressing tumours (INS-1E-hTMEM27, AF 680–8/9-mAb; circles) compared with control tumours (INS-1E, AF 680–8/9-mAb; triangles) and control antibody (INS-1E-hTMEM27, AF 680–anti-HA-mAb; squares). a.u., arbitrary units. (c) Histology of tumours (insulinomas) from the nude mice for antibody localisation. Nuclei were stained with DAPI (blue); AF 488–8/9-mAb (green). Left column depicts images at ×20 magnification, and right column is ×60 magnification. Scale bar = 50 μm
Quantitative analysis of the fluorescence intensity in all these tumours revealed that label uptake was highest 24 h after antibody injection in all cases, and the signal from INS-1E-hTMEM27 tumours was significantly higher than in the controls (p = 0.0001). Thereafter, the fluorescence intensity decreased in all cases. At 3 days after antibody injection, the fluorescence intensity in the INS-1E-hTMEM27 tumours was sevenfold and 17-fold higher than in the controls (INS-1E and INS-1E-hTMEM27 and anti-HA-mAb, respectively, p = 0.0001; Fig. 2b).
The in vivo studies were validated microscopically using cryo-sections derived from tumours collected 3 days after antibody injection. The INS-1E-hTMEM27 tumours showed a strong signal with the AF 488–8/9-mAb, but none with AF 488–anti-HA-mAb (Fig. 2c). The INS-1E tumours were negative for AF 488–8/9-mAb as well (Fig. 2c). In terms of antibody specificity, these observations are consistent with in vitro findings in the cell lines (ESM Fig. 1) and in vivo fluorescence measurements (Fig. 2a, b).
In vivo PET imaging for antibody specificity in mice with subcutaneous insulinomas expressing hTMEM27
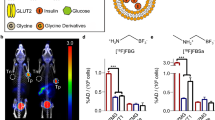
In view of potential translational applications of 8/9-mAb-based beta cell imaging, we performed PET imaging of the mice bearing either subcutaneous INS-1E-hTMEM27 or INS-1E tumours. 89Zr was chosen as radionuclide on the basis of its half-life of 78 h (3 days), which is appropriate for the pharmacokinetic properties of 8/9-mAb (Fig. 2d). Hence, 8/9-mAb was radiolabelled with 89Zr ([89Zr]-8/9-mAb). Binding specificity was tested in vivo using INS-1E-hTMEM27 and INS-1E tumour-bearing mice, which were administered with 150 MBq [89Zr]-8/9-mAb. The animals were subjected to PET imaging 24 h and 72 h after injection. Whole-body images of these animals revealed significantly higher activity in INS-1E-hTMEM27 tumours than INS-1E tumours (Fig. 3a and ESM Fig. 2). At 72 h after injection, the %ID/g in INS-1E-hTMEM27 tumours was six to seven times higher than in control tumours (p = 0.001, Fig. 3b) and comparable to the results obtained using AF-labelled antibody, indicating specific tracer accumulation due to expression of the molecular target and a similar performance of the two imaging methods.

[89Zr]–8/9-mAb targets hTMEM27 in vivo. (a) PET of beta cell tumour-bearing nude mice, 3 days after [89Zr]–8/9-mAb injection. (b) Radioactive uptake shown as %ID/g (p = 0.001). Maximum uptake was seen in tumour (a, middle arrow) compared with liver (a, top arrow) or site of tail injection (a, bottom arrow). Tumour uptake was much higher in INS-1E-hTMEM tumour bearing mice
In vivo targeting of beta cells in mouse pancreas using 8/9-mAb
To evaluate in vivo binding of 8/9-mAb to its target, hTMEM27, a biodistribution study was carried out using AF 680–8/9-mAb. After fluorescent antibody injections in mice, fluorescence reflectance images were recorded at multiple wavelengths and spectrally deconvoluted to discriminate the AF 680 signal from non-specific background fluorescence [28]. Normalised AF 680 signal intensities in an abdominal ROI in RIP-hTMEM27-tg and control mice recorded 72 h after antibody administration were compared (Fig. 4a). The subsequent analysis revealed a 3.3-fold higher fluorescence intensity in the RIP-hTMEM27-tg mice (Fig. 4b; p = 0.01).

8/9-mAb hTMEM27 antibody specifically targets mouse pancreatic beta cells in vivo. (a) RIP-hTMEM-tg and control mice (n = 3), 3 days after AF 680–8/9-mAb injection and spectral deconvolution showing different fluorescence retention. (b) Quantification of the normalised fluorescent intensities from these mice († p = 0.01). (c) Ex vivo near-infrared imaging of mouse (n = 3) organs (L, liver; K, kidney; S, spleen; P, pancreas) from (a), 3 days after i.v. injection of AF 680–8/9-mAb. (d) Fluorescence intensity in organs from transgenic (black bar) and control (white bar) mice; p value for pancreas is 0.084. (e) Ex vivo imaging of mouse organs harvested 3 days after injection of AF 488–8/9-mAb. The images were unmixed (spectral deconvolution) to separate antibody signal (in green) from tissue background (red). Islet uptake of the fluorescently labelled antibody can be seen in pancreas from RIP-h-TMEM27 mice as green hot spots (arrowheads). (f) Quantification of the mean fluorescent signal intensity/exposure time of pancreases from RIP-hTMEM27-tg and control mice (‡ p = 0.0001). (g) Histological analysis of the pancreas from RIP-hTMEM27-tg and C57BL6 (control) mice after i.v. tail vein injection of AF 488–8/9-mAb, showing antibody uptake in the islet beta cells in RIP-hTMEM27-tg mouse pancreas, while control mouse pancreatic sections stained negative. The sections were further stained for insulin (red). The AF 488–8/9-mAb staining colocalised with insulin staining for pancreatic beta cells. Scale bar = 20 μm
To investigate antibody biodistribution in vivo, organs were harvested from AF 680–8/9-mAb-injected RIP-hTMEM27-tg and control mice. Subsequently, ex vivo analysis was performed using fluorescence reflectance imaging including spectral deconvolution (Fig. 4c). The fluorescent signal in isolated pancreas from RIP-hTMEM27-tg mice displayed a striking difference in texture from the control mice. In RIP-hTMEM27-tg mice, the highest fluorescence uptake was detected in dot-like structures, a pattern that is reminiscent of the murine islet distribution along pancreatic ducts (Fig. 4c). This distinct fluorescent signal was absent from pancreas derived from control mice, displaying only a diffuse intensity distribution (Fig. 4c). The normalised pancreatic fluorescence signal was 0.021 ± 0.014 in RIP-hTMEM27-tg mice compared with 0.009 ± 0.0006 in control animals, although this difference did not reach statistical significance (p = 0.08) because of higher degree of variation in the hTMEM27-associated signal. No significant difference in fluorescence intensity was found in the other organs (Fig. 4d).
To further assess the enriched pancreatic signal, magnified images of pancreas derived from RIP-hTMEM27-tg mice showed antibody-associated fluorescence localised to islet-like structures (Fig. 4e), marked as fluorescent hot spots. In contrast, the control mouse pancreas showed minor background signal in the equivalent regions, with the only perceivable signal arising from non-pancreatic connective tissue at the periphery (Fig. 4e). Quantitative analysis of these images revealed that the fluorescent intensity in the hotspots observed in transgenic mice was sevenfold higher than the diffuse signal in the corresponding regions of control mice (Fig. 4f; p = 0.0001).
To elucidate the origin of the fluorescent hot spots, we microscopically analysed pancreatic sections from organs taken 72 h after AF 488–8/9-mAb injection. Co-staining of these sections with an insulin-specific antibody identified the hot spots as islets comprising pancreatic beta cells in the RIP-hTMEM27-tg mice (Fig. 4g), whereas no AF 488–8/9-mAb staining was observed in islet regions of the control mice (Fig. 4g).
Discussion
State-of-the-art beta cell imaging strategies [30, 31] have to be able to monitor beta cells in a longitudinal manner. This requires identification of molecular entities that are highly specific for beta cells, which can be targeted using molecular imaging probes. Ideally, these targets should not only reflect the beta cell functional status, but should also be preserved throughout a wide patient population. A promising candidate that meets these specifications is the cell surface N-glycoprotein, TMEM27, which is exclusively expressed on pancreatic beta cells and kidney [19]. In addition, TMEM27 has a functional relevance in beta cell proliferation and insulin secretion [19, 32], and, although its levels were found to be decreased in islets derived from diabetic donors compared with healthy controls, it was still present in these patients [33, 34].
In our approach of targeting TMEM27, we characterised a mAb against hTMEM27, 8/9-mAb, which specifically labels human pancreatic beta cells and parts of human kidney, as an imaging tool. We used fluorescent labelling of antibodies in combination with FMT as an experimental approach, and radiolabelling in combination with PET for potential translational applications (Fig. 2), using hTMEM27-expressing insulinoma xenograft nude mice and RIP-hTMEM27-tg mice as well as models for the human beta cell. The former revealed a high signal-to-background contrast in subcutaneous tumours 1 day after injection of AF 680–8/9-mAb (Fig. 2a–c), and similarly we observed high label retention in the pancreatic islets of the RIP-hTMEM27-tg mice (Fig. 1d), comparable to the findings derived from human pancreatic sections (Fig. 1a). This study thus represents a novel beta cell assessment method, namely an antigen–antibody pair-mediated (humanised) beta cell imaging approach. The observation that transcript levels of hTMEM27 in islets from human cadaver donors were ten times lower than in RIP-hTMEM27-tg mice may indicate that the sensitivity of the antibody-mediated approach will have to be improved for clinical applications. One possible way to further increase the antibody-mediated hTMEM27 signal is to administer a beta-secretase (BACE) inhibitor before and during the antibody treatment, thereby preventing TMEM27 degradation by BACE2 and significantly increasing antigen availability, since BACE2 inhibition has been shown to increase TMEM27 signals at least 3–4-fold in isolated human islets [33].
The signal generated at the target site is essentially governed by the number of binding sites expressed by the target and by the binding affinity and bioavailability of the probe. Unspecific signals are largely determined by the rate of probe elimination from the circulation, which also dictates its retention in the extracellular space. Fluorescence microscopy revealed that the beta cell-specific fluorescence signal originated from probe molecules in the beta cells, indicating internalisation of 8/9-mAb (Figs 1, 2 and 4). Internalisation contributed to longer retention of labelled probe in the beta cells (both in tumours and pancreatic islets) and potentially also led to accumulation due to probe trapping associated with amplification of the fluorescence signal. TMEM27 is an ideal target for such an accumulation, as, at any given point in time, only 10% of the TMEM27 population is found at the cell surface [35], similar to other signalling receptors, indicating vibrant trafficking and internalisation. Our study may also suggest that dynamically trafficking cell surface proteins are potentially powerful targets for antibody-based imaging approaches. In spite of high specific binding of the 8/9-mAb-based probe, the low fraction of beta cells in pancreatic tissue requires specific local enrichment of an imaging probe by a factor of at least 100 compared with the exocrine tissue, blood and adjacent organs, in order to enable reliable assessment of changes in beta cells in vivo. This value may be reached with further technological development such as: (1) optimising the probe’s pharmacokinetic properties; (2) using antibody fragments or antibodies with even higher affinity for hTMEM27; (3) using antibodies that will only bind to the extracellular part of TMEM27, which stays membrane associated even after cleavage (human amino acid residues 126–141) and therefore may lead to lower background levels, as binding to shed circulating TMEM27 will be avoided; (4) using a BACE inhibitor, which prevents hTMEM27 degradation, thereby increasing antigen availability.
Nevertheless, fluorescence imaging constitutes an attractive imaging modality for research purposes, as it uses stable labels suitable for monitoring animals over extended periods of time and allows direct validation through fluorescence histology, and translation to clinics can be readily achieved by replacing the fluorescent label with a radionuclide label.
In summary, we have demonstrated that selective targeting and imaging of beta cells using an antibody-mediated approach is feasible in vivo, which is relevant with regard to both preclinical and clinical applications, despite current technological limitations. The fact that high antibody retention has been found in the beta cells of human pancreatic sections and humanised beta cells in vivo makes 8/9-mAb a potential candidate for clinical application, and hTMEM27 an attractive antigen as an imaging target.
Abbreviations
- %ID:
-
Percentage of injected dose
- AF:
-
Alexa Fluor
- BACE:
-
Beta-secretase
- FMT:
-
Fluorescence molecular tomography
- HA:
-
Haemagglutinin
- mAb:
-
Monoclonal antibody
- PET:
-
Positron emission tomography
- RIP:
-
Rat insulin promoter
- ROI:
-
Region of interest
- TMEM27:
-
Transmembrane protein 27
References
Aston-Mourney K, Proietto J, Morahan G, Andrikopoulos S (2008) Too much of a good thing: why it is bad to stimulate the beta cell to secrete insulin. Diabetologia 51:540–545
Chang-Chen KJ, Mullur R, Bernal-Mizrachi E (2008) Beta-cell failure as a complication of diabetes. Rev Endocr Metab Disord 9:329–343
Edlund H (2001) Factors controlling pancreatic cell differentiation and function. Diabetologia 44:1071–1079
Gerich JE (1999) Is insulin resistance the principal cause of type 2 diabetes? Diabetes Obes Metab 1:257–263
Sachdeva MM, Stoffers DA (2009) Minireview: meeting the demand for insulin: molecular mechanisms of adaptive postnatal beta-cell mass expansion. Mol Endocrinol 23:747–758
Karaca M, Magnan C, Kargar C (2009) Functional pancreatic beta-cell mass: involvement in type 2 diabetes and therapeutic intervention. Diabetes Metab 35:77–84
Buchanan TA (2003) Pancreatic beta-cell loss and preservation in type 2 diabetes. Clin Ther 25(Suppl B):B32-46
Lingohr MK, Buettner R, Rhodes CJ (2002) Pancreatic beta-cell growth and survival: a role in obesity-linked type 2 diabetes? Trends Mol Med 8:375–384
Hossain P, Kawar B, El Nahas M (2007) Obesity and diabetes in the developing world: a growing challenge. N Engl J Med 356:213–215
Lin M, Lubag A, McGuire MJ et al (2008) Advances in molecular imaging of pancreatic beta cells. Front Biosci 13:4558–4575
Schneider S (2008) Efforts to develop methods for in vivo evaluation of the native beta-cell mass. Diabetes Obes Metab 10(Suppl 4):109–118
Souza F, Freeby M, Hultman K et al (2006) Current progress in non-invasive imaging of beta cell mass of the endocrine pancreas. Curr Med Chem 13:2761–2773
Virostko J, Jansen ED, Powers AC (2006) Current status of imaging pancreatic islets. Curr Diab Rep 6:328–332
Simpson NR, Souza F, Witkowski P et al (2006) Visualizing pancreatic beta-cell mass with [11C]DTBZ. Nucl Med Biol 33:855–864
Denis MC, Mahmood U, Benoist C, Mathis D, Weissleder R (2004) Imaging inflammation of the pancreatic islets in type 1 diabetes. Proc Natl Acad Sci U S A 101:12634–12639
Mankoff DA, Eary JF, Link JM et al (2007) Tumor-specific positron emission tomography imaging in patients: [18F]fluorodeoxyglucose and beyond. Clin Cancer Res 13:3460–3469
Cassidy PJ, Radda GK (2005) Molecular imaging perspectives. J R Soc Interface 2:133–144
Leblond F, Davis SC, Valdes PA, Pogue BW (2010) Pre-clinical whole-body fluorescence imaging: review of instruments, methods and applications. J Photochem Photobiol B 98:77–94
Akpinar P, Kuwajima S, Krutzfeldt J, Stoffel M (2005) Tmem27: a cleaved and shed plasma membrane protein that stimulates pancreatic beta cell proliferation. Cell Metab 2:385–397
Zhang H, Wada J, Hida K et al (2001) Collectrin, a collecting duct-specific transmembrane glycoprotein, is a novel homolog of ACE2 and is developmentally regulated in embryonic kidneys. J Biol Chem 276:17132–17139
Wang H, Iynedjian PB (1997) Modulation of glucose responsiveness of insulinoma beta-cells by graded overexpression of glucokinase. Proc Natl Acad Sci U S A 94:4372–4377
Merglen A, Theander S, Rubi B, Chaffard G, Wollheim CB, Maechler P (2004) Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology 145:667–678
Köhler G, Milstein C (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256:495–497
Perk LR, Vosjan MJ, Visser GW et al (2010) p-Isothiocyanatobenzyl-desferrioxamine: a new bifunctional chelate for facile radiolabeling of monoclonal antibodies with zirconium-89 for immuno-PET imaging. Eur J Nucl Med Mol Imaging 37:250–259
Hohn A, Zimmermann K, Schaub E, Hirzel W, Schubiger PA, Schibli R (2008) Production and separation of “non-standard” PET nuclides at a large cyclotron facility: the experiences at the Paul Scherrer Institute in Switzerland. Q J Nucl Med Mol Imaging 52:145–150
Stuker F, Baltes C, Dikaiou K et al (2011) Hybrid small animal imaging system combining magnetic resonance imaging with fluorescence tomography using single photon avalanche diode detectors. IEEE Trans Med Imaging 30:1265–1273
Ntziachristos V, Weissleder R (2001) Experimental three-dimensional fluorescence reconstruction of diffuse media by use of a normalized Born approximation. Opt Lett 26:893–895
Gao X, Cui Y, Levenson RM, Chung LW, Nie S (2004) In vivo cancer targeting and imaging with semiconductor quantum dots. Nat Biotechnol 22:969–976
Ntziachristos V (2006) Fluorescence molecular imaging. Annu Rev Biomed Eng 8:1–33
Holmberg D, Ahlgren U (2008) Imaging the pancreas: from ex vivo to non-invasive technology. Diabetologia 51:2148–2154
Ahlgren U, Gotthardt M (2010) Approaches for imaging islets: recent advances and future prospects. Adv Exp Med Biol 654:39–57
Fukui K, Yang Q, Cao Y et al (2005) The HNF-1 target collectrin controls insulin exocytosis by SNARE complex formation. Cell Metab 2:373–384
Esterhazy D, Stutzer I, Wang H et al (2011) Bace2 is a beta cell-enriched protease that regulates pancreatic beta cell function and mass. Cell Metab 14:365–377
Altirriba J, Gasa R, Casas S et al (2010) The role of transmembrane protein 27 (TMEM27) in islet physiology and its potential use as a beta cell mass biomarker. Diabetologia 53:1406–1414
Esterhazy D, Akpinar P, Stoffel M (2012) Tmem27 dimerisation, deglycosylation, plasma membrane depletion, and the extracellular Phe-Phe motif are negative regulators of cleavage by Bace2. Biol Chem 393:473–484
Acknowledgements
The authors acknowledge K. Zimmermann, PSI, Villigen, Switzerland for zirconium generation, C. Germanier, IBT ETH, Zurich, Switzerland for help with fluorescence measurements, and the Juvenile Diabetes Research Foundation (JDRF) for human islets from cadavers.
Funding
This work was in part supported by the ‘Systems biology of the beta cell’ collaboration between the Center of Competence for Systems Biology and Metabolic disease (CC-SPMD), ETH, Zurich, F. Hoffmann-La Roche, Basel, Switzerland and the Collaborative European Effort to Develop Diabetes Diagnostics (CEED3).
Duality of interest
This project was funded by F. Hoffmann-La Roche, Basel, Switzerland, but no financial conflict was involved.
Contribution statement
DV was responsible for the conception and design of the study, data collection, analysis and interpretation, drafting and revising the article. MR, MS, RS, CM and WK were responsible for the conception and design of the study and revising the article critically. HW, DE, KD, CD, MH, EF and HM were responsible for data collection and analysis and revising the article. FS, JR and RK were responsible for study design (contributing study reagents and tools), data analysis, and drafting the Methods section of the article. All authors approved the final version of the manuscript to be published.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table 1
Primer pairs to amplify PDX-1 cDNA and hTMEM27 cDNA (PDF 65.6 KB)
Fig. 1
8/9-mAb targets hTMEM27 in vitro. The beta cell type insulinoma cell line, INS-1E, served as the parental line for generating the stable cells expressing hTMEM27 (INS-1E-hTMEM27). Cultured cells were incubated with Alexa Fluor 488-8/9-mAb overnight and subsequently fixed. Fluorescence microscopy of 8/9-mAb immunostained β-cells (Alexa Fluor 488, green) and nuclear staining (Dapi, blue) showed specific uptake in INS-1E-hTMEM27 cells, while there was no uptake by non transfected INS-1E cells. No uptake was seen in the INS-1E-hTMEM27 cells either when using the control antibody, Alexa Fluor 488-anti-HA-mAb (bottom). a) 20× magnification, b) 60× magnification. Scale bar = 20 μm (PDF 38.7 KB)
Fig. 2
In vivo PET imaging of 8/9-mAb binding specificity to mouse insulinomas expressing hTMEM27. INS-1E-TMEM27 and INS-1E insulinoma cell-lines were used to make tumors in nude mice. 89Zr-8/9-mAb was injected in the tumor bearing mice intravenously, followed by PET imaging three days (72 h) after injection. Tumors expressing hTMEM27 had the antibody uptake throughout the tumor depth (INS-1E-hTMEM27) while poor or low uptake was seen in the control insulinomas (Ins-1E) (PDF 122 KB)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Vats, D., Wang, H., Esterhazy, D. et al. Multimodal imaging of pancreatic beta cells in vivo by targeting transmembrane protein 27 (TMEM27). Diabetologia 55, 2407–2416 (2012). https://doi.org/10.1007/s00125-012-2605-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2605-2