
Abstract
Aims/hypothesis
Carbohydrate-free diet prevents hyperglycaemia and beta cell destruction in the New Zealand Obese (NZO) mouse model. Here we have used a sequential dietary regimen to dissociate the effects of obesity and hyperglycaemia on beta cell function and integrity, and to study glucose-induced alterations of key transcription factors over 16 days.
Methods
Mice were rendered obese by feeding a carbohydrate-free diet for 18 weeks. Thereafter, a carbohydrate-containing diet was given. Plasma glucose, plasma insulin and total pancreatic insulin were determined, and forkhead box O1 protein (FOXO1) phosphorylation and the transcription factors pancreatic and duodenal homeobox 1 (PDX1), NK6 homeobox 1 protein (NKX6.1) and v-maf musculoaponeurotic fibrosarcoma oncogene family, protein A (avian) (MAFA) were monitored by immunohistochemistry for 16 days.
Results
Dietary carbohydrates produced a rapid and continuous increase in plasma glucose in NZO mice between day 2 and 16 after the dietary challenge. Hyperglycaemia caused a dramatic dephosphorylation of FOXO1 at day 2, followed by a progressive depletion of insulin stores. The loss of beta cells was triggered by apoptosis (detectable at day 8), associated with reduction of crucial transcription factors (PDX1, NKX6.1 and MAFA). Incubation of isolated islets from carbohydrate-restricted NZO mice or MIN6 cells with palmitate and glucose for 48 h resulted in a dephosphorylation of FOXO1 and thymoma viral proto-oncogene 1 (AKT) without changing the protein levels of both proteins.
Conclusions/interpretation
The dietary regimen dissociates the effects of obesity (lipotoxicity) from those of hyperglycaemia (glucotoxicity) in NZO mice. Obese NZO mice are unable to compensate for the carbohydrate challenge by increasing insulin secretion or synthesising adequate amounts of insulin. In response to the hyperglycaemia, FOXO1 is dephosphorylated, leading to reduced levels of beta cell-specific transcription factors and to apoptosis of the cells.
Similar content being viewed by others
Introduction
Obese rodents with susceptibility for hyperglycaemia such as the C57BKS/J-db/db (diabetic) and the New Zealand Obese (NZO) mouse have been studied as models for the human obesity-associated type 2 diabetes [1–3]. These strains present a progressive failure of insulin-secreting beta cells, and a severely decompensated glucose homeostasis with glucosuria and blood glucose levels >20 mmol/l. By conventional cross-breeding experiments, genomic segments (diabetogenic quantitative trait loci) were identified that are responsible for the decompensation of glucose homeostasis [4–8]. As predicted earlier [2, 9], these diabetogenic alleles were contributed from the obese as well as from the lean (background) strains. More recently, three candidate genes for beta cell failure in obese mice, Sorcs1, Lisch-like (Ildr2) and Zfp69 [10–12], and one candidate suppressor of diabetes, Tbc1d1 [13], have been identified by positional cloning.
It is generally accepted that beta cell malfunction is caused by the following scenario. Obesity induces ectopic fat accumulation in the pancreas, thereby causing apoptosis of beta cells (‘lipotoxicity’) [14–16]. Data from NZO mice showing that dietary fat, in combination with the presence of diabetogenic alleles, markedly increases the prevalence of diabetes are consistent with such a scenario [6]. In addition, the prevalence of diabetes in NZO is reduced when fat oxidation in muscle is stimulated by disruption of Tbc1d1 [13]. However, lipotoxicity does not appear to be sufficient for the destruction of the beta cell. Carbohydrate-restricted diets fully prevented beta cell destruction in both NZO and db/db mice [17, 18] despite an extreme insulin resistance and a marked inflammatory state of adipose tissue [19]. This finding is consistent with a previously suggested scenario in which postprandial hyperglycaemia (‘glucotoxicity’) plays an essential role in the pathogenesis of islet cell failure [20, 21].
Hyperglycaemia produces glucotoxicity for the beta cell through oxidative stress caused by formation of reactive oxygen species [22]. This mechanism has been investigated in cultured beta cell lines, and is assumed to initially involve specific transcription factors controlling the insulin gene, such as pancreatic and duodenal homeobox 1 (PDX1) and v-maf musculoaponeurotic fibrosarcoma oncogene family, protein A (avian) (MAFA), and subsequently the proliferation of the cells [23, 24]. The two scenarios, lipotoxicity and glucotoxicity, are not mutually exclusive but may function in combination [25, 26].
In the present study, we employed the NZO mouse model to dissociate the effects of dietary lipids and dietary carbohydrates in vivo, and to study the time course of beta cell failure. NZO mice were exposed to a carbohydrate-free diet until the age of 18 weeks in order to produce obesity and insulin resistance without hyperglycaemia or diabetes. Thereafter, carbohydrates were added to the diet, and variables of beta cell function were monitored for 16 days. Our data indicate that the dietary carbohydrates rapidly produced hyperglycaemia, paralleled by dephosphorylation of forkhead box O1 protein (FOXO1), and followed by internalisation of glucose transporter type 2 (GLUT2) and progressive decrease of the pancreatic insulin content. In a second phase of the decompensation, plasma insulin levels decreased sharply, further aggravating hyperglycaemia, and nuclear staining of the transcription factors PDX1, NK6 homeobox 1 protein (NKX6.1) and MAFA decreased markedly. These results indicate that the dietary regimen of initial carbohydrate restriction and subsequent refeeding dissociates lipotoxicity and glucotoxicity. In addition, they suggest a scenario in which a hyperglycaemia-induced loss of FOXO1 plays a crucial role in a progressive malfunction of the transcriptional machinery of the beta cell.
Methods
Experimental animals
Male NZO/HIBomDife mice (R. Kluge, German Institute of Human Nutrition, Nuthetal, Germany) from our own colony were housed in groups of five per cage (type II macrolon) at a temperature of 21 ± 1°C with a 12 h light–dark cycle (lights on at 06:00 hours). Animals had free access to food and water, and were kept in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals. All experiments were approved by the Ethics Committee of the State Ministry of Agriculture, Nutrition and Forestry (State of Brandenburg, Germany).
Diets and study design
From weaning at the age of 3 weeks onwards, mice received a carbohydrate-free pelleted diet purchased from Altromin (custom made by the manufacturer, Lage, Germany), containing (wt/wt) 30.2% fat and 26.4% protein with a total metabolisable energy of 15.3 kJ/g. Fat content normalised per total energy was 72.2% and protein content was 27.8%. Because of the soft texture of the diet, mice had access to wooden gnawing sticks in order to avoid excessive teeth growth.
At the age of 18 weeks, mice were randomised to either a carbohydrate-containing diet or continued on a carbohydrate-free diet for up to an additional 16 days (Fig. 1). The ingredients of the diets are given in the Electronic supplementary material (ESM) Table 1 and percentages of macronutrients plus energy density are given in ESM Table 2. Blood samples were collected at the indicated days from the tail vein of fed mice between 09:00 hours and 11:00 hours. Blood glucose was determined with an Ascensia ELITE XL glucose meter (Bayer Health Care, Leverkusen, Germany). Plasma insulin was assayed with the Mouse Insulin Ultrasensitive ELISA kit (DRG Instruments, Marburg, Germany). Body composition (fat and lean mass) was measured before the start of the diet change by nuclear magnetic resonance with an EchoMRI Whole Body Composition Analyzer (Echo Medical Systems, Houston, TX, USA) as previously described [19, 27, 28].

Carbohydrate refeeding causes rapid onset of hyperglycaemia and delayed decompensation of insulin secretion in NZO mice. a Study design for the application of carbohydrate-restricted (−CH) and carbohydrate-containing diets (+CH). Until week 18, obesity and insulin resistance was produced by application of a carbohydrate-free diet. Thereafter, a high-carbohydrate (white squares) or a carbohydrate-free diet (black squares) was given for 16 days. b, c Time course of (b) blood glucose and (c) plasma insulin after the diet change. Samples were collected at the indicated time points, and data represent mean±SEM of at least ten mice
Pancreatic insulin content
For detection of total pancreas insulin, whole pancreases were homogenised in ice-cold acid ethanol (0.1 mol/l HCl in 70% ethanol) and incubated 24 h at 4°C. After centrifugation (16,000 g, 10 min) insulin was detected in the supernatant fraction with the Mouse Insulin Ultrasensitive ELISA kit (DRG Instruments).
Immunohistochemistry of pancreatic islets
Pancreatic tissue excised immediately after exsanguination was fixed in 4% formaldehyde for 24 h, embedded in paraffin according to standard procedures, and longitudinal serial sections (2 μm, at three sampling intervals of 100 μm) were prepared.
For immunostaining of insulin, sheep anti-insulin antiserum (1:6,000; Vector Laboratories, Burlingame, CA, USA) was used, followed by biotinylated donkey anti-sheep IgG antiserum (1:800, Jackson ImmunoResearch Labs, Newmarket, UK). The secondary antibody was detected with streptavidin–biotin–alkaline peroxidase complex (StreptAB-complex/HRP; Dako, Hamburg, Germany) and diaminobenzidine (DAB) as substrate according to the manufacturer’s specifications.
Proinsulin was immunostained with mouse anti-proinsulin antibody (1:50; GS-9A8 Developmental Studies Hybridoma Bank, Iowa City, IA, USA), followed by biotinylated donkey anti-mouse IgG antiserum (1:800, Jackson ImmunoResearch Labs), and detection with DAB as substrate.
For immunostaining of glucagon, phosphorylated FOXO1 (pFOXO1), PDX1, NKX6.1, caspase-3 and MAFA, rabbit anti-glucagon antiserum (1:2,000; DAKO Diagnostika, Hamburg, Germany), rabbit anti-pFOXO1 (Thr24)/FoxO3a (Thr32) antibody (1:50; Cell Signaling), rabbit anti-PDX1 (1:1,000, Upstate Cell Signaling Solutions, Charlottesville, VA, USA), mouse anti-NKX6.1 (1:2,000, F55A10, Developmental Studies Hybridoma Bank, Iowa City, IA, USA), rabbit anti-active caspase 3 (1:100; Promega, Mannheim, Germany) or rabbit anti-MAFA antiserum (1:1,000, Bethyl Labs, Montgomery, AL, USA) was used, respectively. The primary antibodies were detected with Histofine Anti-Rabbit Immuno-POD Polymer for mouse tissues (Nichirei Biosciences, Tokyo, Japan) and DAB as peroxidase (POD) substrate after Histofine incubation.
GLUT2 was detected with rabbit antiserum against the C-terminus of rat GLUT2 (1:1,000) [29] and Alexa Fluor 488 labelled goat anti-rabbit-IgG antiserum (1:200, Invitrogen, Karlsruhe, Germany) as secondary antibody which was visualised by stimulation of fluorescence at 488 nm.
Microscopic investigation and photo documentation were performed with the combined light and fluorescence microscope ECLIPSE E-100 (Nikon, Düsseldorf, Germany) in combination with the video camera CCD-1300CB (Vosskühler, Osnabrück, Germany) and the Analysis System LUCIA (Nikon).
Quantitative analysis of islet morphology
Longitudinal sections of pancreas co-stained with anti-insulin and anti-glucagon antiserum were analysed with the Lucia G image analysis software (Laboratory Imaging, Prague, Czech Republic) via an unbiased stereological approach. In three to six sections from each mouse, a total of 35 islets were analysed with regard to number of immunoreactive cells (insulin and glucagons) and number of PDX-, NKX6.1- and MAFA-positive nuclei. Mean values derived from each individual mouse were used to calculate means ± SEM of three to six mice in each group.
In vitro experiments with isolated islets from NZO mice
Islets of 18 week old NZO mice were isolated by ductal collagenase distension/digestion of the pancreas using a modified method of Gotoh et al. [30]. Briefly, 3 ml of Collagenase-P (Roche, Mannheim, Germany), 1 mg/ml in Hanks’ buffered salt solution (HBSS) containing 25 mmol/l HEPES and 0.5% (wt/vol.) BSA, were injected in the pancreas via the common bile duct. The perfused pancreas was digested in 2 ml of collagenase solution for 11 min at 37°C. Islets were collected by hand-picking and pre-incubated for 24 h in RPMI 1640 medium containing 11 mmol/l glucose, 10% FCS, 100 U/ml penicillin, 100 U/ml streptomycin in humidified 5% CO2, 95% air at 37°C. Islets were treated with different glucose concentrations (11, 27.6, 33.2 and 38.7 mmol/l) in the presence or absence of 0.3 mmol/l palmitic acid for 48 h in humidified 5% CO2, 95% air at 37°C (50 islets/group).
Western blot experiments
Protein was extracted with a cell lysis buffer (20 mmol/l Tris, 150 mmol/l NaCl, 1 mmol/l EDTA, 1 mmol/l EGTA, 1% triton-X-100, 2.5 mmol/l Na4P2O7, 1 mmol/l β-glycerolphosphate, 1 mmol/l Na3VO4, 1 mmol/l NaF) and an additional proteinase inhibitor cocktail tablet (per 50 ml, Roche). Protein samples (15 μg) were separated by electrophoresis through 10% polyacrylamide, 0.1% SDS gels, and transferred to polyvinylidine fluoride membranes, followed by immunoblotting. Immunodetection was developed with ECL Advance (Amersham Biosciences, Buckinghamshire, UK) and imaged with a C09 116 CCD camera (PEQLAB, Erlangen, Germany). Antibodies used were: polyclonal anti-FOXO1 (C29H4), anti-pFOXO1 (Thr24)/FoxO3a (Thr32), anti-phosphorylated-thymoma viral proto-oncogene 1 (AKT) (Ser473) and AKT antibody (Cell Signaling, Boston, MA, USA). At least three blots were performed for each experiment with each blot reprobed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (anti-GAPDH, Applied Biosystems, Foster City, CA, USA) as a loading control.
Cell culture of mouse insulinoma cells
MIN6 cells were maintained in Dulbecco’s modified Eagle’s medium containing 5 mmol/l glucose, with 10% FCS, 100 units/ml penicillin, 100 μg/ml streptomycin and 100 μg/ml l-glutamine in humidified 5% CO2, 95% air at 37°C [31]. For the experiment MIN6 cells were treated with different glucose concentrations (5, 10, 25 and 50 mmol/l) in the presence or absence of 0.3 mmol/l palmitate for 48 h.
Statistical analysis
Differences in body weight and blood glucose under the different dietary regimens were tested for statistical significance with a one-way analysis of variance (one-way ANOVA) after verification of normal distribution, followed by a post hoc test (homogeneous variances by Bonferroni type and inhomogeneous variances by Games–Howell type). All other data sets were tested with the non-parametric Mann–Whitney U test. The calculations were performed with the software package SPSS 14.0 (SPSS, Chicago, IL, USA), and differences were considered significant at p < 0.05.
Results
After weaning at 3 weeks of age, all mice were fed a carbohydrate-free pelleted diet containing (wt/wt) 30.2% fat and 26.4% protein until the age of 18 weeks (Fig. 1a). This dietary regimen caused massive obesity (body weight 79.8 ± 1.2 g; body fat 31.7 ± 0.7 g) and hyperinsulinaemia (plasma insulin 1,229 ± 100 pmol/l). Thereafter, mice were randomised into two groups receiving either the high-fat carbohydrate-free diet [19] or a diet containing (wt/wt) 40% carbohydrate. The composition of the diets is given in ESM Table 1. After the diet change (day 0) serum variables of glucose metabolism were monitored over a period of 16 days. As is illustrated in Fig. 1b,c, NZO mice on the carbohydrate-containing diet became hyperglycaemic (>15 mmol/l) as early as 2 days after the diet change, and hyperglycaemia continued to increase throughout the observation period (>23 mmol/l at day 16). Plasma insulin levels increased until day 8, but decreased dramatically thereafter. In contrast, non-fasted blood glucose levels in control NZO mice on the carbohydrate-free diet remained below 11 mmol/l.
In parallel experimental series, pancreases of mice were isolated at the indicated time points in order to monitor insulin, proinsulin and glucagon content as well as the number of insulin-containing cells. Figure 2 shows staining of insulin (upper panel), and glucagon (middle panel), and proinsulin (bottom) immunoreactivity in islets from mice on the different diets (representative samples). Starting at day 4, a marked reduction in the insulin content of islets was observed. The morphometric analysis (Fig. 3a) indicated a significant reduction of the number of insulin-positive cells per islet (day 8–16). At day 32, few insulin-positive cells were detectable in islets of carbohydrate-challenged mice (ESM Fig. 1). Thus, the immunohistochemical analysis was consistent with the conclusion that beta cells are depleted of insulin between days 4 and 16, and that this depletion accounts for the marked rise in blood glucose levels during this time span. By staining of pancreatic sections with an anti-caspase-3 antibody we detected a significant increase in caspase-3-positive islet cells beginning at day 8 after carbohydrate exposure (Fig. 3b,c), indicating that beta cells undergo apoptosis when obese mice are exposed to carbohydrates.

Immunohistochemistry of insulin, glucagon and proinsulin in pancreatic sections of NZO mice after a diet change from a carbohydrate-free (−CH) to a carbohydrate-containing diet (+CH). Sections from pancreas embedded in paraffin were haematoxylin-stained and immunostained with the respective antisera and developed with DAB as described in the Methods. Scale bars, 50 μm

Reduction of the number of insulin-positive cells. a Morphometric determination of the relative number of cells per μm2 islet area after the diet change from a carbohydrate-free (white bars) to a carbohydrate-containing (black bars) diet. From each mouse, 35 islets in one to six pancreatic sections were evaluated in a blinded procedure. b Immunostaining of activated caspase-3 in islets of NZO mice treated with (+CH) or without (−CH) carbohydrates for 8 and 16 days. c Quantification of caspase-3-positive cells from NZO mice fed the indicated diets. Data represent mean±SEM of three to six mice, calculated from the mean values of the individual mice. *p < 0.05 for –CH vs +CH at the indicated time point
As one of the limiting steps in the synthesis of insulin is the conversion of proinsulin, we visualised proinsulin in beta cells with the aid of an antibody against a peptide corresponding with the proteolytic cleavage site; this antibody detects proinsulin in the presence of insulin. As is illustrated in Fig. 2 (lower panel), immunoreactive proinsulin appeared unaltered from day 0 until day 8 after the diet change. After day 8, immunoreactive proinsulin decreased to almost undetectable levels, in parallel with the decrease in plasma insulin. This finding is consistent with the conclusion that the synthesis of proinsulin, but not its processing, was reduced in mice after refeeding of carbohydrates.
In order to validate that data obtained by immunohistochemistry, we determined the insulin content of total pancreas by immunoassay of acidic ethanol extracts (Fig. 4). A significant decrease of the insulin content in response to the carbohydrate-containing diet was detected as early as 4 days after the diet change. Thus, a gradual decrease in total pancreatic insulin appeared to precede the fall in plasma insulin levels.

Carbohydrate-containing diet causes decrease of pancreatic insulin in NZO mice. A carbohydrate-free diet was given until week 18 in order to produce obesity and insulin resistance. Thereafter, a carbohydrate-containing (black bars) or a carbohydrate-free diet (white bars) was given for 16 days. Whole pancreas was dissected at the indicated time points, and insulin was extracted and determined as described in the Methods. Data represent mean±SEM of six mice; *p < 0.05 for carbohydrate-containing vs carbohydrate-free diet
We and others have previously reported that GLUT2 abundance is reduced and its subcellular distribution is altered in NZO mice [19, 32]. Thus, we studied the time course of these alterations after feeding the carbohydrate-containing diet. As is illustrated in ESM Fig. 2, GLUT2 was predominantly localised in plasma membranes of beta cells at day 0, and at all subsequent time points, when mice received the carbohydrate-free diet. In contrast, GLUT2 was redistributed to intracellular compartments in islets from carbohydrate-fed mice. This re-distribution started at day 4 after the diet change, clearly after hyperglycaemia had established (day 2).
As FOXO1 is an important effector of insulin action in pancreatic beta cells and links insulin signalling to the regulation of beta cell mass [33], we investigated levels of pFOXO1 during the carbohydrate intervention. Strikingly, immunoreactivity of pFOXO1 was dramatically decreased as early as 2 days after changing the diet from carbohydrate-free to a carbohydrate containing diet (Fig. 5a).

Reduced levels of pFOXO1 after carbohydrate intervention. a Immunohistochemistry of pFOXO1 in pancreatic sections of NZO mice after a diet change from a carbohydrate-free (−CH) to a carbohydrate-containing diet (+CH). Sections embedded in paraffin at the indicated day were stained with haematoxylin and anti-pFOXO1 and subsequently developed with DAB as described in Methods. Bars: 50 μm. b Western blot analysis of lysates of isolated islets from carbohydrate-free fed NZO mice after in vitro incubation with the indicated glucose concentrations in the absence or presence of 0.3 mmol/l palmitate. c, d Quantification of western blot products of lysates from three independent experiments as shown in b. c Ratio of pFOXO1/total FOXO1 and d ratio of pAKT/total AKT. Values represent mean±SEM; *p ≤ 0.05, † p = 0.06 for absence (white bars) vs presence (black bars) of 0.3 mmol/l palmitate at the indicated glucose concentration
In order to provide direct evidence for the causal role of hyperglycaemia (glucotoxicity), we incubated isolated islets from carbohydrate-restricted NZO mice with different glucose concentrations (11.1, 27.6, 33.2 and 38.7 mmol/l) in the absence or presence of 0.3 mmol/l palmitate for 48 h. Lysates of the islets were analysed by western blotting with anti-FOXO1 and anti-pFOXO1 antibodies (Fig. 5b, c). Treatment of islets with the combination of glucose (11.1 to 38.7 mmol/l) and palmitate resulted in reduced levels of pFOXO1, whereas the total amount of FOXO1 protein was not altered. A similar effect was obtained for AKT; its phosphorylation was markedly reduced after incubation of islets with glucose concentrations of ≥27.6 mmol/l when 0.3 mmol/l palmitate was present (Fig. 5b, d). These data suggest that pFOXO1 and pAKT were reduced by exposure of islets to high glucose concentration, and that this effect was dependent on the presence of palmitate.
In order to verify this finding in a second system, we used mouse insulinoma cells (MIN6) and incubated them with increasing concentrations of glucose (5, 10, 25 and 50 mmol/l) in the presence and absence of palmitate (0.3 mmol/l) for 48 h. As shown in Fig. 6, levels of pFOXO1 decreased slightly when the cells were incubated with 10 mmol/l glucose in the presence of 0.3 mmol/l palmitate for 48 h. A further increase in the glucose concentration up to 50 mmol/l totally abolished pFOXO1. Under these conditions, the levels of pAKT were also significantly reduced. In contrast, exposure of MIN6 cells with increasing concentrations of glucose in the absence of palmitate did not influence the phosphorylation status of FOXO1 or AKT. In order to test whether palmitate alone affects phosphorylation of FOXO1 and AKT, we incubated MIN6 cells with increasing concentrations of palmitate in the absence or presence of 50 mmol/l glucose. Only the exposure of MIN6 cells with glucose plus palmitate resulted in a dose-dependent decrease of the amount of pFOXO1 and pAKT, whereas incubation with palmitate alone did not show differences (Fig. 6d–f).

Reduced levels of pFOXO1 and pAKT in MIN6 cells in response to glucose and palmitate exposure. a Western blot analyses of lysates from MIN6 cells incubated with the indicated glucose concentrations in the absence or presence of 0.3 mmol/l palmitate for 48 h were performed with the indicated antibodies as described in Methods. b, c Quantification of western blot products of lysates from three independent experiments as shown in a: pFOXO1/total FOXO1 (b) and ratio of pAKT/total AKT (c). *p ≤ 0.05 for absence (white bars) vs presence (black bars) of palmitate at the indicated glucose concentration. d Western blot analysis of lysates from MIN6 cells incubated with the indicated palmitate concentrations in the absence or presence of 50 mmol/l glucose for 48 h. e, f Quantification of western blot products of lysates from three independent experiments as shown in d: Ratio of pFOXO1/total FOXO1 (e) and ratio of pAKT/total AKT (f). Values represent mean±SEM; *p ≤ 0.05 for low glucose (white bars) vs high glucose (black bars) at the indicated palmitate concentration
According to the early dephosphorylation of FOXO1, the nuclear staining of PDX1 (Fig. 7a, b), NKX6.1 (Fig. 7c,d) and MAFA (ESM Fig. 3) was decreased later (day 8–16), in parallel with the decrease in plasma insulin levels.

Immunohistochemistry and quantification of PDX1 (a, b) and NKX6.1 (c, d) in pancreatic sections of NZO mice after a diet change from carbohydrate-free (−CH) (white bars) to carbohydrate-containing diet (+CH) (black bars). (a, c) Sections embedded in paraffin at the indicated day were haematoxylin-stained and immunostained with the respective antiserum and developed with DAB as described in Methods. Scale bars, 50 μm. (b, d) Quantifications of nuclei positive for PDX1 (b) and NKX6.1 (d) were performed for three to six mice per group; *p ≤ 0.05 for –CH vs +CH at the indicated time points
Discussion
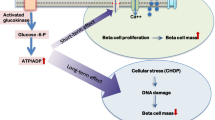
In this study, we present an experimental setup that allows monitoring of beta cell failure initiated by dietary carbohydrates for 2–16 days. By an initial carbohydrate restriction and subsequent refeeding, the consequences of lipotoxicity and glucotoxicity were dissociated. During carbohydrate restriction, NZO mice developed obesity and severe insulin resistance, but no hyperglycaemia. Thereafter, exposure to dietary carbohydrates rapidly produced a biphasic pattern of beta cell failure (Fig. 8). First, an initial hyperglycaemia, revealing an insufficiency of the pancreas to secrete adequate amounts of insulin, was paralleled by a dramatic dephosphorylation of FOXO1. This hyperglycaemia produced internalisation of GLUT2 and initiated a progressive depletion of insulin stores. In a second phase (beginning at day 8), plasma insulin levels fell dramatically, leading to complete decompensation of glucose homeostasis at day 16, induction of apoptosis, and thereby to the loss of beta cells (day 32). In parallel, crucial transcription factors (PDX1, NKX6.1, MAFA) were reduced. This sequence of events is consistent with a scenario in which insulin synthesis is severely affected, and beta cell destruction is ultimately caused by the progressive hyperglycaemia (glucotoxicity).

Schematic presentation of the time course of events leading to beta cell loss in NZO mice after carbohydrate refeeding
Among the markers of beta cell function we monitored, pFOXO1 was the first to respond to the dietary change in that its immunoreactivity was markedly reduced at the earliest time point studied (day 2). This alteration was detected by immunohistochemistry, which is a qualitative method not allowing quantification. Furthermore, the in vivo experiments suggest, but do not prove, a causal role of the hyperglycaemia. Thus, we obtained direct evidence for the effect of hyperglycaemia on FOXO1 by isolation of islets and incubation with different glucose concentrations in the absence and presence of palmitate. With this approach, we provided direct evidence for role of hyperglycaemia (glucotoxicity) in the AKT–FOXO1 pathway, because we detected reduced levels of pFOXO1 and pAKT in islets treated with high glucose concentrations in the presences of palmitate (Fig. 5b). This finding was confirmed in a second system, MIN6 cells, which exhibited a reduction in pFOXO1 and pAKT when treated with glucose plus palmitate but not with palmitate alone (Fig. 6), clearly demonstrating that not fatty acids alone but glucose, in combination with fatty acids, produces inactivation of the AKT–FOXO1 pathway.
FOXO1 is a transcription factor that is excluded from nuclei upon phosphorylation by AKT/PKB via the insulin/insulin-like growth factor-I pathway [34–36]; a consequence of this exclusion is the rapid induction of the insulin gene, and a stimulation of beta cell proliferation. Disruption of AKT–FOXO1 signalling might be responsible for the apoptosis of beta cells (Fig. 3b,c). Our data do not show which apoptotic pathway is induced, and do not provide direct evidence for a causal role of FOXO1 in beta cell death. However, one can speculate that a dephosphorylation of AKT and FOXO1 downregulates survival pathways and/or induces transcription of apoptotic proteins such as BCL2-associated X protein (BAX) [37]. In addition, as it was suggested that dephosphorylated FOXO1 inhibits the transcription of Pdx1 through competing the binding sites with FOXA2 [33, 38] one could speculate that FOXO1 binds to the Pdx1 promoter and inhibits PDX1 and finally insulin production in response to hyperglycaemia. However, we also do not provide direct evidence for this kind of regulation. Accordingly, we observed a reduced content of PDX1 and other downstream transcription factors (NKX6.1 and MAFA) at later time points of carbohydrate intervention. Chronic hyperglycaemia was previously shown to induce a progressive loss of beta cell phenotype and of insulin secretory function with decreased production of beta cell-specific transcripts such as insulin, NKX6.1, hepatic nuclear factor-α (HNFα) and GLUT2 [39]. Our finding might indicate that an early modification induced by exposure of beta cells to elevated concentrations of glucose is the alteration of the phosphorylation status of FOXO1 protein which finally results in a ‘de-differentiation’ of the beta cells.
It should be noted that, in addition to hyperglycaemia, other confounding factors such as insulin resistance have contributed to the beta cell failure in NZO mice. NZO mice receiving the carbohydrate-restricted high-fat diet were severely insulin resistant, as shown by euglycaemic–hyperinsulinaemic clamp experiments [19]. This severe insulin resistance, together with the dietary carbohydrates, produced a metabolic challenge that could not be compensated by the beta cell. Furthermore, the glycaemic control decompensated rapidly after refeeding carbohydrates. This finding indicates an inability of the beta cell to secrete adequate amounts of insulin which has developed in the absence of dietary carbohydrates. Consequently, lipid accumulation in beta cells could be responsible for their initial malfunction. However, these considerations do not alter the conclusion that hyperglycaemia was required for apoptosis and loss of beta cells, although it may not have been sufficient. The model we present here is suitable for the identification of the lipid-induced abnormalities in beta cells, and for their dissociation from the ‘toxic’ effect of elevated blood glucose.
The protective effect of the carbohydrate-free diet has been observed in two mouse strains, NZO and db/db [17–19]. In both strains, beta cell failure was associated with a marked decrease in the immunoreactivity of the GLUT2 [18, 19]. Internalisation and reduced abundance of GLUT2 in beta cells of NZO mice and in other models of diabetes (Chinese hamster and Psammomys obesus) has been reported earlier to precede the loss of insulin immunoreactivity [32, 40, 41]. Here, a reduction of GLUT2 immunoreactivity in plasma membranes was detected at day 4, apparently after the onset of hyperglycaemia. Thus, internalisation of GLUT2 is a consequence of hyperglycaemia rather than its cause.
Elevated proinsulin levels in serum and beta cells have been reported for another rodent model of type 2 diabetes, Psammomys obesus [42]. Thus, it was suggested that a block in the conversion of the pro-hormone to insulin is the cellular mechanism of inadequate insulin secretion in this model. In our model, beta cells were depleted of proinsulin and insulin in parallel, suggesting that the synthesis of proinsulin, rather than its processing, is inadequate.
In summary, our data present a novel experimental model for the in vivo monitoring of alterations leading to beta cell failure in obese mice. The dietary regimen of carbohydrate restriction and refeeding dissociates the effects of obesity (lipotoxicity) from those of hyperglycaemia (glucotoxicity) in NZO mice. With this model, we show that a dietary carbohydrate challenge causes an early and dramatic decrease in pFOXO1. Obese NZO mice are unable to compensate the challenge by increased insulin secretion, and to synthesise adequate amounts of insulin. In a second phase, the progressive hyperglycaemia depletes beta cells of insulin and causes loss of transcription factors that are critical for their function and survival.
Abbreviations
- AKT:
-
Thymoma viral proto-oncogene 1
- DAB:
-
Diaminobenzidine
- FOXO1:
-
Forkhead box O1 protein
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- GLUT2:
-
Glucose transporter type 2
- MAFA:
-
v-Maf musculoaponeurotic fibrosarcoma oncogene family, protein A (avian)
- NKX6.1:
-
NK6 homeobox 1 protein
- NZO:
-
New Zealand Obese
- p:
-
Phosphorylated
- PDX1:
-
Pancreatic and duodenal homeobox 1
References
Herberg L, Coleman DL (1977) Laboratory animals exhibiting obesity and diabetes syndromes. Metabolism 26:59–98
Herberg L, Leiter EH (2001) Obesity/diabetes in mice with mutations in the leptin or leptin receptor genes. In: Sima AAF, Shafrir E (eds) Frontiers in animal diabetes research, vol. 2: animal models of diabetes. Harwood Academic Publishers, Amsterdam, pp 63–107
Ortlepp JR, Kluge R, Giesen K et al (2000) A metabolic syndrome of hypertension, hyperinsulinemia, and hypercholesterolemia in the New Zealand obese (NZO) mouse. Eur J Clin Investig 30:195–202
Leiter EH, Reifsnyder PC, Flurkey K, Partke HJ, Junger E, Herberg L (1998) NIDDM genes in mice. Deleterious synergism by both parental genomes contributes to diabetic thresholds. Diabetes 47:1287–1295
Plum L, Kluge R, Giesen K, Altmüller J, Ortlepp JR, Joost HG (2000) Type-2-diabetes-like hyperglycaemia in a backcross model of New Zealand obese (NZO) and SJL mice: characterization of a susceptibility locus on chromosome 4 and its relation with obesity. Diabetes 49:1590–1596
Plum L, Giesen K, Kluge R et al (2002) Characterization of the diabetes susceptibility locus Nidd/SJL in the New Zealand obese (NZO) mouse: islet cell destruction, interaction with the obesity QTL Nob1, and effect of dietary fat. Diabetologia 45:823–830
Reifsnyder PC, Leiter EH (2002) Deconstructing and reconstructing obesity-induced diabetes (diabesity) in mice. Diabetes 51:825–832
Schmidt C, Gonzaludo NP, Strunk S et al (2008) A meta-analysis of QTL for diabetes related traits in rodents. Physiol Genomics 34:42–53
Coleman DL (1978) Obese and diabetes: two mutant genes causing diabetes–obesity syndromes in mice. Diabetologia 14:141–148
Clee SM, Yandell BS, Schueler KM et al (2006) Positional cloning of Sorcs1, a type 2 diabetes quantitative trait locus. Nat Genet 38:688–693
Dokmanovic-Chouinard M, Chung WK, Chevre JC et al (2008) Positional cloning of "Lisch-Like", a candidate modifier of susceptibility to type 2 diabetes in mice. PLoS Genet 4:e1000137
Scherneck S, Nestler M, Vogel H et al (2009) Positional cloning of zinc finger domain transcription factor Zfp69, a candidate gene for obesity-associated diabetes contributed by mouse locus Nidd/SJL. PLoS Genet 5:e1000541
Chadt A, Leicht K, Deshmukh A et al (2008) Tbc1d1 mutation in lean mouse strain confers leanness and protects from diet-induced obesity. Nat Genet 40:1354–1359
Unger RH (2003) Mini review: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology 144:5159–5165
Li N, Frigerio F, Maechler P (2008) The sensitivity of pancreatic beta-cells to mitochondrial injuries triggered by lipotoxicity and oxidative stress. Biochem Soc Trans 36:930–934
Chang-Chen KJ, Mullur R, Bernal-Mizrachi E (2008) Beta-cell failure as a complication of diabetes. Rev Endocr Metab Disord 9:329–943
Leiter EH, Coleman DL, Ingram DK, Reynolds MA (1983) Influence of dietary carbohydrate on the induction of diabetes in C57BL/KsJ-db/db diabetes mice. J Nutr 113:184–195
Mirhashemi F, Kluth O, Scherneck S et al (2008) High-fat, carbohydrate-free diet markedly aggravates obesity but prevents beta cell loss and diabetes in the obese, diabetes-susceptible db/db strain. Obesity Facts 1:292–297
Jürgens H, Neschen S, Ortmann S et al (2007) Development of diabetes in obese, insulin-resistant mice: essential role of dietary carbohydrate in ß-cell destruction. Diabetologia 50:1481–1489
Leahy JL, Bonner-Weir S, Weir G (1992) Beta-cell dysfunction induced by chronic hyperglycaemia. Current ideas on mechanism of impaired glucose-induced insulin secretion. Diabetes Care 15:442–455
Yki-Jarvinen H (1992) Glucose toxicity. Endocr Rev 13:415–431
Weir GC, Laybutt DR, Kaneto H, Bonner-Weir S, Sharma A (2001) Beta-cell adaptation and decompensation during the progression of diabetes. Diabetes 50(Suppl 1):S154–S159
Poitout V, Hagman D, Stein R, Artner I, Robertson RP, Harmon JS (2006) Regulation of the insulin gene by glucose and fatty acids. J Nutr 136:873–876
Harmon JS, Stein R, Robertson RP (2005) Oxidative stress-mediated, post-translational loss of MAFA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. J Biol Chem 280:11107–11113
Poitout V, Robertson RP (2008) Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr Rev 29:351–366
Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen S, Eizirik DL (2005) Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 54(Suppl 2):S97–S107
Tinsley FC, Taicher GZ, Heiman ML (2004) Evaluation of a quantitative magnetic resonance method for mouse whole body composition analysis. Obes Res 12:150–160
Jürgens HS, Schürmann A, Kluge R et al (2006) Hyperphagia, lower body temperature, and reduced running wheel activity precede development of morbid obesity in New Zealand obese mice. Physiol Genomics 13:234–241
Hellwig B, Brown FM, Schürmann A, Shanahan MF, Joost HG (1992) Localization of the binding domain of the inhibitory ligand forskolin in the glucose transporter GLUT4 by photolabeling, proteolytic cleavage and a site-specific antiserum. Biochim Biophys Acta 1111:178–184
Gotoh M, Maki T, Kiyoizumi T, Satomi S, Monaco AP (1985) An improved method for isolation of mouse pancreatic islets. Transplantation 40:437–438
Lacy PE, Kostianovsky M (1967) Method for the isolation of intact islets of Langerhans from the rat pancreas. Diabetes 16:35–39
Chankiewitz E, Peschke D, Herberg L et al (2006) Did the gradual loss of GLUT2 cause a shift to diabetic disorders in the New Zealand obese mouse (NZO/Hl)? Exp Clin Endocrinol Diabetes 114:262–269
Kitamura T, Nakae J, Kitamura Y et al (2002) The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest 110:1839–1847
Kitamura T, Ido Kitamura Y (2007) Role of FoxO proteins in pancreatic beta cells. Endocr J 54:507–515
Kops GJ, de Ruiter ND, De Vries-Smits AM, Powell DR, Bos JL, Burgering BM (1999) Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature 398:630–634
Nakae J, Park BC, Accili D (1999) Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive pathway. J Biol Chem 274:15982–15985
Matheny RW Jr, Adamo ML (2009) Current perspectives on Akt Akt-ivation and Akt-ions. Exp Biol Med 234:1264–1270
Shao S, Fang Z, Yu X, Zhang M (2009) Transcription factors involved in glucose-stimulated insulin secretion of pancreatic beta cells. Biochem Biophys Res Commun 384:401–404
Laybutt DR, Glandt M, Xu G et al (2003) Critical reduction in beta-cell mass results in two distinct outcomes over time. Adaptation with impaired glucose tolerance or decompensated diabetes. J Biol Chem 278:2997–3005
Jörns A, Tiedge M, Sickel E, Lenzen S (1996) Loss of GLUT2 glucose transporter expression in pancreatic beta cells from diabetic Chinese hamsters. Virchows Arch 428:177–185
Jörns A, Tiedge M, Ziv E, Shafrir E, Lenzen S (2002) Gradual loss of pancreatic beta-cell insulin, glucokinase and GLUT2 glucose transporter immunoreactivities during the time course of nutritionally induced type-2 diabetes in Psammomys obesus (sand rat). Virchows Arch 440:63–69
Leibowitz G, Yuli M, Donath MY et al (2001) Beta-cell glucotoxicity in the Psammomys obesus model of type 2 diabetes. Diabetes 50(Suppl 1):S113–S117
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (GK1208) and the German Ministry of Education and Research (DZD:01GI0922). The authors are indebted to A. Teichmann for expert technical assistance.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
O. Kluth and F. Mirhashemi contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
Immunohistochemistry of insulin in pancreatic sections of NZO mice 32 days after a diet change from carbohydrate-free (−CH) to carbohydrate-containing diet (+CH). Sections from pancreas embedded in paraffin were haematoxylin-stained and immunostained with the anti-insulin antiserum and developed with DAB as described in Methods. Adjacent sections were stained with haematoxylin/eosin (HE) (PDF 217 kb)
ESM Fig. 2
Immunohistochemistry of GLUT2 in pancreatic sections of NZO mice after a change from carbohydrate-free (−CH) to carbohydrate-containing diets (+CH). Sections embedded in paraffin at the indicated day were stained with anti-GLUT2 antiserum and subsequently with a fluorescence-labelled Alexa Fluor secondary antibody, respectively, as described in Methods. Scale bars, 50 μm (PDF 210 kb)
ESM Fig. 3
Immunohistochemistry of MAFA in pancreatic sections of NZO mice after a dietary change from carbohydrate-free (−CH) to carbohydrate-containing (+CH) diet. Sections embedded in paraffin at the indicated day were haematoxylin-stained and immunostained with the anti-MAFA antiserum and developed with DAB as described in Methods. Scale bars, 50 μm. Quantification of MAFA-positive nuclei was performed for six mice per group (lower panel) (PDF 264 kb)
ESM Table 1
Composition of the carbohydrate-containing (+CH) and carbohydrate-free (−CH) experimental diets given to 18-week-old NZO mice for 16 days (PDF 78 kb)
ESM Table 2
Macronutrient content and energy density of the carbohydrate-containing (+CH) and carbohydrate-free (−CH) experimental diets given to 18-week-old NZO mice for 16 days (PDF 74 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kluth, O., Mirhashemi, F., Scherneck, S. et al. Dissociation of lipotoxicity and glucotoxicity in a mouse model of obesity associated diabetes: role of forkhead box O1 (FOXO1) in glucose-induced beta cell failure. Diabetologia 54, 605–616 (2011). https://doi.org/10.1007/s00125-010-1973-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-010-1973-8