
Abstract
Aims/hypothesis
IL-6 is a proinflammatory cytokine associated with the pathogenesis of hepatic diseases. Metformin is an anti-diabetic drug used for the treatment of type 2 diabetes, and orphan nuclear receptor small heterodimer partner (SHP, also known as NR0B2), a transcriptional co-repressor, plays an important role in maintaining metabolic homeostasis. Here, we demonstrate that metformin-mediated activation of AMP-activated protein kinase (AMPK) increases SHP protein production and regulates IL-6-induced hepatic insulin resistance.
Methods
We investigated metformin-mediated SHP production improved insulin resistance through the regulation of an IL-6-dependent pathway (involving signal transducer and activator of transcription 3 [STAT3] and suppressor of cytokine signalling 3 [SOCS3]) in both Shp knockdown and Shp null mice.
Results
IL-6-induced STAT3 transactivation and SOCS3 production were significantly repressed by metformin, adenoviral constitutively active AMPK (Ad-CA-AMPK), and adenoviral SHP (Ad-SHP), but not in Shp knockdown, or with the adenoviral dominant negative form of AMPK (Ad-DN-AMPK). Chromatin immunoprecipitation (ChIP), co-immunoprecipitation (Co-IP) and protein localisation studies showed that SHP inhibits DNA binding of STAT3 on the Socs3 gene promoter via interaction and colocalisation within the nucleus. Upregulation of inflammatory genes and downregulation of hepatic insulin signalling by acute IL-6 treatment were observed in wild-type mice but not in Shp null mice. Finally, chronic IL-6 exposure caused hepatic insulin resistance, leading to impaired insulin tolerance and elevated gluconeogenesis, and these phenomena were aggravated in Shp null mice.
Conclusions/interpretation
Our results demonstrate that SHP upregulation by metformin may prevent hepatic disorders by regulating the IL-6-dependent pathway, and that this pathway can help to ameliorate the pathogenesis of cytokine-mediated metabolic dysfunction.
Similar content being viewed by others


Introduction
Proinflammatory cytokines, including IL-1, IL-6 and TNF-α, are associated with insulin resistance and metabolic syndromes, including obesity, type 2 diabetes and dyslipidaemia [1]. The proinflammatory cytokine IL-6 is a well-known regulator of the acute phase reaction in response to hepatotoxins such as alcohol and other pathophysiological changes resulting from obesity-related insulin resistance [2]. IL-6 has been identified as a predictor or pathogenic marker of hepatic diseases and also activates a family of proteins known as suppressors of cytokine signalling 3 (SOCS3) via the phosphorylation of Janus kinase 2 (JAK2) and signal transducer and activator of transcription 3 (STAT3) [1, 3]. IL-6-induced SOCS3 protein production inhibits hepatic insulin signalling via several mechanisms, including the direct blockage of the insulin receptor and insulin receptor substrate-1,2 (IRS-1,2) activation and degradation [3–6]. Several studies have shown that IL-6 administration increases whole-body insulin sensitivity in rodent models, and overexpression of human IL-6 promotes energy expenditure or systemic insulin sensitivity in a diet-induced obesity mouse model [7, 8]. While these findings demonstrate a positive role for IL-6 in insulin resistance, other groups have shown that IL-6 causes a deterioration of insulin resistance, leading to elevated blood glucose levels, hepatic glucose production and impaired insulin tolerance under both in vitro and in vivo conditions [9–11]. Therefore, the physiological role of IL-6 in the aetiology of insulin resistance remains controversial.
Metformin, a glucose-lowering agent, reduces hepatic glucose production, hepatic triacylglycerol and NEFA levels in obese mice, and also ameliorates hyperglycaemia [12, 13]. Metformin also increases insulin sensitivity via upregulation of insulin signalling through insulin receptor and IRS-1,2 phosphorylation [14]. We have recently shown that metformin and fenofibrate inhibit hepatic gluconeogenic genes and the fibrotic marker gene, plasminogen activator inhibitor-1 (Pai-1, also known as Serpine1) via activation of AMP-activated protein kinase (AMPK) in the liver [15, 16]. AMPK is a major intracellular energy sensor and a master regulator of glucose and lipid homeostasis [13, 17]. AMPK is activated by a variety of physiological processes, including the intracellular AMP/ATP ratio, physiological stresses, and the activity of certain pharmacological agents [17].
The small heterodimer partner (SHP, also known as NR0B2) is an atypical orphan nuclear receptor, which functions as a transcriptional co-regulator by directly interacting with other nuclear receptors and/or transcription factors [15, 18, 19]. SHP regulates the transcription of target genes involved in a variety of metabolic pathways and plays a pivotal role in the maintenance of metabolic homeostasis, including glucose, lipid and bile acid metabolism [19]. Our previous studies demonstrated that SHP production is induced by pharmacological agents, such as AMPK activators, that suppress hepatic metabolic disorders [15, 16]. However, the effects of AMPK activators on SHP protein production and their subsequent role in improving the proinflammatory cytokine-mediated dysfunctions of hepatic insulin resistance have yet to be clearly elucidated.
The present study has identified the effect of acute and chronic IL-6 treatment on hepatic insulin resistance and demonstrated that the induction of SHP by metformin ameliorates hepatic insulin resistance via downregulation of the IL-6-dependent pathway under both in vivo and in vitro conditions. The results of our current study suggest that the regulation of SHP by AMPK activators may provide a novel pathway that can improve the pathological progression of hepatic disorders through cytokines, as well as a potential therapeutic strategy for the prevention of cytokine-mediated metabolic dysfunction.
Methods
Animal experiments
C57BL/6J (WT) and congenic Shp null mice (B6.129/Sv-Shp tm1) were used for the experiments as previously described [16]. At the end of the specified treatment/feeding periods, mice were killed with CO2, and the liver tissues were collected. As for IL-6 and/or insulin stimulation experiment, both WT and Shp null mice at 12 weeks of age (Korea Research Institute of Bioscience and Biotechnology [KRIBB], Daejeon, Republic of Korea) were treated with oral metformin (200 mg/kg), and injected intraperitoneally with recombinant human IL-6 (1 μg/kg) after 6 h. The mice were then treated with insulin (1 U/kg) 1 h after the IL-6 injection. Finally, 20 min after insulin treatment, the mice were killed and their liver tissues were collected as described above. For chronic IL-6 treatment, IL-6 was intraperitoneally injected into mice at a daily dose of 2 μg/kg body weight. After 2 weeks of exposure, the mice were killed, and liver tissues and blood samples were collected as described above. Plasma glucose levels and insulin concentrations were measured by Glucostix AccuCheck (Roche Diagnostics, Mannheim, Germany) and Mouse Insulin ELISA (ALPCO Diagnostics, Salem, NH, USA), respectively, in accordance with the recommended protocols. All animal studies and protocols were approved and carried out by the Institutional Animal Use and Care Committee (IAUCC) of KRIBB.
Metabolic variables
Plasma glucose was measured using a glucometer (Roche) in blood collected from the tail of mice as previously described [15, 20]. For the insulin tolerance test, mice fasted for 4 h were administered intraperitoneally with insulin (0.5 U/kg), and glucose concentrations were measured at 0, 15, 30, 60, 90 and 120 min. For the glucose tolerance test, a single dose of 2.5 μg/kg glucose was injected intraperitoneally after 14 h fasting. Blood glucose was measured under the same conditions.
Materials and methods
Information on the materials and methods used in this study is shown in the electronic supplementary material (ESM) Methods section.
Statistical analysis
Data are expressed as means ± SEM. Analysis of variance was used to determine significant differences, via Student’s t tests and/or ANOVA. Values of p < 0.05 were considered to be statistically significant.
Results
Metformin inhibits IL-6-mediated STAT3 transactivation through induction of an SHP-dependent pathway
As reported previously, several AMPK activators (metformin, fenofibrate, hepatocyte growth factor [HGF] and sodium arsenite [NaArs]) have a significant effect on the upregulation of SHP production both in vivo and in vitro [15, 16, 21, 22]. Therefore, we tested whether the induction of SHP by metformin regulates the inhibition of the IL-6-mediated signalling pathway in primary hepatocytes. In primary rat hepatocytes, IL-6-induced STAT3 phosphorylation at Tyr705 was significantly repressed individually through metformin-induced SHP protein production via the activation of AMPK and the adenoviral overexpression of Shp (Ad-Shp) in a dose-dependent manner (Fig. 1a, b). To test whether metformin or SHP regulates Stat3 gene promoter activity, we performed transient transfection assays using a reporter gene containing multiple STAT3 binding sites (m67-Luc). The promoter activity increased by IL-6 or JAK2/STAT3 was reduced significantly by metformin and SHP in a dose-dependent manner (Fig. 1c, d). To confirm that the induction of SHP by metformin regulates IL-6-stimulated STAT3 transactivity, we evaluated the effect of SHP on IL-6-induced SOCS3 protein production in primary hepatocytes. Metformin significantly repressed IL-6-induced STAT3 transactivation and SOCS3 abundance through upregulation of SHP, whereas endogenous knockdown of Shp with adenoviral siRNA Shp (Ad-si Shp) or oligonucleotide siRNA Shp (si Shp) rescued the effect (Fig. 1e, f). Overall, these results demonstrate that metformin has an important effect on the downregulation of the IL-6-mediated signal pathway via the induction of SHP production.

Metformin inhibits IL-6-induced STAT3 transactivation in rat primary hepatocytes. a, b Rat primary hepatocytes were cultured under serum-free conditions for 24 h. After 12 h of pretreatment with metformin (Met, a), the cells were treated with IL-6 for 30 min at the indicated dose (20 ng/ml). Rat primary hepatocytes were infected with Ad-Shp (b) at a multiplicity of infection (MOI) of 30 or 60 for 36 h. After 36 h of infection, the cells were treated with IL-6 (20 ng/ml). Whole cell extracts (40 μg/lane) were assessed by western blot analysis with the indicated antibodies and then normalised to an internal control (β-actin and/or total forms [non-phosphorylated proteins]). c, d HepG2 cells were transiently cotransfected with the m67-Luc reporter gene and Shp or Tel-Jak2 (100 ng) or Stat3 (100 ng) or in combination, and treated for 1 h with IL-6 (20 ng/ml) in cells either non-pretreated or pretreated with metformin for 12 h. Luciferase activity was normalised to β-galactosidase activity to correct for variations in transfection efficiency. * p < 0.05 compared with untreated control, † p < 0.05 compared with IL-6-treated or Tel-Jak2 and Stat3 cotransfected cells. e Rat primary hepatocytes were infected with 60 MOI of GFP-tagged Ad-si Shp for 36 h. After infection with Ad-si Shp, the cells were treated with IL-6 (20 ng/ml) for 30 min after 12 h of metformin pretreatment. Whole cell extracts (40 μg/lane) were isolated and analysed by immunoblotting with the indicated antibodies and then normalised to an internal control (β-actin/total form [non-phosphorylated protein]). f HepG2 cells were transfected with the oligonucleotide siRNA Shp (si Shp) and control si Scram. After 36 h of transfection, the cells were transiently transfected with the m67-Luc reporter gene and subsequently treated with IL-6 (20 ng/ml) in the presence or absence of metformin for 12 h. Luciferase activity was normalised to β-galactosidase activity to correct for transfection efficiency. All data are representative of at least three independent experiments. Data are expressed as fold activations relative to the controls (±SEM). * p < 0.05 compared with untreated controls, † p < 0.05 compared with IL-6-treated cells, ‡ p < 0.005 compared with IL-6- and Met-treated cells
AMPK inhibits IL-6-mediated Socs3 mRNA expression via the induction of Shp
To determine whether AMPK-mediated expression of Shp is involved in the regulation of Socs3 mRNA expression, we assessed the effects of AMPK and SHP using northern blot analysis. We demonstrated previously that AMPK induces Shp mRNA expression under both in vivo and in vitro conditions but not with the dominant negative form of Ampk (DN-Ampk) [15, 16, 21, 22]. As expected, overexpression of Ampk using an adenoviral constitutively active form of Ampk (Ad-CA-Ampk) significantly increased Shp mRNA expression in primary hepatocytes (Fig. 2a), whereas overexpression using Ad-DN-Ampk did not (Fig. 2c). IL-6-stimulated Socs3 mRNA levels were reduced markedly by Ad-CA-Ampk or Ad-Shp in a dose-dependent manner (Fig. 2a, b). Metformin decreased the induction of Socs3 mRNA levels by IL-6, whereas Ad-DN-Ampk and Ad-si Shp reversed the metformin-mediated inhibition of IL-6-induced Socs3 mRNA levels relative to controls (Fig. 2c, d). Overall, these results demonstrate that IL-6-mediated Socs3 mRNA expression is suppressed by the AMPK–SHP-dependent pathway.

IL-6 mediated induction of Socs3 mRNA expression is repressed by the AMPK–SHP pathway in hepatocytes. a, b Rat primary hepatocytes were infected with Ad-CA-Ampk and Shp for 36 h, then exposed for 1 h to IL-6 (20 ng/ml). The total RNA (30 μg/lane) was isolated from the hepatocytes and used for northern blot analysis. Shp and Socs3 mRNA levels were normalised to glyceraldehyde-3-phosphate dehydrogenase (Gapdh) mRNA expression. c, d Rat primary hepatocytes were infected with Ad-DN-Ampk, Ad-Scram and Ad-si Shp for 36 h, and then treated with either IL-6 (20 ng/ml) for 1 h or with various concentrations of metformin for 12 h. Total RNA (30 μg/lane) was extracted from hepatocytes and used for northern blot analysis. Shp and Socs3 mRNA levels were normalised to an internal control (Gapdh)
Metformin inhibits IL-6-mediated STAT3 occupancy on the Socs3 gene promoter through induction of SHP
To confirm whether metformin, AMPK and SHP regulate the transcriptional activity of Socs3, we performed transient transfection assays using a reporter harbouring the Socs3 gene promoter in HepG2 cells. The increase in Socs3 promoter activity by IL-6 was markedly repressed by metformin, CA-Ampk and Shp in a dose-dependent manner but not by DN-Ampk (Fig. 3a) or Shp knockdown (Fig. 3b). Overall, these results demonstrate that IL-6-induced Socs3 promoter activity is repressed by the AMPK–SHP pathway. Previous reports suggest that the −72 and −64 bp proximal STAT consensus element (TTCCAGGAA) is essential for cytokine- and growth hormone-induced SOCS3 transactivation [23, 24]. As expected, our results demonstrated that IL-6-activated Socs3 gene promoter activity was inhibited by either metformin or SHP in a dose-dependent manner, whereas this activity was completely lost in mutant STAT-binding sites (Fig. 3c). These observations indicate that the STAT-binding site required for the IL-6 response is located within the region between −72 and −64 bp on the Socs3 gene promoter.

Induction of SHP by metformin inhibits IL-6-mediated Socs3 transcriptional activity. a HepG2 cells were transiently cotransfected with the Socs3-Luc reporter gene and CA-Ampk, DN-Ampk or Shp and subsequently treated with IL-6 in the presence of metformin for 12 h under the conditions shown. Luciferase activity was normalised to β-galactosidase activity to correct for variations in transfection efficiency. * p < 0.05, † p < 0.05 and ‡ p < 0.05 compared with untreated control, IL-6-treated cells and IL-6- and metformin (Met)-treated cells, respectively. b HepG2 cells were transfected with si Shp and control si Scram. After 36 h of transfection, the cells were cotransfected with the indicated reporter gene and then treated with IL-6 and metformin. Luciferase activity was normalised to β-galactosidase activity to correct for transfection efficiency. The results shown are representative of at least three independent experiments. All data are indicated as fold activations relative to the control (±SEM). * p < 0.05, † p < 0.005 and ‡ p < 0.05 compared with untreated control, IL-6-treated cells, and IL-6- and Met-treated cells, respectively. c Schematic diagrams of WT and mutant Socs3 promoter constructs from −72 to −64 bp as indicated (upper). HepG2 cells were cotransfected with the WT (black bars) and mutant (mt, white bars) forms of the Socs3 reporter and Shp (lower). After 36 h of transfection, the cells were treated with IL-6 (20 ng/mL) and the indicated doses of metformin. Luciferase activity was normalised to β-galactosidase activity to correct for transfection efficiency. All data represent the means of three independent experiments. All data are shown as fold activations relative to the control (±SEM). * p < 0.05, † p < 0.05 compared with untreated control and IL-6-treated cells, respectively. d ChIP assay: rat primary hepatocytes were infected for 36 h with Ad-si Shp and Ad-Scram, and then treated for 30 min with IL-6 and for 12 h with metformin at the indicated concentrations. Input represents 10% of purified DNA in each sample. Cell extracts were immunoprecipitated with anti-STAT3 (non-phosphorylated STAT3) and anti-SHP antibodies, and purified DNA samples were employed for PCR with primers binding to the specific proximal (left) and non-specific distal (right) regions on the Socs3 gene promoter. All data are representative of at least three independent experiments
In accordance with this experiment, we investigated whether the increase in SHP by AMPK in primary rat hepatocytes repressed the DNA binding activity of STAT3 on the Socs3 gene promoter using chromatin immunoprecipitation (ChIP) assays. As expected, endogenous STAT3 directly bound to the proximal (−215/−33) site following IL-6 treatment, and this activity was completely eliminated by either Ad-SHP or Ad-CA-AMPK. However, Ad-DN-AMPK restored the STAT3 binding activity in the proximal Socs3 gene promoter following IL-6 and metformin treatment, as indicated by the transient transfection assay. Moreover, the non-specific distal region (−1,698/−1,485) of the SocS3 gene promoter was unable to recruit this protein under all conditions (ESM Fig. 1). We performed the ChIP assay with anti-STAT3 antibody in primary hepatocytes to further confirm the inhibition of DNA binding activity of endogenous STAT3 protein on the Socs3 gene promoter by SHP. Endogenous STAT3 was directly bound to the proximal region following IL-6 treatment, and this activity was completely eliminated when the cells were treated with metformin. However, Ad-si Shp treatment returned the protein complex to the proximal promoter following IL-6 and metformin treatment, in comparison with the control Ad-Scram (Fig. 3d, left). Moreover, the non-specific distal region of the Socs3 gene promoter was unable to recruit this protein under all conditions (Fig. 3d, right). Neither endogenous SHP nor control IgG bound to the Socs3 gene promoter under both proximal and distal conditions (Fig. 3d, bottom). Collectively, these results demonstrate that the AMPK-mediated induction of SHP reduces IL-6-stimulated Socs3 transcriptional activity via the repression of DNA binding activity of STAT3 on the Socs3 gene promoter.
SHP physically interacts and colocalises with STAT3
To confirm the endogenous interaction between SHP and STAT3, a co-immunoprecipitation (Co-IP) assay was conducted using AML-12 cells. Endogenous SHP and STAT3 proteins strongly interacted with each other as a result of both IL-6 and metformin treatment compared with controls or IL-6 alone (Fig. 4a). We also assessed the endogenous interaction of these two proteins in mouse liver samples using anti-phospho-STAT3 and STAT3 (non-phospho-STAT3) antibodies under the indicated conditions. As anticipated, these two proteins strongly interacted with each other after combination treatment in comparison with the controls or IL-6 alone (Fig. 4b, c). Moreover, we performed in vivo glutathione S-transferase (GST)-pull down assays by cotransfecting mammalian expression vectors encoding either Gst (also known as Mgst1) alone or Gst-Shp together with Flag-Stat3 into HepG2 cells. GST-SHP strongly interacted with Flag-STAT3 but not with the control GST alone (ESM Fig. 2a), which was consistent with the results of the Co-IP assay. Next, we used domain mapping to define the interaction site. Flag-STAT3 proteins interacted with GST-SHP, whereas this protein interaction was not observed for the STAT3 protein in which the DNA binding domain (DBD) had been deleted (ESM Fig. 2b). Overall, these results indicate that SHP interacts directly with STAT3 under both in vitro and in vivo conditions, and that the DBD of STAT3 is essential for the interaction with SHP.

Direct interaction between SHP and STAT3. a AML-12 cells were treated for 30 min with IL-6 and for 12 h with metformin at the indicated concentrations. Protein extractions from hepatocytes were immunoprecipitated (IP) using phospho-STAT3 antibody then blotted with SHP antibody. Levels of phospho-STAT3, STAT3 and SHP from 10% of lysates were analysed by western blotting (WB) with specific antibodies. b, c WT mice were injected with IL-6 (1 μg/kg body weight) for 1 h and metformin (200 mg/kg body weight) for 6 h at the concentrations shown. Co-IP assays with liver extracts (n = 4) showed a functional association between SHP and STAT3. Tissue extracts were immunoprecipitated with either phospho-STAT3 or STAT3 (non-phosphorylated STAT3) antibody, and blotted with SHP antibody. Levels of phospho-STAT3, STAT3 and SHP from 10% of input were analysed by immunoblotting. d AML-12 cells were treated with IL-6 (20 ng/mL) for 30 min in the presence of metformin for 12 h. Cytosolic and nuclear protein fractions were extracted as described in the Methods section, and the protein fractions were analysed by SDS-PAGE. The protein fractions from hepatocytes were used for western blot analysis using the indicated antibodies and the purities of the protein fractions were verified by the cytosolic marker β-tubulin and the nuclear marker, lamin A/C. All data are representative of at least three independent experiments. e Subcellular localisation of SHP and STAT3. HepG2 cells were transiently transfected with pEGFP-Shp and pcDNA3/Flag-Stat3, then treated for 30 min with IL-6 (20 ng/ml). The merged image depicts the colocalisation of SHP and STAT3. TO-PRO-3 is specific nuclear dye
To elucidate the correlation between STAT3 phosphorylation or nuclear translocation and the increase in SHP stimulated by metformin, we separated the cytosolic and nuclear protein fractions after exposure of hepatocytes to IL-6 and metformin under the indicated conditions [25]. The level of STAT3 phosphorylation or translocation induced by IL-6 increased significantly in the nucleus, but its effects were repressed in the nuclear fraction, owing to the higher induction of SHP by metformin relative to the cytosolic fraction (Fig. 4d). These results suggest that the increase in SHP by metformin is indicative of a potent regulator of IL-6-stimulated STAT3 phosphorylation or translocation. To determine the subcellular localisation of SHP and STAT3, confocal microscopic analysis was performed after transfecting pEGFP-Shp and pcDNA3/Flag-Stat3, with or without IL-6, into hepatocytes. As expected, STAT3 was detected predominantly in the nucleus with IL-6 and was distributed in whole cells without IL-6, whereas the GFP-SHP protein was localised mainly in the nucleus, which is consistent with the results of the fractionation analysis. Furthermore, the merged image shows that these proteins were colocalised within the nucleus upon IL-6 treatment (Fig. 4e). Collectively, these results clearly demonstrate that SHP physically interacts with STAT3 in the nucleus.
Metformin-induced SHP protein production improves hepatic insulin receptor signalling
Several studies have demonstrated that IL-6 and SOCS3 induce hepatic insulin resistance via the inhibition of hepatic insulin receptor signalling [3–6, 26, 27]. To determine the correlation between the IL-6-induced inhibition of hepatic insulin receptor signalling and the increase in SHP following metformin treatment, we assessed the role of SHP in the IL-6-mediated suppression of IRS-1 and AKT activity in primary hepatocytes. Insulin-dependent AKT phosphorylation was markedly inhibited by IL-6-induced STAT3 phosphorylation and SOCS3 protein production, and this activity was reversed by the induction of SHP via metformin or Ad-Shp (Fig. 5a, b). Interestingly, IL-6-mediated SOCS3 protein production significantly repressed insulin-stimulated IRS-1 and AKT phosphorylation, and the activation of insulin-signalling-related genes was restored by the inhibition of SOCS3 via metformin-induced SHP protein production, but not in Shp knockdown (knocked down with Ad-siRNA Shp; Fig. 5c). Overall, these results indicate that the induction of SHP by metformin improves hepatic insulin signalling by inhibiting the IL-6-mediated signalling pathway.

Metformin improves IL-6-mediated dysfunction of hepatic insulin receptor signalling by SHP. a, b Rat primary hepatocytes were treated for 12 h with metformin and infected for 36 h with Ad-Shp, then exposed to insulin for 20 min and IL-6 for 1 h at the indicated concentrations. Whole cell extracts were isolated and analysed using western blot analysis with various antibodies. The protein levels were normalised to an internal control (β-actin and/or non-phosphorylated proteins). c Rat primary hepatocytes were infected with 60 MOI of Ad-si Shp and Ad-Scram for 36 h. After infection, the cells were treated with a combination of insulin, IL-6 and metformin. Whole cell extracts were isolated and analysed by immunoblotting with the indicated antibodies, then normalised to an internal control (β-actin and/or non-phosphorylated forms). All data are representative of at least three independent experiments
Metformin-induced SHP protein production regulates inflammatory genes and insulin receptor signalling by acute IL-6 treatment in the liver
As demonstrated previously, metformin has a significant effect on the downregulation of IL-6-mediated STAT3 transactivation and SOCS3 protein production via the induction of SHP in primary hepatocytes (Figs 1, 2 and 5). To confirm whether IL-6-stimulated inflammatory genes are altered by metformin-induced SHP production, we administered IL-6 and metformin to both WT and Shp null mice. As anticipated, metformin-induced SHP protein production via AMPK activation in WT mice, and the increase in STAT3 phosphorylation and SOCS3 content by IL-6 was markedly repressed by metformin-induced SHP protein production in WT mice (Fig. 6a). However, metformin had no repressive effects on the IL-6-mediated pathway in Shp null mice relative to WT mice (Fig. 6a). Under the same conditions, the induction of SHP by metformin markedly decreased IL-6-induced inflammatory genes (Saa1, Socs3, Il-6 [also known as Il6] and Tnf-α [also known as Tnf]) and fibrotic marker gene (Pai-1) expression in wild-type mice but not in Shp null mice (Fig. 6b–g). Interestingly, Shp null mice displayed elevated basal mRNA levels of all genes involved in inflammation relative to WT mice (Fig. 6b–g). Overall, these results demonstrate that upregulation of inflammatory genes by acute IL-6 exposure was repressed by metformin-induced SHP protein production via AMPK activation in vivo. Moreover, to determine the role of SHP on hepatic insulin receptor signalling in vivo, we injected insulin, IL-6 and metformin into WT and Shp null mice. Insulin-dependent phosphorylation of the insulin receptor, IRS-1 and AKT was repressed by IL-6-induced SOCS3, and metformin-stimulated SHP protein production significantly reversed IL-6-mediated inhibition of hepatic insulin receptor signalling in WT mice but not in Shp null mice (Fig. 6h–m). Overall, these results indicate that the induction of SHP by metformin improves inflammation and hepatic insulin receptor signalling by regulating an acute IL-6-mediated signal pathway under in vivo conditions.

Metformin-induced SHP regulates hepatic insulin action and inflammation genes by acute treatment of IL-6 in the liver. a–g WT (black bars) and Shp null (white bars) mice were injected with IL-6 (1 μg/kg body weight) for 1 h and metformin (200 mg/kg body weight) for 6 h at the concentrations shown. Tissues were extracted from liver tissues in the indicated groups, and then assessed using western blot analysis and RT-PCR with various antibodies and primers, respectively. The protein levels were normalised to total antibodies and/or β-actin levels. Inflammation markers were normalised to an internal control (β-actin). All mice were separated into experimental groups (n = 4 mice per group). * p < 0.05, † p < 0.05 and ‡ p < 0.05 compared with untreated WT mice, IL-6-treated WT mice and IL-6- and Met-treated WT mice, respectively. h–m WT (black bars) and Shp null (white bars) mice were injected with insulin (1 U/kg body weight) for 20 min, IL-6 (1 μg/kg body weight) for 1 h, and metformin (200 mg/kg body weight) for 6 h. Tissues were obtained from liver tissues in the indicated groups, then analysed by western blot analysis with various antibodies. i–m In the blots obtained in (h), protein production was normalised to total antibody and/or β-actin levels. All mice were separated into experimental groups (n = 4 mice per group). * p < 0.05, † p < 0.05, ‡ p < 0.05 and § p < 0.05 compared with untreated WT mice, insulin-injected WT mice, IL-6- and insulin-injected WT mice, and IL-6-, insulin-, and Met-injected WT mice
SHP ameliorates hepatic insulin resistance caused by chronic IL-6 treatment
Previous studies have shown that chronic IL-6 treatment causes hepatic insulin resistance as a result of elevated gluconeogenic genes, hepatic glucose production and impaired insulin sensitivity under both in vivo and in vitro conditions [9–11, 26, 27]. Based on these findings, we hypothesised that chronic IL-6 exposure increases hepatic glucose output and expression of gluconeogenic genes in primary hepatocytes. To test this hypothesis, we first elucidated the critical role of SHP in the IL-6-mediated glucose production and expression of gluconeogenic regulators in primary hepatocytes. The expression of glucose 6-phosphatase (G6pase, also known as G6pc) and phosphoenolpyruvate carboxykinase (Pepck) mRNA increased significantly with chronic IL-6 exposure and decreased in response to metformin in control (Ad-Scram), but not in Shp knockdown (Fig. 7a–c). As expected, the increase in glucose production by IL-6 was markedly reduced by metformin treatment relative to the control (Ad-Scram), but not in Shp knockdown (Fig. 7d). Taken together, these results strongly suggest that the stimulatory effect of chronic IL-6 on hepatic gluconeogenesis is altered by SHP.

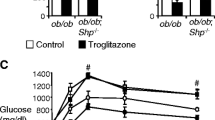
Chronic treatment of IL-6-mediated hepatic insulin resistance is altered by SHP. a–c Rat primary hepatocytes were infected with Ad-Scram (black bars) and Ad-si Shp (white bars) for 36 h and then treated with either IL-6 (20 ng/ml) for 1 h or 24 h in the absence or presence of metformin for 12 h at the concentrations shown. Total RNA was extracted from hepatocytes and used for RT-PCR. Shp, G6pase and Pepck mRNA levels were normalised to an internal control (β-actin). * p < 0.05 and † p < 0.05 compared with untreated control or acute and chronic IL-6 treated cells, respectively. d Under the same conditions, measurement of glucose output was performed using glucose-free media supplemented with the gluconeogenic substrates sodium lactate (20 mmol/l) and sodium pyruvate (1 mmol/l), as described in the ESM Methods. * p < 0.05 and † p < 0.05 compared with untreated control or acute and chronic IL-6 treated cells, respectively. e–i Chronic exposure by intraperitoneally injecting IL-6 into WT and Shp null mice at a daily dose of 2 μg/kg body weight. After 2 weeks of exposure, the mice were killed and liver tissues or blood samples were collected. Tissues were extracted from liver tissues in the indicated groups, and then assessed using RT-PCR (e–i), feeding blood glucose levels (j,k), and insulin tolerance test (l,m). e–i Total RNA was isolated for RT-PCR analysis from WT (black bars) and Shp null (white bars) mice. Socs3, Pepck, G6pase, Pgc-1α and Gk mRNA levels were normalised to an internal control with β-actin. All mice were separated into experimental groups (n = 3–5 mice per group). * p < 0.05 and † p < 0.05 compared with untreated WT mice or IL-6 treated WT mice, respectively. j,k Blood glucose concentrations in WT (j) and Shp null (k) fed mice were measured 2 weeks after administration of saline solution (control, black bars) or IL-6 (white bars) as described above. All mice were separated into experimental groups (n = 3 mice per group). * p < 0.05 compared with Shp null mice. l,m WT (l) and Shp null (m) mice were injected for 10 days with saline solution (control, circles) or IL-6 (squares). Mice were then fasted for 4 h before an insulin tolerance test. Insulin was administered intraperitoneally at a dose of 0.5 U/kg body weight. Blood glucose concentrations were measured from the tail vein at the indicated time periods. All mice were separated into experimental groups (n = 4–5 mice per group). * p < 0.05 compared with untreated Shp null mice
Finally, we evaluated the physiological effect of chronic IL-6 exposure on glucose homeostasis and insulin sensitivity in WT and Shp null mice. The expression of Pepck, G6pase and Socs3 mRNA was significantly elevated by IL-6 treatment in the liver of WT and Shp null mice (Fig. 7e–i). However, chronic IL-6 treatment had no effect on peroxisome proliferator-activated receptor (PPAR)γ coactivator-1α (Pgc-1α, also known as Ppargc1a) mRNA expression in the liver of WT and Shp null mice (Fig. 7e–i). Consistent with a previous report [26], chronic IL-6 exposure significantly decreased glucokinase (Gk) mRNA expression in WT mice, and this phenomenon was not observed in Shp null mice (Fig. 7e–i). Elevated glucose tolerance and impaired insulin tolerance signify insulin sensitivity. Blood glucose levels were significantly elevated by IL-6 treatment in Shp null mice, but not in WT mice (Fig. 7j, k). After chronic IL-6 treatment, Shp null mice had markedly impaired insulin sensitivity relative to controls (Fig. 7l, m), whereas glucose tolerance was not different in WT and Shp null mice (data not shown). Chronic IL-6 treatment also significantly increased blood glucose and insulin levels under fasting and feeding conditions in Shp null mice compared with WT mice (data not shown). Overall, these findings strongly suggest that SHP plays a critical role in the improvement in IL-6-induced insulin resistance.
Discussion
In this study, we have demonstrated that metformin-induced SHP protein production ameliorates hepatic insulin resistance by regulating the STAT3-dependent pathway, gluconeogenesis and insulin sensitivity under both in vitro and in vivo conditions. However, these effects of metformin were blocked by Shp knockdown in primary hepatocytes and in the liver of Shp null mice. Following the results of this study, we propose that the metformin–AMPK–SHP pathway may prevent hepatic metabolic disorders related to insulin resistance by improving insulin sensitivity and glucose homeostasis, and inhibiting the cytokine-mediated signal pathway.
The role of IL-6 remains controversial and involves dual functions in cytokine-associated insulin resistance [3–8]. Recent reports have suggested that IL-6 has a positive effect on insulin signalling in individual tissues [8, 28]. However, most previous reports have demonstrated that cytokine-mediated insulin resistance in the liver ensues from STAT3 activation and the induction of SOCS3, and the subsequent inhibition of insulin signalling [3–6, 26, 27]. Therefore, the correlation between the AMPK–SHP network and the dysregulation of hepatic insulin receptor signalling by proinflammatory cytokines has yet to be fully elucidated. In this study, we observed that the IL-6-induced inhibition of hepatic insulin receptor signalling is mediated by an AMPK–SHP-dependent pathway under both in vitro and in vivo conditions. Indeed, our results demonstrate that the improvement of hepatic insulin receptor signalling was mediated by the AMPK–SHP pathway by suppressing cytokine-mediated genes in primary hepatocytes and IL-6 and/or insulin stimulation experiments (Figs 5 and 6). These results lead us to speculate that the AMPK activators that upregulate SHP protein production may provide beneficial effects on hepatic insulin resistance by regulating the insulin signalling pathway via the IL-6–STAT3–SOCS3 pathway.
Indeed, several previous studies have demonstrated that chronic IL-6 exposure elevates hepatic glucose production by inducing gluconeogenic genes in primary hepatocytes as well as increasing blood glucose levels in rodents and humans [9–11, 29, 30]. The physiological function of IL-6 in metabolic disorders is contradictory and incompletely resolved. These findings have caused us to evaluate the physiological effect of chronic IL-6 exposure on hepatic insulin resistance in WT and Shp null mice to identify the role of SHP in IL-6-mediated failure of glucose homeostasis. Our results strongly suggest that chronic IL-6 treatment causes insulin resistance, leading to elevated expression of gluconeogenic regulators, glucose production and blood glucose level, and impaired insulin tolerance, consistent with previous results [9–11, 29, 30]. Moreover, chronic IL-6 treatment markedly impaired insulin sensitivity in Shp null mice (Fig. 7). Therefore, SHP plays a critical role in improving IL-6-induced insulin resistance. However, we cannot exclude the possibility that IL-6 may depend on other regulatory processes such as the actions of insulin and glucagon, glycogenolysis, and the involvement of other regulatory hormones and insulin-response tissues.
Two signalling pathways are activated during IL-6 signalling and action: first, the JAK2/STAT3 pathway and second, the Src homology 2-containing tyrosine phosphatase (SHP-2)/ ERK/MAPK pathway. The IL-6-induced activation of the JAK/STAT pathway in both immune and non-immune-associated cell types leads to elevated SOCS3 production [3, 31]. IL-6 rapidly increases AMPK activity in skeletal muscle and adipose tissue, under both in vivo and in vitro conditions [32, 33]. However, our study demonstrated that IL-6 treatment did not result in any significant increase in AMPK phosphorylation in vitro or in vivo (Figs 1 and 6). This finding suggests that IL-6 and AMPK activation are not positively correlated in the liver, which is consistent with the findings of a previous report [34].
Previous studies from our group have shown that metformin, HGF and NaArs repress the expression of hepatic gluconeogenic regulators via the AMPK–SHP pathway [15, 21, 22]. In the present study, we demonstrated that AMPK-mediated induction of SHP improves insulin receptor signalling by repressing IL-6-mediated STAT3 transactivation and SOCS3 levels and attenuating the cytokine-mediated signalling pathway in both a rodent model and in primary hepatocytes (Figs 1, 2, 5 and 6). Accordingly, activators of AMPK, including HGF, NaArs and several natural products, may prove quite advantageous, because of the induction of SHP through the AMPK-dependent pathway and improving hepatic insulin receptor signalling by suppressing the cytokine-mediated pathway.
Recent reports have demonstrated that the AMPK activators, 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), metformin and adiponectin, repress the IL-6-mediated inflammatory response by downregulating STAT3 phosphorylation in HepG2 cells, although the molecular mechanisms underlying this phenomenon have yet to be elucidated in detail [34, 35]. The findings of our study strongly suggest a novel mechanism between the induction of SHP production by AMPK activators and IL-6-induced STAT3 transactivation by regulating a direct interaction and blocking DNA binding. However, we cannot clarify metformin-mediated inhibition of STAT3 phosphorylation because SHP could interact with both phospho-STAT3 and STAT3 (non-phospho-STAT3) (Fig. 4a–c). Therefore, we suggest that the decrease in phospho-STAT3 levels may be due to increased interaction of SHP and non-phospho-STAT3. On the other hand, we are currently unable to dismiss the possibility that another molecular pathway exists between the IL-6-induced STAT3–SOCS3 pathway and AMPK-mediated SHP protein production in the liver; it will be a matter for future studies to elucidate the underlying mechanisms of these phenomena in detail. Finally, our findings indicate that IL-6 is a major stimulus leading to insulin resistance, whereas the increase of SHP by metformin improves hepatic insulin resistance by regulating the IL-6–STAT3–SOCS3 network (Fig. 8).

IL-6 is a major stimulus leading to insulin resistance. Metformin, a known activator of SHP via the AMPK pathway, directly represses the IL-6-induced STAT3–SOCS3 pathway and improves hepatic insulin resistance via an IL-6-dependent pathway
In conclusion, our results suggest that upregulation of SHP by metformin represents a novel pathway in relation to hepatic metabolic disorders and may be an important mediator of improved cytokine-induced insulin resistance in the liver. Moreover, we speculate that the metformin–AMPK–SHP pathway may ameliorate the pathogenesis of cytokine-mediated metabolic dysfunction. Therefore, as we described in the novel schematic model shown in Fig. 8, inhibition of STAT3 by SHP may provide new insights into the beneficial effects of cytokine-induced insulin resistance and may also help in the development of novel therapeutic agents for treating insulin resistance in the future.
Abbreviations
- Ad-CA-Ampk :
-
Adenoviral constitutively active Ampk
- Ad-DN-Ampk :
-
Adenoviral dominant negative Ampk
- Ad-si Shp :
-
Adenoviral siRNA Shp
- AMPK:
-
AMP-activated protein kinase
- ChIP:
-
Chromatin immunoprecipitation
- Co-IP:
-
Co-immunoprecipitation
- DBD:
-
DNA binding domain
- GST:
-
Glutathione S-transferase
- HGF:
-
Hepatocyte growth factor
- JAK2:
-
Janus kinase 2
- KRIBB:
-
Korea Research Institute of Bioscience and Biotechnology
- MOI:
-
Multiplicity of infection
- SHP:
-
Small heterodimer partner
- siRNA:
-
Small interfering RNA
- SOCS3:
-
Suppressor of cytokine signalling 3
- STAT3:
-
Signal transducer and activator of transcription 3
- WT:
-
Wild-type
References
Tilg H, Moschen AR (2008) Insulin resistance, inflammation, and non-alcoholic fatty liver disease. Trends Endocrinol Metab 19:371–379
Carter-Kent C, Zein NN, Feldstein AE (2008) Cytokines in the pathogenesis of fatty liver and disease progression to steatohepatitis: implications for treatment. Am J Gastroenterol 103:1036–1042
Ueki K, Kondo T, Tseng YH, Kahn CR (2004) Central role of suppressors of cytokine signaling proteins in hepatic steatosis, insulin resistance, and the metabolic syndrome in the mouse. Proc Natl Acad Sci USA 101:10422–10427
Senn JJ, Klover PJ, Nowak IA, Mooney RA (2002) Interleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes 51:3391–3399
Torisu T, Sato N, Yoshiga D et al (2007) The dual function of hepatic SOCS3 in insulin resistance in vivo. Genes Cells 12:143–154
Ueki K, Kondo T, Kahn CR (2004) Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol Cell Biol 24:5434–5446
Holmes AG, Mesa JL, Neill BA et al (2008) Prolonged interleukin-6 administration enhances glucose tolerance and increases skeletal muscle PPARα and UCP2 expression in rats. J Endocrinol 198:367–374
Sadagurski M, Norquay L, Farhang J, D’Aquino A, Copps K, White A (2010) Human IL-6 enhances leptin action in mice. Diabetologia 53:525–535
Blumberg D, Hochwald S, Brennan MF, Burt M (1995) Interleukin-6 stimulates gluconeogenesis in primary cultures of rat hepatocytes. Metabolism 44:145–146
Ritchie DG (1990) Interleukin-6 stimulates hepatic glucose release from prelabeled glycogen pools. Am J Physiol 258:E57–E64
Tsigos C, Papanicolaou DA, Kyrou I, Defensor R, Mitsiadis CS, Chrousos GP (1997) Dose-dependent effects of recombinant human interleukin-6 on glucose regulation. J Clin Endocrinol Metab 82:4167–4170
Lin HZ, Yang SQ, Chuckaree C, Kuhajda F, Ronnet G, Diehl AM (2000) Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nat Med 6:998–1003
Zhou G, Myers R, Li Y et al (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108:1167–1174
Yuan L, Ziegler R, Hamann A (2003) Metformin modulates insulin post-receptor signaling transduction in chronically insulin-treated HepG2 cells. Acta Pharmacol Sin 24:55–60
Kim YD, Park KG, Lee YS et al (2008) Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase-dependent regulation of the orphan nuclear receptor SHP. Diabetes 57:306–314
Chanda D, Lee CH, Kim YH et al (2009) Fenofibrate differentially regulates plasminogen activator inhibitor-1 gene expression via adenosine monophosphate-activated protein kinase-dependent induction of orphan nuclear receptor small heterodimer partner. Hepatology 50:880–892
Zhang BB, Zhou G, Li C (2009) AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab 9:407–416
Lee YS, Chanda D, Sim J, Park YY, Choi HS (2007) Structure and function of the atypical orphan nuclear receptor small heterodimer partner. Int Rev Cytol 261:117–158
Chanda D, Park JH, Choi HS (2008) Molecular basis of endocrine regulation by orphan nuclear receptor small heterodimer partner. Endocr J 55:253–268
Um JH, Park SJ, Kang H et al (2010) AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes 59:554–563
Chanda D, Li T, Song KH et al (2009) Hepatocyte growth factor family negatively regulates hepatic gluconeogenesis via induction of orphan nuclear receptor small heterodimer partner in primary hepatocytes. J Biol Chem 284:28510–28521
Chanda D, Kim SJ, Lee IK, Shong M, Choi HS (2008) Sodium arsenite induces orphan nuclear receptor SHP gene expression via AMP-activated protein kinase to inhibit gluconeogenic enzyme gene expression. Am J Physiol Endocrinol Metab 295:368–379
Barclay JL, Anderson ST, Waters MJ, Curlewis JD (2007) Regulation of suppressor of cytokine signaling 3 (SOC3) by growth hormone in pro-B cells. Mol Endocrinol 21:2503–2515
Gatto L, Berlato C, Poli V et al (2004) Analysis of SOCS-3 promoter responses to interferon gamma. J Biol Chem 279:13746–13754
Shah M, Patel K, Fried VA, Sehgal PB (2002) Interactions of STAT3 with caveolin-1 and heat shock protein 90 in plasma membrane raft and cytosolic complexes. Preservation of cytokine signaling during fever. J Biol Chem 277:45662–45669
Klover PJ, Zimmers TA, Koniaris LG, Mooney RA (2003) Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes 52:2784–2789
Senn JJ, Klover PJ, Nowak IA et al (2003) Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J Biol Chem 278:13740–13746
Wunderlich FT, Ströhle P, Könner AC et al (2010) Interleukin-6 signaling in liver-parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab 12:237–249
Banzet S, Koulmann N, Simler N et al (2009) Control of gluconeogenic genes during intense/prolonged exercise: hormone-independent effect of muscle-derived IL-6 on hepatic tissue and PEPCK mRNA. J Appl Physiol 107:1830–1839
Stith RD, Luo J (1994) Endocrine and carbohydrate responses to interleukin-6 in vivo. Circ Shock 44:210–215
Kristiansen OP, Mandrup-Poulsen T (2005) Interleukin-6 and diabetes: the good, the bad, or the indifferent? Diabetes 54:S114–S124
Kelly M, Gauthier MS, Saha AK, Ruderman NB (2009) Activation of AMP-activated protein kinase by interleukin-6 in rat skeletal muscle: association with changes in cAMP, energy state, and endogenous fuel mobilization. Diabetes 58:1953–1960
Carey AL, Steinberg GR, Macaulay SL et al (2006) Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes 55:2688–2697
Nerstedt A, Johansson A, Andersson CX, Cansby E, Smith U, Mahlapuu M (2010) AMP-activated protein kinase inhibits IL-6-stimulated inflammatory response in human liver cells by suppressing phosphorylation of signal transducer and activator of transcription 3 (STAT3). Diabetologia 53:2406–2416
Sun H, Zhang Y, Gao P et al (2011) Adiponectin reduces C-reactive protein expression and downregulates STAT3 phosphorylation induced by IL-6 in HepG2 cells. Mol Cell Biochem 347:183–189
Acknowledgements
We would like to thank L. Hennighausen (National Institutes of Health, Bethesda, MD, USA) and S.-Y. Choi (Chonnam National University Medical School, Gwangju, Republic of Korea) for their critical suggestions and helpful discussions.
Funding
This work was supported by the National Creative Research Initiatives grant from the Korean Ministry of Education, Science and Technology (20110018305), a grant from the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare & Family Affairs, Republic of Korea (A100588), and the Future-based Technology Development Program (BIO Fields) through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (20100019512) (to H.S. Choi), and by the KRIBB Research Initiative Program of Korea, Republic of Korea (to C.H. Lee).
Contribution statement
YDK contributed to the conception, design and performance of experiments, analysis and interpretation of data and drafting or writing the manuscript; YHK contributed to the design and performance of animal experiments, analysis and interpretation of experimental results and critical review of the manuscript; YMC contributed to the design and performance of experiments, analysis and interpretation of results, and critical review of the manuscript; DKK, SWA, JML, DC and MS contributed to the analysis and interpretation of data, and revision of the manuscript for important intellectual content; CHL and HSC contributed to the conception and design of the experiments and the article, and critical review and revision of the manuscript. All authors have approved the final version of the manuscript to be published.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript
Author information
Authors and Affiliations
Corresponding authors
Additional information
Y. D. Kim and Y. H. Kim contributed equally to this study.
Electronic Supplementary Materials
Below is the link to the electronic supplementary material.
ESM Fig. 1
PDF 73 kb
ESM Fig. 2
PDF 88 kb
ESM Methods
PDF 141 kb
Rights and permissions
About this article
Cite this article
Kim, Y.D., Kim, Y.H., Cho, Y.M. et al. Metformin ameliorates IL-6-induced hepatic insulin resistance via induction of orphan nuclear receptor small heterodimer partner (SHP) in mouse models. Diabetologia 55, 1482–1494 (2012). https://doi.org/10.1007/s00125-012-2494-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2494-4