
Abstract
Aims/hypothesis
Experimental studies have suggested that apoptosis is involved in diabetic embryopathy through oxidative stress. However, the precise mechanism of diabetic embryopathy is not yet clear. Thioredoxin (TRX) is a small, ubiquitous, multifunctional protein, which has recently been shown to protect cells from oxidative stress and apoptosis. Using transgenic mice that overproduce human TRX-1 (TRX-Tg mice), we examined whether oxidative stress is involved in fetal dysmorphogenesis in diabetic pregnancies.
Methods
Non-diabetic and streptozotocin-induced diabetic (DM) female mice were mated with male TRX-Tg mice. Pregnant mice were killed either at day 10 or day 17 of gestation, and viable fetuses and their placentas were recovered, weighed and assessed for gross and histological morphology, biochemical markers and gene expression.
Results
In both wild-type (WT) and transgenic (Tg) groups, fetal and placental weights in the diabetic group were significantly decreased compared with the non-diabetic group. The incidence of malformation was higher in the diabetic group, and was significantly decreased in the TRX-Tg group (DM-WT vs DM-Tg; 28.6% vs 10.4%). Oxidative stress markers such as thiobarbituric acid reactive substances and 8-hydroxy-2′-deoxyguanosine were increased in DM-WT group fetuses but were decreased in fetuses from the DM-Tg group. Furthermore, immunohistochemically assayed apoptosis and cleaved caspase-3 production in embryonic neuroepithelial cells was significantly increased in the DM-WT group, and was significantly decreased in the DM-Tg group.
Conclusions/interpretation
These results indicate that oxidative stress is involved in diabetic embryopathy, and that the antioxidative protein TRX at least partially prevents diabetic embryopathy via suppression of apoptosis.
Similar content being viewed by others


Introduction
Maternal hyperglycaemia during early pregnancy is associated with a considerable risk of malformations in the offspring. At present, almost 80 years after the introduction of insulin therapy, although the rates of most complications have been reduced, the incidence of congenital malformation continues to increase [1]. The malformation rate in the offspring of diabetic mothers is twofold to fivefold higher than that for the normal population, and congenital malformations are the major cause of mortality and morbidity in the offspring of diabetic mothers [1–4]. Therefore, extensive studies have been performed to determine the mechanisms of diabetes-induced embryonic malformations. However, as yet, the underlying mechanisms have not been fully elucidated, especially in in-vivo systems.
In patients with diabetes mellitus, oxygen free radicals are produced in excess, which reportedly plays a role in several diabetic complications, such as nephropathy, neuropathy and retinopathy [5]. It was suggested that maternal oxygen free radicals might be involved in the induction of fetal congenital malformations [6]. Metabolic changes such as hyperglycaemia in the embryo or fetus also cause increased production of reactive oxygen species (ROS) in the embryo, causing the damage [7, 8]. ROS, including oxygen, superoxide and hydrogen peroxide (H2O2), can react with, and damage, various intracellular molecules. It has been suggested that embryonic dysmorphogenesis in a diabetic or hyperglycaemic environment might be induced by the generation of ROS [9] and that ROS-scavenging enzymes can protect diabetic embryos from glucose-induced malformations [10]. Trocino et al. showed that anomalies induced in rat embryos grown under hyperglycaemic conditions are associated with glutathione (GSH) depletion and that GSH esters can attenuate hyperglycaemia-induced abnormalities [11]. The teratogenic effect of diabetic serum was prevented by superoxide dismutase (SOD) and N-acetylcysteine in rat whole embryo culture [12]. These results suggest that redox imbalance can be induced by embryonic hyperglycaemic conditions and that regulation of the redox status is essential for normal embryonic development.
Thioredoxin (TRX) is a small (12 kDa), ubiquitous protein that is known to function as an antioxidant protein, catalysing thiol-disulphide oxidoreduction [13]. The consensus sequence of its active site, Cys-Gly-Pro-Cys, is conserved across species, and the two-cysteine residues can be reversibly oxidised to participate in reversible oxidation–reduction reactions [14]. TRX has been shown to protect cells from oxidative stress and apoptosis [15–17]. Several studies have shown that TRX is induced by various types of stress, such as viral infection, ischaemic insult, ultraviolet light, X-ray irradiation and H2O2 [18]. TRX acts as a scavenger of ROS and can repair proteins oxidised by ROS [13, 18]. In-vitro studies have demonstrated that TRX has a protective effect against the cytotoxicity of ROS [18]. TRX also regulates various intracellular molecules via thiol redox control involving transcription factors such as NF-κB, activator protein 1 (AP-1), redox factor 1 and mitogen-activated kinase [15, 19, 20]. These data suggest that TRX plays a number of important biological roles in both intra- and extracellular compartments.
Apoptosis commonly occurs during a variety of developmental processes in mammals. Recently, it was reported that apoptosis occurs in various tissues of early post-implantation embryos undergoing organogenesis [21, 22]. Furthermore, enhanced production of ROS is reportedly an important stimulus in the induction of mitochondrial transition, caspase activation and apoptosis [23, 24]. Moreover, hyperglycaemia induces apoptosis in early organogenesis in pre- and post-implantation rodent embryos through cell-death effector pathways [25–29]. Also, neural tube defects (NTDs) are associated with apoptosis in the offspring of diabetic mice [30, 31]. Taken together, these findings support the assumption that diabetes-induced inappropriate apoptosis in embryos might lead to NTDs. Therefore, the purpose of the present study was to examine whether oxidative stress is involved in diabetic embryopathy and whether overproduction of the antioxidative human TRX-1 protein by the use of transgenic mice (TRX-Tg mice) can prevent diabetic embryopathy.
Methods
Animals
C57BL/6NCrj (wild-type [WT]) mice were purchased from Charles River Japan (Osaka, Japan) and reared in our laboratory. Transgenic mice overexpressing the human TRX-1 (also known as TXN) gene on a C57BL/6 background were purchased from the Oriental BioService (Kyoto, Japan). The transgenic mice were originally reported by Takagi et al. [20]. In brief, to create TRX-Tg mice, TRX-1 cDNA was inserted between the beta-actin promoter and the beta-actin terminator, and the transgene was microinjected into the pronucleus of fertilised eggs from C57BL/6 mice [20, 32]. The presence of the TRX-1 transgene was confirmed by PCR analysis using mouse genomic DNA as a template and the following oligonucleotide primers: forward primer, 5′-CCGCTCGTCAGACTCCAGC-3′; and reverse primer, 5′-CTGGTTATATTTTCAGAAAACATG-3′ (Sigma-Genosys; Sigma-Aldrich, St Louis, MO, USA). The mice were subjected to a 12 h light/12 h dark schedule and had free access to tap water and a standard laboratory diet (CLEA Rodent Diet CE-7, CLEA JAPAN, Osaka).
Approximately 4 weeks before pregnancy, diabetes was induced in 6-week-old female WT mice by a single intraperitoneal injection of 200 mg/kg body weight streptozotocin (STZ) (Nacalai Tesque, Kyoto, Japan), as previously reported [33] (Fig. 1). STZ was dissolved in 0.2 ml of 0.01 mol/l citrate buffer immediately before use. The mice became hyperglycaemic within 1 week of STZ administration and diabetes mellitus was confirmed when the blood glucose level was more than 16.65 mmol/l (300 mg/dl). Diabetes was treated with subcutaneously implanted insulin pellets (Linshin, Scarborough, ON, Canada) that continuously release insulin. The insulin pellets controlled the diabetes to the extent that the mice gained weight and ovulated normally [30, 31]. Approximately 2–3 weeks after implantation of the insulin pellets, mice became hyperglycaemic, at which time they were mated with heterozygous TRX-Tg males. The blood glucose level at the time of mating was more than 11.1 mmol/l (200 mg/dl), and the blood glucose level at day 5 of gestation was more than 16.65 mmol/l (300 mg/dl; Table 1). Control animals (without diabetes mellitus) that were injected with 0.2 ml of 0.01 mol/l citrate buffer and had been treated with non-insulin pellets were also mated with male TRX-Tg mice. On the morning after mating, a screen for copulation plugs was performed, and if present, this day was considered as day 0 of gestation.

Experimental model of mouse diabetic pregnancy showing a description of induction and treatment of diabetes, and embryo assays. Details of procedures are provided in the Methods
Pregnant mice were anaesthetised with pentobarbital sodium before operation. The uterine horns were cut open and carefully inspected to reveal all implantations, both viable and non-viable, regardless of size. Viable fetuses and their placentas were recovered and weighed, either on day 10 to assay histology, or on day 17 to assay biochemical markers, gene expression and gross fetal malformations. Litter size was from six to eight when maternal weight gain, fetal weight and placental weight were compared. For histological and immunohistochemical studies, embryos were fixed in 4% paraformaldehyde (Nacalai Tesque) in PBS at 4°C overnight. Embryos were then dehydrated in graded concentrations of ethanol, embedded in paraffin and sectioned at a thickness of 5 μm. For examination of fetal malformations at day 17 of gestation, viable fetuses were denoted as malformed when they showed NTDs (including anencephaly, exencephaly, microencephaly and spina bifida), omphalocele, anophthalmia or other gross malformations. The resorbed sites were inspected and the resorptions were divided into two groups: one with no identifiable fetal tissues (essentially only a lump of non-maternal tissue that was denoted as early resorption) and the other with one or more identifiable tissues (placenta and tissue that was often identifiable as embryo–fetus, denoted as late resorption). For RT-PCR and biochemical marker analyses, fetuses and fetal livers were dissected and immediately frozen in liquid nitrogen and stored at −80°C until use. All experimental procedures were approved by the Ethics Committee for Animal Research of the Mie University Graduate School of Medicine.
Immunohistochemical staining
The levels of human and mouse TRX-1 (mTRX-1) proteins in the neuroepithelial cells of embryos at day 10 of gestation were examined immunohistochemically. Briefly, following deparaffinisation and rehydration, the sections were then immunoreacted with mouse anti-hTRX-1 monoclonal antibody or rabbit anti-mTRX-1 polyclonal antibody (Redox Bio Science, Kyoto, Japan) as primary antibody. The absence of endogenous mouse IgG was confirmed in these embryonic tissues before the mouse anti-hTRX-1 monoclonal antibody was used. The sections were incubated with horseradish peroxidase (HRP)-labelled goat anti-mouse IgG or HRP-labelled goat anti-rabbit IgG (MBL, Nagoya, Japan) and HRP was visualised by diaminobenzidine and H2O2.
To evaluate oxidative stress in the neuroepithelial cells of embryos at day 10 of gestation, a marker of oxidative stress, 8-hydroxy-2′-deoxyguanosine (8-OHdG), which is a DNA base-modified product that is induced by oxidative stress, was assayed. This marker was detected with the primary mouse anti-8-OHdG monoclonal antibody (Japan Institute for the Control of Aging, Fukuroi, Japan). The rest of the staining procedure was the same as that used for detection of TRX except that incubation with H2O2 was omitted. The slides were viewed at a magnification of ×40 by a person unaware of the treatment to which each embryo had been exposed. The staining of each embryo was assigned a score between 1 and 5 (1, minimal staining; 5, maximal staining).
We evaluated apoptosis in the neuroepithelial cells of embryos at day 10 of gestation, using an ApopTag Plus Peroxidase In Situ Apoptosis Detection kit according to the manufacturer’s instructions (Millipore Corporation, Billerica, MA, USA). Furthermore, to investigate the factors downstream of the apoptosis pathway, we analysed the expression of a major apoptosis marker, cleaved caspase-3, in the neuroepithelial cells of embryos at day 10 of gestation. We used a rabbit anti-cleaved caspase-3 polyclonal antibody (Cell Signaling Technology, Beverly, MA, USA) and biotinylated goat anti-rabbit IgG (Lab Vision, Fremont, CA, USA) followed by reaction with avidin-biotin complex reagent (VECTASTAIN Elite ABC, Vector Laboratories, Burlingame, CA, USA). Antibody binding sites were visualised with diaminobenzidine.
Quantitative real-time PCR
Total RNA was isolated from the fetuses at day 17 of gestation using Sepasol-RNA1 Super according to the manufacturer’s instructions (Nacalai Tesque). RT of total RNA was conducted according to the manufacturer’s instructions with TaqMan Reverse Transcription Reagents and a TaqMan Gold RT-PCR kit (Applied Biosystems, Foster City, CA, USA). RT-PCR products were analysed by quantitative real-time PCR using TaqMan Gene Expression Assays (Applied Biosystems). Primers and probes for detection of human TRX-1 and mouse Trx-1 were designed using Primer Express 1.0 (PerkinElmer Applied Biosystems, Norwalk, CT, USA) from available sequences (TRX-1 [NM_003329], Trx-1 [NM_01160]), and are shown in Electronic supplementary material (ESM) Table 1. The level of gene expression was normalised to that of acidic ribosomal phosphoprotein P0 (36B4) RNA (Mm01974474_gH). All PCRs were performed using a real-time PCR 7300 system. Gene expression was quantified as the second step of a two-step RT-PCR. Assays were done in a 50 μl singleplex reaction mixture containing TaqMan Universal PCR Master Mix, 20× TaqMan Gene Expression Assay Mix, and cDNA according the manufacturer’s instructions (Applied Biosystems). Reaction conditions consisted of preincubation at 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min.
Measurement of TBARS
Thiobarbituric acid reactive substances (TBARS) are lipid peroxides, and are a major indicator of oxidative stress [34]. TBARS were determined fluorometrically after heating samples with thiobarbituric acid (TBA) [34–36]. Briefly, 100 μl of a tissue homogenate of fetal livers at day 17 of gestation was heated to 95°C for 60 min together with 100 μl of SDS solution and 2.5 ml of TBA/buffer reagent (TBARS Assay Kit, ZeptoMetrix, Buffalo, NY, USA). Standard samples were prepared from malondialdehyde-bis-(dimethyl acetal) (TBARS Assay Kit, ZeptoMetrix). The samples were then centrifuged at 3,000 rpm for 15 min. Fluorescence of the supernatant fraction was measured at 532 nm in a spectrophotometer.
Statistical analyses
The results are expressed as the mean ± SD. Results were analysed using Student’s t test and ANOVA followed by a post hoc test, or a χ 2 test, as appropriate. A p value < 0.05 was considered to be significant.
Results
Maternal status
Details of the variables measured to assay maternal and fetal status in pregnancies of non-diabetic and diabetic mice are shown in Table 1. All the dams used in this study were WT. The pregnancy rate of mice in the streptozotocin-induced diabetic (DM) group was lower than that of mice in the non-DM group. The number of pregnant mice in the non-DM group was 20, and that in the DM group was 28. The litter size in mice in the DM group was significantly smaller than that in mice in the non-DM group. The rate of resorption in the DM group was higher than that in the non-DM group; however, this difference was not statistically significant. The rate of early resorption was similar between the non-DM and DM groups, whereas the rate of late resorption was increased in the DM group. Fewer late resorptions of Tg fetuses than WT fetuses were observed in the DM group. The serum glucose concentration of mice in the DM group was elevated approximately twofold to fivefold compared with that of mice in the non-DM group. The litter size was six to eight when maternal weight gains were compared, because the litter size of mice in the DM group was smaller than that of mice in the non-DM group. The body weight of mice in the DM group was similar to that of mice in the non-DM group on the day of conception. However, the DM group mice gained less weight than the non-DM group mice during pregnancy.
Fetal and placental weight
The weight of fetuses and placentas at day 17 of gestation in non-diabetic and diabetic mice is shown in Table 2. The litter size was six to eight, because the litter size in mice in the DM group was smaller than that of mice in the non-DM group. The number of fetuses or placentas in the non-DM group was 82 in total (WT, 44; Tg, 38), and the number of those in the DM group was 74 (WT; 33, Tg; 41). In both the WT and the Tg groups, fetuses from a diabetic mother were significantly smaller than those from a non-diabetic mother. However, when the Tg group as a whole was compared with the WT group, overexpression of TRX-1 did not affect fetal weight. In both the WT and the Tg groups, placental weight was significantly decreased in the DM group compared with the non-DM group. However, overexpression of TRX-1 did not affect placental weight when the WT and the Tg groups as a whole were compared.
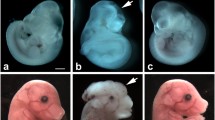
Fetal malformations
The number of fetuses in the non-DM group was 130 in total (WT, 65; Tg, 65), and the number of fetuses in the DM group was 147 (WT, 70; Tg, 77) at day 17 of gestation. No external malformations were observed in either the non-DM-WT or the non-DM-Tg fetuses. By contrast, a significantly higher rate of external malformations was observed in the DM-WT fetuses than that in the non-DM-WT fetuses. When the DM-WT and DM-Tg groups were compared, the malformation rate was significantly lower in the DM-Tg fetuses than in the DM-WT fetuses, as shown in Table 3. The most commonly observed malformations were NTDs (mainly exencephaly), but omphalocele and anophthalmia were also observed. The rate of NTDs in the DM-Tg fetuses was significantly lower than that in the DM-WT fetuses.
Immunohistochemical analysis and mRNA expression of Trx-1 and TRX-1
The production of endogenous mTRX-1 and overproduction hTRX-1 in embryos at day 10 of gestation was analysed histochemically using specific antibodies. The production of mTRX-1 in the DM-Tg embryos was similar to that in the DM-WT embryos. A similar result was obtained in the non-diabetic embryos (data not shown). Intense staining of hTRX-1 was detected in the DM-Tg embryos (n = 4), while hTRX-1 production was not detected in the DM-WT embryos (n = 4; Fig. 2c, d). hTRX-1 production in the non-DM group (n = 3) was the same as that in the DM group (Fig. 2a, b).

Levels of hTRX-1 in non-diabetes mellitus (non-DM) and diabetes mellitus (DM) groups. a–d Immunohistochemical staining for hTRX-1 proteins in the neuroepithelial cells of non-diabetic and diabetic embryos at day 10 of gestation. a Non-diabetic WT embryo; b non-diabetic Tg embryo; c diabetic WT embryo; d diabetic Tg embryo. Magnification is ×400. e Quantification of TRX-1 mRNA expression in fetuses at day 17 of gestation
The expression of Trx-1 and TRX-1 mRNA in fetuses of the DM and non-DM groups at day 17 of gestation was analysed by RT-PCR. The expression of Trx-1 mRNA in the DM-Tg fetuses was similar to that in the DM-WT fetuses. A similar result was obtained in the non-diabetic fetuses (data not shown). The TRX-1 mRNA was intensely expressed in the DM-Tg fetuses (n = 5), whereas its expression was not detected in the DM-WT fetuses (n = 5; Fig. 2e). The TRX-1 expression pattern in the non-DM-Tg and WT groups (n = 3) was the same as that in the DM group (Fig. 2e).
Biochemical analysis of oxidative stress
Oxidative stress was assayed by TBARS analysis. The TBARS concentration in fetal livers at day 17 of gestation from DM mothers was higher than that from non-DM mothers (Fig. 3a). The TBARS concentration of the DM-WT group was approximately twofold higher than that of the non-DM-WT group. Although TRX did not significantly affect the TBARS concentration in non-diabetic fetal livers, the TBARS concentration in fetal livers from diabetic pregnancies was decreased by TRX, but this change was not significant.

a Concentration of TBARS in fetal livers at day 17 of gestation. Statistical analyses were made by ANOVA followed by a post hoc test. *p < 0.05. b–e Immunohistochemical staining for 8-OHdG in the neuroepithelial cells of non-diabetic (non-DM) and diabetic (DM) embryos at day 10 of gestation. b Non-diabetic WT embryo; c non-diabetic Tg embryo; d diabetic WT embryo; e diabetic Tg embryo. Magnification is ×400. f Evaluation of immunostaining of 8-OHdG in neuroepithelial cells of non-diabetic and diabetic embryos at day 10 of gestation. Statistical analyses were made by ANOVA followed by a post-hoc test. *p < 0.05
Immunohistochemical analysis of 8-OHdG
As the analysis of TBARS in the current study has limitation in precision, we also determined oxidative stress by assay of the production of 8-OHdG, a well-described marker of oxidative stress using embryos. Production of 8-OHdG was dramatically increased in the neuroepithelial cells of the DM-WT embryos (n = 10) at day 10 of gestation, compared with the non-DM-WT embryos (n = 12; Fig. 3b, d, f). However, in the DM-Tg embryos (n = 12), production of 8-OHdG was less than that in the DM-WT embryos (n = 12; Fig. 3d–f).
Immunohistochemical analysis of apoptosis and cleaved caspase-3
The frequency of apoptotic cells in the neuroepithelial cells of the DM-WT embryos (n = 4) at day 10 of gestation was higher than that of the DM-Tg and non-DM-WT embryos (n = 4; Fig. 4a). The frequency of apoptotic cells in the neuroepithelial cells was similar between the DM-Tg and non-DM-Tg embryos (n = 4; Fig. 4a). Staining of cleaved caspase-3 was weak in TRX-Tg embryos and was almost negative in both non-DM and DM-Tg embryos at day 10 of gestation (n = 6; Fig. 4c, e). While cleaved caspase-3 staining was observed in both non-DM and DM-WT embryos, the number of cells that were positively stained for cleaved caspase-3 was much greater in the DM-WT embryos (n = 4) than in the non-DM-WT embryos (n = 6; Fig. 4b, d, f).

a Evaluation of staining of apoptosis in neuroepithelial cells of non-diabetic and diabetic embryos at day 10 of gestation. Statistical analyses were made by ANOVA followed by a post hoc test. *p < 0.05. b–e Immunohistochemical staining of cleaved caspase-3 in the neuroepithelial cells of non-diabetic (non-DM) and diabetic (DM) embryos at day 10 of gestation. b Non-diabetic WT embryo; c non-diabetic Tg embryo; d diabetic WT embryo; e diabetic Tg embryo. Magnification is ×400. f Quantification of caspase-3-positive cells expressed as a percentage of all counted cells in a field. Statistical analyses were made by ANOVA followed by a post hoc test. *p < 0.05
Discussion
Most studies on the mechanisms of diabetes-induced embryonic malformations have been performed in in-vitro systems. However, the mechanisms that underlie congenital malformations in in-vivo systems have not been fully elucidated. In the present study, we investigated the effect of TRX, an antioxidative and anti-apoptotic protein, on the morphological and biochemical outcome of diabetic pregnancy in mice. This study used TRX-Tg mice and was a completely in-vivo system. In this experiment, maternal mice were WT, and Tg and WT fetuses were exposed to hyperglycaemia under the same uterine conditions. Our results demonstrated that transgenic mice embryos that overproduce human TRX-1 were significantly more resistant to the teratogenic effects of hyperglycaemia than WT embryos in the same litter. External malformations, mostly NTDs, were used as outcome variables as indicators of teratogenicity. The majority of malformations in the DM group were exencephaly. Fetuses of diabetic mice displayed a high incidence of malformations affecting the neural tube, with the midbrain and hindbrain structures being the regions most often affected [30]. Many fetuses exhibited fusion defects, especially exencephaly. The incidence of exencephaly in the total DM group (WT and Tg) was 14.3%. By contrast, the rate of exencephaly in the DM-Tg fetuses was significantly lower than that in the DM-WT fetuses.
Our report is the first evidence that TRX-1 can protect against the teratogenic effects of hyperglycaemia in an in-vivo system. The protective role of TRX may be related to its ability to modulate oxidative stress, and the increase of TRX may be also associated with improved redox capacity in the fetal tissues. In fact, it has been suggested that TRX and/or the redox status modified by TRX, plays a crucial role in biological protection against oxidative stress during development and in various physiological states in adults [20, 37]. For instance, TRX-Tg mice have been reported to be more resistant to a variety of oxidative stresses and to survive longer than WT mice [32]. In addition, it has been reported that there are no differences in the expression of antioxidant enzymes such as Cu/Zn-SOD, Mn-SOD and glutathione peroxidase between TRX-Tg mice and WT mice [20, 37]. In addition, pancreatic beta-cell-specific overproduction of TRX in NOD mice, produced using an insulin promoter, prevents autoimmune and STZ-induced diabetes [38]. More recently, Kobayashi et al. showed that TRX-Tg embryos were significantly more resistant to the teratogenic effects of hyperoxia than WT embryos [39]. TRX protein production begins in embryonic tissues from day 8 to 10 gestation, which is the time that organogenesis commences and the time that metabolic state of embryos becomes active [40]. Thus, the TRX protein may contribute to the regulation or the modulation of oxidative stress in embryos. In this regard, it is interesting to note that homozygous mutant mouse embryos that lack the Trx gene die shortly after implantation [41], which corresponds to the stage when the TRX protein expression begins in WT embryos. These previous findings suggest that the protective effects of TRX against teratogenesis of early mouse embryos that we observed in the present study may be due to TRX-mediated attenuation of the embryopathic effects of oxidative stress. In addition to the results of TBARS analysis, when examined at the organogenetic period of development (day 8 to 10 of gestation), the level of oxidative stress, as measured by the production of 8-OHdG, was found to be markedly increased in the tissues of the DM-WT embryos, but not in the DM-Tg embryos. These results suggest that TRX protects against oxidative stress.
We found that the rate of early resorption in the non-DM and DM groups was similar in frequency. In contrast, there was a higher rate of late resorptions in the DM groups than in the non-DM groups, and a lower rate in the DM-Tg group compared with the DM-WT group. Evidently a large number of late resorptions were induced by maternal diabetes, and these were decreased by TRX. The cause of these late resorptions may be related to the increased rate of severe lethal malformations. The difference of resorption rate in early and late pregnancy implies that the aetiological processes of early resorptions are different from those of late resorptions.
It has been suggested that apoptosis is involved in diabetes-induced embryonic dysmorphogenesis [22, 26–29]. In addition, diabetes-induced inappropriate apoptosis in embryos during neurulation has been demonstrated to be one of the mechanisms leading to neural tube defects [31]. We therefore assayed apoptosis in the embryos of DM and non-DM, WT or TRX-Tg mice during organogenesis, by histochemical analysis of apoptotic cells and cleaved caspase-3. In the apoptotic cascade, procaspase-3 is cleaved after mitochondrial cytochrome c release and therefore, the presence of cleaved caspase-3 can be used as a marker for apoptosis [42]. Activation of caspase-3 results in nuclear degradation and cellular morphological change. Anti-caspase-3 staining of embryos at day 10 of gestation showed increased apoptosis in the DM-WT embryos, whereas few caspase-3 positive cells were detected in the non-DM and DM-Tg embryos. The results of the current study indicate that TRX decreases the occurrence of apoptosis in diabetic embryos. It has been shown that oxidative stress induces cytochrome c release from mitochondria and activation of caspases, p53 and kinases, including apoptosis signal-regulating kinase 1 (ASK1), c-Jun N-terminal kinase and p38 mitogen-activated protein kinase [16]. TRX inhibits apoptosis signalling not only by scavenging intracellular ROS in cooperation with the GSH system, but also by inhibiting the activity of ASK1 and p38. Therefore, TRX may block these steps in the signalling of TRX.
The results of the present study strongly suggest that TRX, an antioxidative protein, plays a crucial role in biological protection against oxidative stress induced by hyperglycaemia. Thus, antioxidative and anti-apoptotic proteins may prove useful in preventive treatment of congenital malformations caused by maternal diabetes. Furthermore, as the TRX-Tg mice that were used for our experiments provide a completely in-vivo system, this system is a useful model for assessment of the pathophysiological mechanism of diabetic embryopathy in vivo. Further studies on the signalling pathways of oxidative stress and apoptosis are required to completely elucidate the mechanism of diabetic embryopathy.
Abbreviations
- DM group:
-
Streptozotocin-induced diabetic group
- GSH:
-
Glutathione
- HRP:
-
Horseradish peroxidase
- NTDs:
-
Neural tube defects
- 8-OHdG:
-
8-Hydroxy-2′-deoxyguanosine
- ROS:
-
Reactive oxygen species
- SOD:
-
Superoxide dismutase
- STZ:
-
Streptozotocin
- TBA:
-
Thiobarbituric acid
- TBARS:
-
Thiobarbituric acid reactive substances
- Tg:
-
Transgenic
- TRX-1:
-
Thioredoxin-1
- TRX-Tg mice:
-
Transgenic mice overproducing human TRX-1
- WT:
-
Wild-type
References
Kuhl C, Coustan D, Kitzmiller J, Philipps A, Binder C, Schneider H (1998) Report on the 28th Annual Meeting of the Diabetic Pregnancy Study Group. Diabetologia 41(Suppl):8–14
Hanson U, Persson B, Thunell S (1990) Relationship between haemoglobin A1C in early type 1 (insulin-dependent) diabetic pregnancy and the occurrence of spontaneous abortion and fetal malformation in Sweden. Diabetologia 33:100–104
Suhonen L, Hiilesmaa V, Teramo K (2000) Glycaemic control during early pregnancy and fetal malformations in women with type 1 diabetes mellitus. Diabetologia 43:79–82
Inge M, Harold W, Gerard H (2004) Risk of complications of pregnancy in women with type 1 diabetes: nationwide prospective study in the Netherlands. BMJ 328:915–919
Wolff SP (1993) Diabetes mellitus and free radicals. Br Med Bull 49:642–652
Eriksson UJ, Borg LAH (1993) Diabetes and embryonic malformations: role of substrate-induced free-oxygen radical production in dysmorphogenesis in cultured rat embryos. Diabetes 42:411–419
Reece EA, Homko CJ, Wu YK, Wiznitzer A (1998) The role of free radicals and membrane lipids in diabetes-induced congenital malformations. J Soc Gynecol Investig 5:178–187
Eriksson UJ, Cederberg J, Wentzel P (2003) Congenital malformations in offspring of diabetic mothers—animal and human studies. Rev Endocr Metab Disord 4:79–93
Eriksson UJ, Borg LA, Forsberg H, Styrud J (1991) Diabetic embryopathy. Studies with animal and in vitro models. Diabetes 40(Suppl 1.2):94–98
Eriksson UJ, Borg LAH (1991) Protection by free oxygen radical scavenging enzymes against glucose induced embryonic malformations in vitro. Diabetologia 34:325–331
Trocino RA, Akazawa S, Ishibashi M et al (1995) Significance of glutathione depletion and oxidative stress in early embryogenesis in glucose-induced rat embryo culture. Diabetes 44:992–998
Wentzel P, Thunberg L, Eriksson UJ (1997) Teratogenic effect of diabetic serum is prevented by supplementation of superoxide dismutase and N-acetylcysteine in rat embryo culture. Diabetologia 40:7–14
Holmgren A (1985) Thioredoxin. Ann Rev Biochem 54:237–271
Homgren A (1989) Thioredoxin and glutaredoxin systems. J Biol Chem 264:13963–13966
Saitoh M, Nishitoh H, Fujii M et al (1998) Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 17:2596–2606
Ueda S, Masutani H, Nakamura H, Tanaka T, Ueno M, Yodoi J (2002) Redox control of cell death. Antioxid Redox Signal 4:405–414
Yoshida T, Oka S, Masutani H, Nakamura H, Yodoi J (2003) The role of thioredoxin in the aging process: involvement of oxidative stress. Antioxid Redox Signal 5:563–570
Nakamura H, Nakamura K, Yodoi J (1997) Redox regulation of cellular activation. Annu Rev Immunol 15:351–369
Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J (1997) AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc Natl Acad Sci USA 94:3633–3638
Takagi Y, Mitsui A, Nishiyama A et al (1999) Overexpression of thioredoxin in transgenic mice attenuates focal ischemic brain damage. Proc Natl Acad Sci USA 96:4131–4136
Sun F, Akazawa S, Sugahara K (2002) Apoptosis in normal rat embryo tissues during early organogenesis: the possible involvement of Bax and Bcl-2. Arch Histol Cytol 65:145–157
Sun F, Kawasaki E, Akazawa S et al (2005) Apoptosis a nd its pathway in early post-implantation embryos of diabetic rats. Diab Res Clin Pract 67:110–118
Korsmeyer SJ, Yin X-M, Oltvai ZN, Veis-Novack DJ, Linette GP (1995) Reactive oxygen species and the regulation of cell death by the Bcl-2 gene family. Biochem Biophys Acta 1271:63–66
Kroemer G (1997) The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med 3:614–620
Moley K, Chi M, Knudson C, Korsmeyer S, Mueckler M (1998) Hyperglycemia induces apoptosis in pre-implantation embryos through cell death effector pathways. Nat Med 4:1421–1424
Akazawa S (2005) Diabetic embryopathy: studies using a rat embryo culture system and an animal model. Congenit Anom 45:73–79
Zhao Z, Reece EA (2005) Experimental mechanisms of diabetic embryopathy and strategies for developing therapeutic interventions. J Soc Gynecol Investig 12:549–557
Reece EA, Wu YK, Zhao Z, Dhanasekaran D (2006) Dietary vitamin and lipid therapy rescues aberrant signaling and apoptosis and prevents hyperglycemia-induced diabetic embryopathy in rats. Am J Obstet Gynecol 194:580–585
Gareskog M, Cederberg J, Eriksson UJ, Wentzel P (2007) Maternal diabetes in vivo and high glucose concentration in vitro increases apoptosis in rat embryos. Reprod Toxicol 23:63–74
Phelan SA, Ito M, Loeken MR (1997) Neural tube defects in embryos of diabetic mice: role of Pax-3 gene and apoptosis. Diabetes 46:1189–1197
Loeken MR (2006) Advances in understanding the molecular causes of diabetes-induced birth defects. J Soc Gynecol Investig 13:2–10
Mitsui A, Hamuro J, Nakamura H et al (2002) Overexpression of human thioredoxin in transgenic mice controls oxidative stress and life span. Antioxid Redox Signal 4:693–696
Punareewattana K, Sharova LV, Li W, Ward DL, Holladay SD (2003) Reduced birth defects caused by maternal immune stimulation may involve increased expression of growth promoting genes and cytokine GM-CSF in the spleen of diabetic ICR mice. Int Immunopharmacol 3:1639–1655
Cederberg J, Siman CM, Eriksson UJ (2001) Combined treatment with vitamin E and vitamin C decreased oxidative stress and improves fetal outcome in experimental diabetic pregnancy. Pediatr Res 49:755–762
Siman CM, Eriksson UJ (1997) Vitamin C supplementation of the maternal diet reduces the rate of malformation in the offspring of diabetic rats. Diabetologia 40:1416–1424
Cederberg J, Eriksson UJ (2005) Antioxidative treatment of pregnant diabetic rats diminishes embryonic dysmorphogenesis. Birth Defects Res A Clin Mol Teratol 73:498–505
Nakamura H, Mitsui A, Yodoi J (2002) Thioredoxin overexpression in transgenic mice. Methods Enzymol 347:436–440
Hotta M, Tashiro F, Ikegami H et al (1998) Pancreatic beta cell-specific expression of thioredoxin, an antioxidative and antiapoptotic protein, prevents autoimmune and streptozotocin-induced diabetes. J Exp Med 188:1445–1451
Kobayashi-Miura M, Nakamura H, Yodoi J, Shiota K (2002) Thioredoxin, an anti-oxidant protein, protects mouse embryos from oxidative stress-induced developmental anomalies. Free Radic Res 36:949–956
Kobayashi M, Nakamura H, Yodoi J, Shiota K (2000) Immunohistochemical localization of thioredoxin and glutaredoxin in mouse embryos and fetuses. Antioxid Redox Signal 2:653–663
Matsui M, Oshima M, Oshima H et al (1996) Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev Biol 178:179–185
Larisa N, Ryan RW, Robert PE, Garth P (2003) The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol Cell Biol 23:916–922
Acknowledgements
We thank H. Noro for her excellent technical assistance. This work was supported in part by grants for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (No.1865484, 19390428, 18791150 and 20659170), the Smoking Research Foundation, Takeda Scientific Research Foundation, Kanzawa Medical Research Foundation and the Danone Institute of Japan.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below is the link to the electronic supplementary material.
ESM Table 1
Primers and probes used for the detection of Trx-1 and TRX-1 mRNA (PDF 9 kb)
Rights and permissions
About this article
Cite this article
Kamimoto, Y., Sugiyama, T., Kihira, T. et al. Transgenic mice overproducing human thioredoxin-1, an antioxidative and anti-apoptotic protein, prevents diabetic embryopathy. Diabetologia 53, 2046–2055 (2010). https://doi.org/10.1007/s00125-010-1784-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-010-1784-y