
Abstract
Recent clinical studies show that hypoglycaemia is associated with increased risk of death, especially in patients with coronary artery disease or acute myocardial infarction. This paper reviews data from cellular and clinical research supporting the hypothesis that acute hypoglycaemia increases the risk of malignant ventricular arrhythmias and death in patients with diabetes by generating the two classic abnormalities responsible for the proarrhythmic effect of medications, i.e. QT prolongation and Ca2+ overload. Acute hypoglycaemia causes QT prolongation and the risk of ventricular tachycardia by directly suppressing K+ currents activated during repolarisation, a proarrhythmic effect of many medications. Since diabetes itself, myocardial infarction, hypertrophy, autonomic neuropathy and congestive heart failure also cause QT prolongation, the arrhythmogenic effect of hypoglycaemia is likely to be greatest in patients with pre-existent cardiac disease and diabetes. Furthermore, the catecholamine surge during hypoglycaemia raises intracellular Ca2+, thereby increasing the risk of ventricular tachycardia and fibrillation by the same mechanism as that activated by sympathomimetic inotropic agents and digoxin. Diabetes itself may sensitise myocardium to the arrhythmogenic effect of Ca2+ overload. In humans, noradrenaline (norepinephrine) also lengthens action potential duration and causes further QT prolongation. Finally, both hypoglycaemia and the catecholamine response acutely lower serum K+, which leads to QT prolongation and Ca2+ loading. Thus, hypoglycaemia and the subsequent catecholamine surge provoke multiple, interactive, synergistic responses that are known to be proarrhythmic when associated with medications and other electrolyte abnormalities. Patients with diabetes and pre-existing cardiac disease may therefore have increased risk of ventricular tachycardia and fibrillation during hypoglycaemic episodes.
Similar content being viewed by others


Introduction
With the advent of very aggressive lowering of HbA1c values, patients with diabetes are at increased risk of hypoglycaemic episodes [1–3]. Several recent papers suggest that hypoglycaemia is associated with increased mortality rates [3–8], particularly in patients with pre-existent cardiac disease or acute myocardial infarction [3–5]. The Action to Control Cardiovascular Risk in Diabetes (ACCORD) Study [3] found excess mortality rates in diabetic patients under tight glycaemic control who had either multiple risk factors or known coronary artery disease, when compared with conventional glucose control. Since over one third of excess deaths were attributed to unexpected cardiac death, i.e. possible sudden death, the ACCORD study is the first large controlled clinical trial to raise the possibility that ventricular tachycardia and fibrillation are promoted by hypoglycaemia.
These findings fit with prior studies. The modern recognition that malignant arrhythmias might be a consequence of hypoglycaemia started in Europe after the introduction of human insulin in the 1980s. Disturbing reports of deaths at night in young patients with type 1 diabetes led to the term ‘dead in bed syndrome’ [9–13]. Since many autopsies showed no pathological findings, several investigators speculated that hypoglycaemia increases the risk of sudden cardiac death. Pioneering clinical research by S. R. Heller and colleagues [14–21], and by others [22, 23] showed that hypoglycaemia and the associated catecholamine surge increase the QT interval, an electrocardiographic change strongly associated with sudden death in other contexts, particularly with medications.
Cellular research on ventricular myocardium in the 1980s also showed that diabetes itself increases the risk of electrical instability. Action potential prolongation, the substrate for QT prolongation on the electrocardiogram, was first reported by Fein et al. in ventricular papillary muscles from rats with streptozotocin-induced diabetes [24]. The effect is dramatically increased by the addition of hypertension [25]. Shortly thereafter, I and other members of the same research group showed that papillary muscles from this animal model of type 1 diabetes have a greater propensity than normal to develop delayed afterdepolarisations, a form of cellular electrophysiological instability associated with Ca2+ overload, ventricular tachycardia and sudden death [26].
Since then, many papers have been published on the cellular electrophysiological properties of ventricular muscle in animal models of cardiovascular disease and diabetes, as well as on hypoglycaemia itself. These studies show that the effect of acute hypoglycaemia, particularly in the setting of diabetes and pre-existent heart disease, is indistinguishable at a cellular level from that of medications known to be associated with arrhythmias and sudden death. Hypoglycaemia can therefore be considered a proarrhythmic event. The purpose of this paper is to review cellular and clinical research supporting the hypothesis that hypoglycaemia increases the risk of ventricular arrhythmias and sudden death.
The two proarrhythmic mechanisms and their relevance to hypoglycaemia
Proarrhythmic responses are not random. Almost all proarrhythmic medications cause one or both of the following: (1) longer action potential duration; and (2) higher myoplasmic Ca2+ concentration. Both of these changes convert normally quiescent cells into spontaneously active cells that have abnormal pacemaker activity; both changes are also likely to occur during hypoglycaemia.
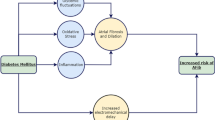
When action potentials become too long, ventricular cells fail to repolarise and may subsequently generate recurrent spontaneous action potentials from depolarised takeoff potentials via reactivation of Ca2+ channels, a process called early afterdepolarisation (Fig. 1) If such non-driven action potentials propagate to normal myocardium, they cause ventricular tachycardia. Since repolarisation of ventricular action potentials causes T waves on the ECG, longer action potentials cause QT interval prolongation, which is recognised as a marker of the propensity to develop early afterdepolarisations and ventricular tachycardia, as described in a review by D. M. Roden [27].

Proposed mechanism of proarrhythmic events in patients with cardiovascular disease, diabetes and hypoglycaemia. The two main mechanisms for proarrhythmia involve longer action potentials, which cause QT prolongation on the ECG and lead to proarrhythmic early afterdepolarisations (EAD, arrows) and Ca2+ overload, which causes increased contractility and leads to proarrhythmic non-driven action potentials following rapid pacing stimulations (arrowhead) triggered by delayed afterdepolarisations (DAD, arrow). Diabetes itself increases action potential duration and the propensity to develop Ca2+ overload. Other conditions common to patients with cardiovascular disease do the same (small boxes). These reduce reserve for the additional proarrhythmic effects of hypoglycaemia. Acute lowering of extracellular glucose causes additional action potential prolongation, increasing the risk of early afterdepolarisations. The catecholamine surge in response to low glucose leads to longer action potentials in humans and increases the risk of developing Ca2+ overload. Acute reduction in extracellular K+ seen during hypoglycaemic episodes also leads to action potential prolongation and Ca2+ overload. Both pathways interact synergistically to increase the risk of ventricular arrhythmias and sudden death. For details, see text. MI, myocardial infarction. Figure adapted from Nordin [41] with permission from the American Physiological Society and from Nordin and Aronson [99] with permission from Springer Science and Business Media
One major way in which action potentials can be prolonged is by blocking repolarising K+ channels that are activated during the start of the action potential; the two most important K+ channels are IKr and IKs. Genetic mutations in the proteins involved cause congenital QT prolongation, which has a high rate of sudden death. Furthermore, given that many types of medications, including tricyclic antidepressants, antibiotics and other non-cardiac drugs, cause blockade of repolarising K+ channels, occurrence of QT prolongation and sudden death [27] suggest that this is a fundamental pathway for electrical instability.
A second type of proarrhythmic oscillatory activity is associated with Ca2+ overload [28–31]. Increased intracellular Ca2+ concentration in myocardial muscle leads to the development of spontaneous intracellular Ca2+ cycling between the sarcoplasm and the myoplasm, which depolarises the membrane potential by generating electrogenic currents carried primarily by Na–Ca exchange molecules [31]. If strong enough, the oscillations, called delayed afterdepolarisations, can generate non-driven, pacemaker action potentials that can propagate as ventricular tachycardia (Fig. 1). Originally described with digitalis glycosides [28–30], delayed afterdepolarisations have been seen in multiple settings including diabetes itself [26] and beta adrenergic stimulation [32–35]. A large body of evidence links the proarrhythmic effect of inotropic agents, including catecholamines [36], to the development of Ca2+ overload, delayed afterdepolarisations and both ventricular tachycardia and fibrillation [37, 38].
Two important facts are crucial to an understanding of the close relationship between the basic pathophysiological processes described above and hypoglycaemia.
First, action potential prolongation and Ca2+ overload are final common pathways for electrical instability of ventricular muscle, the effect of which is not confined specifically to medications. With regard to action potential prolongation, perhaps the clearest example of this phenomenon is the fact that highly reproducible, completely reversible early afterdepolarisations can be generated simply by injecting tiny amounts of depolarising current, which do not affect any channel proteins directly, but merely delay repolarisation [39–41]. Blockade of repolarising K+ channels is an even more specific final common pathway for electrical instability in genetic mutations and with multiple medications and other interventions [27]. Ca2+ overload is also a final common pathway for electrical instability. Thus delayed afterdepolarisations can be amplified simply by injecting cyclic AMP into ventricular myocytes [32]. These and other studies confirm that action potential prolongation and Ca2+ overload cause electrical instability due to abnormal interactions that are built into the biophysical properties of the channels themselves [40, 41]. Thus, almost any intervention that leads to action potential prolongation or increases intracellular Ca2+ will be proarrhythmic.

Second, multiple interventions that cause action potential prolongation or Ca2+ overload act synergistically to increase the risk of afterdepolarisations, ventricular tachycardia and sudden death. With regard to action potential prolongation, Roden has named this phenomenon ‘reduced repolarisation reserve’ [42]. Furthermore, Ca2+ overload and action potential prolongation can interact synergistically to magnify the risk of electrical instability. One highly relevant example is reduced extracellular K+, which causes Ca2+ loading and longer action potentials. In this setting, Ca2+ oscillations coordinate with and amplify the magnitude of early afterdepolarisations, making them more likely to propagate as ventricular tachycardia [39].
Evidence that hypoglycaemia is a proarrhythmic event in patients with diabetes and cardiovascular disease
The above concepts directly apply to hypoglycaemia in patients with diabetes and pre-existent cardiovascular disease. The evidence is summarised in Fig. 1 and the following sections.
Diabetes induces a proarrhythmic state associated with QT prolongation and increased risk of Ca2+ overload
Diabetes was an independent risk factor for sudden death in several epidemiological studies [43–45]. In one large study [44], diabetes increased the risk of sudden death by an odds ratio of 3.8 in patients with prior history of coronary artery disease. Diabetes also adds to the risk of sudden death in patients with hypertrophy or diastolic dysfunction [46, 47], and is associated with ventricular fibrillation independently of coronary artery disease or heart failure status [48].
Diabetes causes QT prolongation [49–51]. The change in QT interval corresponds to the fact that, at a cellular level, diabetes lengthens action potentials [24, 52] and reduces repolarising of K+ currents [52] in a way similar to proarrhythmic medications. QT prolongation predicts mortality in type 1 [53] and type 2 [54–58] diabetes. In the case of type 1 diabetes, QT prolongation conferred an odds ratio for death of 24.6 (95% CI 6.52–92.85) [53].

Diabetic muscle is also more susceptible than normal to arrhythmias from Ca2+ overload. Exposing muscle from rats [26] or guinea-pigs [59] with streptozotocin-induced diabetes to high Ca2+ solutions with ouabain leads to a greater incidence of delayed afterdepolarisations than in control. This susceptibility may be related to changes in gene expression caused by hyperglycaemia. Diabetes depresses Na-K ATPase activity [60], and this digoxin-like effect reduces the ability of diabetic muscle to control intracellular [Na+] and therefore intracellular [Ca2+].
Many cardiovascular diseases are also associated with QT prolongation and Ca2+ overload, and therefore increase risk of proarrhythmic effects during hypoglycaemia
In addition to the effect of diabetes itself, action potential prolongation and QT prolongation occur in a variety of other cardiac conditions commonly seen in patients with diabetes. Hypertrophy [61, 62], myocardial infarction [63, 64] and congestive heart failure [65, 66], all of which are known to increase the risk of sudden death, are also known to cause action potential and QT prolongation. Each is associated with reduction of repolarising K+ channels [65–67]. These processes reduce repolarisation reserve in patients with cardiovascular disease and at risk of hypoglycaemia. Moreover, patients with congestive heart failure frequently have increased levels of catecholamines [68] and may be on inotropic agents, both of which cause Ca2+ overload and increase the risk of sudden death.
Hypoglycaemia causes QT prolongation by a cellular mechanism identical to that of proarrhythmic drugs
In addition to the studies of Heller and colleagues [14–21], at least five other groups have confirmed that hypoglycaemia itself is associated with lengthening of the QT interval. [22, 23, 69–71]. Zhang et al [72] showed that hypoglycaemia inhibits current through the human ether-a-go-go related gene channel, which generates IKr, one of the main repolarising K+ channels in human myocytes. Interestingly, the same team also showed that hyperglycaemia impairs current flow through the same channels, but by a different mechanism. These experiments directly link hypoglycaemia to the mechanism of drug-induced proarrhythmia [27]. Hypoglycaemia, moreover, has been repeatedly associated with a reduction in the serum K+ concentration. Hypokalaemia lengthens the QT interval [73, 74] and is also associated with an increase in rates of sudden death [75].

The catecholamine surge following hypoglycaemic episodes is also proarrhythmic
During hypoglycaemia, the adrenal medulla secretes noradrenaline and adrenaline, and the sympathetic nervous system increases alpha and beta stimulation [76, 77]. Increased catecholamine activity, both endogenous and through medications, has long been recognised to increase cardiac death [36, 78–80]. Catecholamines cause early and delayed afterdepolarisations in myocytes from guinea pigs and dogs [33, 34]. Exposure of ventricular myoctes isolated from human end-stage failing hearts to noradrenaline also causes delayed afterdepolarisations [37], longer action potentials [81] and early afterdepolarisations [81]. Corresponding to these basic experiments, catecholamines cause longer QT intervals in humans [19, 82]. Furthermore, adrenaline infusion lowers serum K+ [17]. The effect of adrenaline on QT interval corrected for heart rate (QTc) is reduced by holding serum K+ constant, but remains highly significant [17]. Thus, the catecholamine surge in hypoglycaemia is likely to be highly arrhythmogenic.

Clinical evidence that hypoglycaemia increases the risk of sudden death
No definitive clinical study has yet proved that hypoglycaemia increases the risk of arrhythmias. However, there is strong and increasing circumstantial evidence from several sources that hypoglycaemia increases the risk of cardiovascular sudden death.
First, case reports and small case series have shown that the incidence of sudden death during sleep in patients with type 1 diabetes is three to four times that of the non-diabetic population [15]. Second, several recent large studies have shown a close association between hypoglycaemia and death [6–8]. The NICE-SUGAR Study [6] demonstrated that tight control of glucose (target range 4.5–6.1 mmol/l) for patients in intensive care units led to a significantly higher death rate than in patients with more conventional control. The incidence of hypoglycaemia in the tight control group was over ten times higher than in the control group (6.8% vs 0.5%). Furthermore, death from cardiovascular causes was statistically higher in the tight control group. Two other studies have directly linked episodes of hypoglycaemia with increased risk of death in hospitalised patients with diabetes [7] and in outpatients [8]. Both studies showed that the risk of death was directly related to the severity of hypoglycaemia, suggesting a causal relationship. In addition, risk of death is increased within 24 h of a hypoglycaemic event [8], suggesting an acute event such as an arrhythmia as the cause.
A third set of studies has shown a striking risk of low serum glucose on admission to hospital affecting 30 day outcome in patients admitted for ST elevation myocardial infarction [4, 5]. In one study [4], the overall death rate was 4.6% in patients with admission glucose <4.5 mmol/l compared with 1.0% for patients with glucose between 4.5 and 7.0 mmol/l (Fig. 2). Even more striking was the rate of death in patients with glucose <4.5 mmol/l and a thrombolysis in myocardial infarction score of five or more (i.e. severe cardiac compromise), namely 22.6%. This was more than 11 times the death rate of patients with glucose values between 4.5 and 7.0 mm/l, and much higher than any other glucose level, including glucose >11 mmol/l. Low admission glucose increased mortality rates in patients with and without diabetes, suggesting the effect is unlikely to be related only to medications used to treat diabetes. These results were confirmed in a second study [5], which used a different database of patients with ST elevation myocardial infarctions. Neither study documented the mode of death. Thus there is no information to evaluate a direct relationship between hypoglycaemia on admission and sudden death. Nevertheless, other experimental evidence suggests that hypoglycaemia might exacerbate the already high probability of developing ventricular arrhythmias in a setting of reduced repolarisation reserve caused by myocardial infarction [63, 64].

Plots of death or recurrent myocardial infarction as a function of admission serum glucose value in (a) all patients and (b) diabetic patients in a study of 4,224 cases of acute ST elevation myocardial infarction [4]. In the total cohort (a), a U-shaped relationship can be seen, with a higher rate in patients with admission glucose <4.5 mmol/l than in those with higher values (p < 0.001). b In patients with diabetes, the risk associated with low admission glucose was even more prominent (p < 0.001). c Death at 30 days plotted as a function of initial glucose level and thromblysis in myocardial infarction risk score (TRS) on presentation. Higher TRS indicates more advanced cardiac disease and haemodynamic instability at the time of the myocardial infarction. The highest risk of death (22.6%) was in patients admitted with serum glucose <4.5 mmol/l and TRS >4. Figure modified from Pinto et al. [4] with permission from Elsevier

The final source of data supporting the hypothesis that hypoglycaemia increases risk of sudden death is the ACCORD study [3], which tested the hypothesis that cardiovascular complications of diabetes could be reduced by very aggressive lowering of HbA1c levels. The study recruited over 10,000 patients, all of whom had either documentation of or multiple risk factors for coronary artery disease. The design included two groups: one that aimed to reduce HbA1c values to less than 7%, and the other, a tight glycaemic control group, that aimed for a reduction to less than 6%. The ACCORD study was stopped early when a significant risk of increased death was found in the second group (tight control, with the goal of HbA1c of 6%). The actual mean values of HbA1c were 6.4% in the tight control group and 7.4% in the standard control group. The risk of death from cardiovascular causes was increased by 35% (p = 0.02). The excess levels of cardiovascular deaths were spread through several categories, but 46% (19/41) were listed as unexpected cardiac deaths. No other associations in the study design (e.g. specific medications) were found to be significant. The incidence of hypoglycaemia requiring assistance was over three times higher in the tight control group. Despite the increase in hypoglycaemic episodes in the tight control group, preliminary non-prespecified exploratory analysis did not identify hypoglycaemia as the cause of increased death in the ACCORD study. However, the number of deaths was relatively small.

Other recent studies have reinforced the findings of the ACCORD study. In a retrospective cohort study from the UK General Practice Research Database, Currie et al [83] showed that the adjusted hazard ratio for mortality in patients with type 2 diabetes was significantly higher in the lowest decile of HbA1c (median 6.4%, 6.1–6.6) than in the decile with lowest hazard (median 7.5%, 7.5–7.6%) and higher than any other decile of HbA1c below 10%. The cause of death was not determined in this study, but the authors speculated that hypoglycaemia was a likely cause. In the VADT study [84], another randomised study evaluating intensive vs conventional treatment for type 2 diabetes, subgroup analysis of the cause of death in patients in the intensive treatment arm demonstrated a strong trend towards increased rates of sudden death (1.2% vs 0.4%, p = 0.07). In contrast, the ADVANCE study [85] did not show an increased risk of death with intensive treatment. However, the rate of hypoglycaemia was much lower than in ACCORD.
Are there possible antiarrhythmic effects of diabetes or hypoglycaemia that might ameliorate the proarrhythmic response?
From the perspective of cellular electrophysiology, the only way the multiple proarrhythmic effects of hypoglycaemia could be negated would be through equally powerful anti-arrhythmic effects induced by the hypoglycaemic state.
The parasympathetic response induced by hypoglycaemia may be antiarrhythmic. Parasympathetic stimulation, by slowing heart rate, may reduce the amount of Ca2+ loading from the sympathetic surge of hypoglycaemia. Bradycardia has been recorded in patients during hypoglycaemic episodes [71], while studies in animal models show that direct stimulation of the vagus nerve [86] and exposure to choline [87] or pilocarpine [88] reduce ventricular arrhythmias during occlusion of the left anterior descending coronary artery. The effect may be due to a reduction in Ca2+ overload and delayed afterdepolarisations. However, slowing of the heart rate increases the risk of early afterdepolarisations and torsade de pointes in patients with QT prolongation [27]. Thus, the overall effect of parasympathetic stimulation is not clear.
A second possible condition that may reduce the proarrhythmic effect of hypoglycaemia is the blunting of autonomic responses seen in diabetic patients with autonomic neuropathy or following recurrent episodes of hypoglycaemia. Lee et al. [19] showed that QTc prolongation due to hypoglycaemia is reduced in patients with severe autonomic neuropathy and impaired baroreflex sensitivity, primarily due to reduced levels of adrenaline during hypoglycaemic episodes. The same basic response would apply to reduced sympathetic responses to recurrent episodes of hypoglycaemia [89, 90]. However, patients with diabetes and autonomic neuropathy have longer QT and QTc intervals than controls [57, 91], and autonomic neuropathy itself is associated with an increase in sudden cardiac death in diabetes [57]. Therefore, patients with diabetic autonomic neuropathy have less repolarisation reserve [42] and are likely to be more susceptible to other causes of QT prolongation, such as hypoglycaemia, even if the catecholamine response is blunted.
Summary and recommendations
Hypoglycaemia, particularly when seen in the setting of cardiac disease, creates multiple changes that are proarrhythmic. Diabetes [26, 49–51] and cardiovascular complications [61–68] increase the likelihood of pre-existent QT prolongation and Ca2+ overload, thus reducing reserve against further conditions that lead to each of these two. Hypoglycaemia [14–23] and the sympathetic response [19, 33, 34, 37, 82] create additional QT prolongation and Ca2+ overload. The two proarrhythmic pathways are known to interact in a synergistic way to increase the risk of arrhythmias [39, 42].
From a basic science perspective, the evidence that associates hypoglycaemia and the secondary catecholamine response with sudden death is very strong. Hypoglycaemia causes the specific changes in K+ currents [72] and Ca2+ loading [26, 59] that have been repeatedly associated with the development of malignant ventricular arrhythmias and sudden death from medications [36, 78–80]. While parasympathetic activation and autonomic neuropathy may attenuate the proarrhythmic effect of hypoglycaemia by reducing the effect of the catecholamine surge [19, 86–88] both cause other changes that may themselves be proarrhythmic [27, 57, 71]. Recurrent episodes of hypoglycaemia also blunt the catecholamine response [89, 90], but multiple proarrhythmic changes are still induced by direct effects of hypoglycaemia.
No large epidemiological study has yet definitively confirmed or disproved the hypothesis that hypoglycaemia is a proarrhythmic event leading to an increase in malignant ventricular tachyarrhythmias and sudden death. However, studies have repeatedly suggested that hypoglycaemia is particularly dangerous for patients with pre-existent heart disease or other risks for sudden death [3–8], especially severe myocardial infarction [4].
Despite the association between hypoglycaemia and sudden death, there is no definitive evidence that ventricular tachycardia and fibrillation are more common in patients with hypoglycaemia. One recent study of 24 patients with type 1 diabetes [71] did show that ventricular ectopy developed during hypoglycaemic episodes, including one case of ventricular couplets in a patient whose QTc during the episode was 560 ms, considered to be dangerously long.
With regard to medications, the link between death and QT prolongation or Ca2+ overload has sometimes been delayed or revealed only in large epidemiological studies. This is particularly true in populations at risk of death from other causes. For example, the well recognised danger of torsade de pointes ventricular tachycardia in patients on methadone was overlooked for over 45 years because deaths were attributed to the known risk of respiratory depression and overdose [92]. In patients with cardiovascular disease, the risk of sudden death is high, and this is independent of glucose control. It is possible that the additional risk of death from hypoglycaemia has been similarly obscured. And since the effect of hypoglycaemia is evanescent, fatal arrhythmias may be even more difficult to prove than in the case of medications, where the risk is more constant.
Several papers now urge caution when lowering blood glucose in patients with known heart disease. These include recommendations made at the original press conference following the release of the ACCORD study data [93], as well as a recent position statement from the American Diabetes Association, in conjunction with the American College of Cardiology and the American Heart Association [94]. The position statement specifically recommends considering less stringent HbA1c goals than 7% for patients with advanced macrovascular disease.
These recommendations, however, also emphasise that tight control of glucose is highly beneficial for patients with diabetes. Indeed, follow-up studies from long-term cohorts with type 1 [95] and type 2 [96] diabetes have demonstrated that early aggressive lowering of HbA1c levels leads to a reduction of myocardial infarction and other measures of macrovascular disease 10 or more years later.
Taken together, the data suggest that it is appropriate to lower HbA1c levels aggressively for patients with early diabetes and at low risk of myocardial infarction or coronary artery disease. For patients who already have structural heart disease, including hypertrophy, prior myocardial infarctions, autonomic neuropathy, systolic heart failure or QT prolongation, less tight control may be appropriate. The overall strategy should be to reach the lowest HbA1c that can be achieved and maintained safely. Determining the precise values will require further research.
How can we test the hypothesis that hypoglycaemia increases the risk of sudden death? Evaluation of large databases was crucial for confirming the association between many medications and sudden death [97], as most recently shown for atypical antipsychotic medications, which cause QT prolongation [98]. Given the scope of the problem, this should be a high priority for clinical research in the future. Few questions are more important for the practice of medicine today.
Abbreviations
- ACCORD:
-
Action to Control Cardiovascular Risk in Diabetes
- QTc:
-
QT interval corrected for heart rate
References
The DCCT Research Group (1991) Epidemiology of severe hypoglycaemia in the Diabetes Control and Complications Trial. Am J Med 90:450–459
Egger M, Davey Smith G, Stettler C, Diem P (1997) Risk of adverse effects of intensified treatment in insulin-dependent diabetes mellitus: a meta-analysis. Diabet Med 14:919–928
The Action to Control Cardiovascular Risk in Diabetes Study Group, Gerstein HC, Miller ME, Byintgton RP et al (2008) Effects of intensive glucose lowering in type 2 diabetes. New Engl J Med 358:2545–2559
Pinto DS, Skolnick AH, Kirtane AJ, for the TIMI Study Group et al (2005) U-shaped relationship of blood glucose with adverse outcomes among patients with ST-segment elevation myocardial infarction. J Am Coll Cardiol 46:178–180
Pinto DS, Kirtane AJ, Pride YB, for the CLARITY-TIMI 28 Investigators et al (2008) Association of blood glucose with angiographic and clinical outcomes among patients with ST-segment elevation myocardial infarction (from the CLARITY-TIMI-28 Study). Am J Cardiol 101:303–307
The NICE-SUGAR Study Investigators (2009) Intensive vs conventional glucose control in critically ill patients. N Engl J Med 360:1283–1297
Turchin A, Matheny ME, Shubina M, Scanlon JV, Greenwood B, Pendergrass ML (2009) Hypoglycaemia and clinical outcomes in patients with diabetes hospitalized in the general ward. Diabetes Care 32:1153–1157
Moen MF, Zhan M, Hsu VD et al (2009) Frequency of hypoglycaemia and its significance in chronic kidney disease. Clin J Am Soc Nephrol 4:1121–1127
Tattersall RB, Gill GV (1991) Unexplained deaths of type 1 diabetic patients. Diabet Med 8:49–58
Thordarson H, Sovik O (1995) Dead in bed syndrome in young diabetic patients in Norway. Diabet Med 12:782–787
Sartor G, Dahlquist G (1995) Short-term mortality in childhood onset insulin-dependent diabetes mellitus: a high frequency of unexpected deaths in bed. Diabet Med 12:607–611
Edge JA, Ford-Adams ME, Dunger DB (1999) Causes of death in children with insulin dependent diabetes 1990–1996. Arch Dis Child 81:318–323
Heller S (1999) Dead in bed. Diabet Med 16:782–785
Marques JL, George E, Peacey SR et al (1997) Altered ventricular repolarization during hypoglycaemia in patients with diabetes. Diabet Med 14:648–654
Heller SR (2002) Abnormalities of the electrocardiogram during hypoglycaemia: the cause of the dead in bed syndrome? Int J Clin Pract Suppl 129:27–32
Robinson RT, Harris ND, Ireland RH, Lee S, Newman C, Heller SR (2003) Mechanisms of abnormal cardiac repolarization during insulin-induced hypoglycaemia. Diabetes 52:1469–1474
Lee S, Harris ND, Robinson RT, Yeoh L, Macdonald IA, Heller SR (2003) Effects of adrenaline and potassium on QTc interval and QT dispersion in man. Eur J Clin Invest 33:93–98
Murphy NP, Ford-Adams ME, Ong KK et al (2004) Prolonged cardiac repolarisation during spontaneous nocturnal hypoglycaemia in children and adolescents with type 1 diabetes. Diabetologia 47:1940–1947
Lee SP, Yeoh L, Harris ND et al (2004) Influence of autonomic neuropathy on QTc interval lengthening during hypoglycaemia in type 1 diabetes. Diabetes 53:1535–1542
Robinson RT, Harris ND, Ireland RH, Macdonald IA, Heller SR (2004) Changes in cardiac repolarization during clinical episodes of nocturnal hypoglycaemia in adults with type 1 diabetes. Diabetologia 47:312–315
Lee SP, Harris ND, Robinson RT et al (2005) Effect of atenolol on QTc interval lengthening during hypoglycaemia in type 1 diabetes. Diabetologia 48:1269–1272
Eckert B, Agardh CD (1998) Hypoglycaemia leads to an increased QT interval in normal men. Clin Physiol 18:570–575
Landstedt-Hallin L, Englund A, Adamson U, Lins PE (1999) Increased QT dispersion during hypoglycaemia in patients with type 2 diabetes mellitus. J Intern Med 246:299–307
Fein FS, Aronson RS, Nordin C, Miller-Green B, Sonnenblick EH (1983) Altered myocardial response to ouabain in diabetic rats: mechanics and electrophysiology. J Mol Cell Card l5:769–784
Fein FS, Capasso J, Aronson RS et al (1984) Combined renovascular hypertension and diabetes in rats: a new preparation of congestive cardiomyopathy. Circulation 70:318–330
Nordin C, Gilat E, Aronson RS (1985) Delayed afterdepolarizations and triggered activity in ventricular muscle in rats with streptozotocin induced diabetes. Circ Res 57:28–34
Roden DM (2004) Drug-induced prolongation of the QT interval. N Engl J Med 350:1013–1022
Ferrier G (1977) Digitalis arrhythmias: role of oscillatory afterpotentials. Prog Cardiovasc Dis 19:459–474
Rosen MR, Gelband H, Merker C, Hoffman BF (1973) Mechanisms of digitalis toxicity. Effects of ouabain on phase four of canine Purkinje fiber transmembrane potentials. Circulation 47:681–689
Kass RS, Tsien RW, Weingart R (1978) Ionic basis of transient inward current induced by strophanthidin in cardiac Purkinje fibres. J Physiol 281:209–226
Fedida D, Noble D, Rankin AC, Spindler AJ (1987) The arrhythmogenic transient inward current iTi and related contraction in isolated guinea-pig ventricular myocytes. J Physiol 392:523–542
Matsuda H, Noma A, Kurachi Y, Irisawa H (1982) Transient depolarization and spontaneous voltage fluctuations in isolated single cells from guinea pig ventricles. Calcium-mediated membrane potential fluctuations. Circ Res 51:142–151
Priori SG, Corr PB (1990) Mechanisms underlying early and delayed afterdepolarizations induced by catecholamines. Am J Physiol 258:H1796–H1805
Volders PG, Kulcsar A, Vos MA et al (1997) Similarities between early and delayed afterdepolarizations induced by isoproterenol in canine ventricular myocytes. Cardiovasc Res 34:348–359
Priori SG, Mantica M, Schwartz PJ (1988) Delayed afterdepolarizations elicited in vivo by left stellate ganglion stimulation. Circulation 78:178–185
Packer M, Carver JR, Rodeheffer RJ et al (1991) Effect of oral milrinone on mortality in severe chronic heart failure. The PROMISE Study Research Group. N Engl J Med 325:1468–1475
Verkerk AO, Veldkamp MW, Baartscheer A et al (2001) Ionic mechanism of delayed afterdepolarizations in ventricular cells isolated from human end-stage failing hearts. Circulation 104:2728–2733
Lubbe WF, Podzuweit T, Opie LH (1992) Potential arrhythmogenic role of cyclic adenosine monophosphate (AMP) and cytosolic calcium overload: implications for prophylactic effects of beta-blockers in myocardial infarction and proarrhythmic effects of phosphodiesterase inhibitors. J Am Coll Cardiol 19:1622–1633
Ming Z, Aronson R, Nordin C (1994) Mechanism of current-induced early afterdepolarizations in isolated guinea pig myocytes. Am J Physiol 267:H1419–H1428
Nordin C, Ming Z (1995) Computer model of current-induced early afterdepolarizations in guinea pig myocytes. Am J Physiol 268:H2440–H2459
Nordin C (1993) Computer model of membrane current and intracellular Ca2+ flux in the isolated guinea pig ventricular myocyte. Am J Physiol 265:H2117–H2136
Roden DM (2006) Long QT syndrome: reduced repolarization reserve and the genetic link. J Int Med 259:59–69
Curb JD, Rodriguez BL, Burchfiel CM, Abbott RD, Chiu D, Yano K (1995) Sudden death, impaired glucose tolerance, and diabetes in Japanese American men. Circulation 91:2591–2595
Escobedo LG, Caspersen CJ (1997) Risk factors for sudden coronary death in the United States. Epidemiology 8:175–180
Jouven X, Desnos M, Guerot C, Ducimetiere P (1999) Predicting sudden death in the population: the Paris Prospective Study I. Circulation 99:1978–1983
Wachtell K, Okin PM, Olsen MH et al (2007) Regression of electrocardiographic left ventricular hypertrophy during antihypertensive therapy and reduction in sudden cardiac death: the LIFE Study. Circulation 116:700–705
Al-Khatib SM, Shaw LK, O’Connor C, Kong M, Califf RM (2007) Incidence and predictors of sudden cardiac death in patients with diastolic heart failure. J Cardiovasc Electrophysiol 18:1231–1235
Movahed MR, Hashemzadeh M, Jamal M (2007) Increased prevalence of ventricular fibrillation in patients with type 2 diabetes mellitus. Heart Vessels 22:251–253
Festa A, D’Agostino R, Rautaharju P, Mykkanen L, Haffner SM (2000) Relation of systemic blood pressure, left ventricular mass, insulin sensitivity and coronary artery disease to QT interval duration in nondiabetic and type 2 diabetic subjects. Am J Cardiol 86:1117–1122
Dekker JM, Feskens EJ, Schouten EG, Klootwijk P, Pool J, Kromhout D (1996) QTc duration is associated with levels of insulin and glucose intolerance. The Zutphen Elderly Study. Diabetes 45:376–380
Brown DW, Giles WH, Greenlund KJ, Valdez R, Croft JB (2001) Impaired fasting glucose, diabetes mellitus, and cardiovascular disease risk factors are associated with prolonged QTc duration. Results from the Third National Health and Nutrition Examination Survey. J Cardiovasc Risk 8:227–233
Lengyel C, Virag L, Biro T et al (2007) Diabetes mellitus attenuates the repolarization reserve in mammalian heart. Cardiovasc Res 73:512–520
Veglio M, Sivieri R, Chinaglia A, Scaglione L, Cavalo-Perin P, for the Neuropathy Study Group of the Italian Society of the Study of Diabetes, Piemonte Affiliate (2000) QT interval prolongation and mortality in type 1 diabetic patients: a 5-year cohort prospective study. Diabetes Care 23:1381–1383
Naas AA, Davidson NC, Thompson C et al (1998) QT and QTc dispersion are accurate predictors of cardiac death in newly diagnosed non-insulin dependent diabetes: cohort study. BMJ 316:745–746
Christensen PK, Gall MA, Major-Pedersen A et al (2000) QTc interval length and QT dispersion as predictors of mortality in patients with non-insulin-dependent diabetes. Scand J Clin Lab Invest 60:323–332
Linnemann B, Janka HU (2003) Prolonged QTc interval and elevated heart rate identify the type 2 diabetic patient at high risk for cardiovascular death. The Bremen Diabetes Study. Exp Clin Endocrinol Diabetes 111:215–222
Ewing DJ, Boland O, Neilson JM, Cho CG, Clarke BF (1991) Autonomic neuropathy, QT interval lengthening, and unexpected deaths in male diabetic patients. Diabetologia 34:182–185
Veglio M, Bruno G, Borra M et al (2002) Prevalence of increased QT interval duration and dispersion in type 2 diabetic patients and its relationship with coronary heart disease: a population-based cohort. J Intern Med 251:317–324
Aonime M, Nobe S, Arita M (1990) The making of diabetic guinea pigs by streptozotocin and high incidence of triggered activity in the ventricular muscle. Jpn J Physiol 40:651–663
Golfman L, Dixon IM, Takeda N, Lukas A, Dakshinamurti K, Dhalla NS (1998) Cardiac sarcolemmal Na(+)–Ca2+ exchange and Na(+)–K+ ATPase activities and gene expression in alloxan-induced diabetes in rats. Mol Cell Biochem 188:91–101
Nordin C, Siri F, Aronson RS (1989) Electrophysiologic characteristics of single myoctyes isolated from hypertrophied guinea-pig hearts. J Mol Cell Cardiol 21:729–739
Yan GX, Rials SJ, Wu Y et al (2001) Ventricular hypertrophy amplifies transmural repolarization dispersion and induces early afterdepolarizations. Am J Physiol Heart Circ Physiol 281:H1968–H1975
Kenigsberg DN, Khanal S, Kowalski M, Krishnan SC (2007) Prolongation of the QTc interval is seen uniformly during early transmural ischemia. J Am Coll Cardiol 49:1299–1305
Friedman PL, Stewart JR, Fenoglio JJ, Wit AL (1973) Survival of subendocardial Purkinje fibers after extensive myocardial infarction in dogs. Circ Res 33:597–611
Beuckelmann DJ, Nabauer M, Erdmann E (1993) Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res 73:379–385
Janse MJ (2004) Electophysiological changes in heart failure and their relationship to arrhythmogenesis. Cardiovasc Res 61:208–217
Volk T, Nguyen TH, Schultz JH, Faulhaber J, Ehmke H (2001) Regional alterations of repolarizing K+ currents among the left ventricular free wall of rats with ascending aortic stenosis. J Physiol 530:443–455
Kaye DM, Lefkovits J, Jennings GL, Bergin P, Broughton A, Esler MD (1995) Adverse consequences of high sympathetic nervous activity in the failing human heart. J Am Col Cardiol 26:1257–1263
Nguyen HT, Ghevondian N, Nguyen ST, Jones TW (2007) Detection of hypoglycemic episodes in children with type 1 diabetes using an optimal Bayesian neural network algorithm. Conf Proc IEEE Eng Med Biol Soc 2007:3140–3143
Due-Anderson R, Hoi-Hansen T, Larroude CE et al (2008) Cardiac repolarization during hypoglycaemia in type 1 diabetes: impact of basal renin–angiotensin system activity. Europace 10:860–867
Gill GV, Woodward A, Casson IF, Weston PJ (2009) Cardiac arrhythmia and nocturnal hypoglycaemia in type 1 diabetes—the ‘dead in bed’ syndrome revisited. Diabetologia 52:42–45
Zhang Y, Han H, Wang J, Wang H, Yang B, Wang Z (2003) Impairment of human ether-a-go-go related gene (HERG) K+ channel function by hypoglycemia and hyperglycemia. Similar phenotypes but different mechanisms. J Biol Chem 278:10417–10426
Chvilicek JP, Hurlbert BJ, Hill GE (1995) Diuretic-induced hypokalaemia inducing torsades de pointes. Can J Anaesth 42:1137–1139
Nordin C, Kohli A, Beca S et al (2006) Importance of hepatitis C coinfection in the development of QT prolongation in HIV-infected patients. J Electrocard 39:199–205
Siscovick DS, Raghunathan TE, Psaty BM et al (1994) Diuretic therapy for hypertension and the risk of primary cardiac arrest. N Engl J Med 330:1852–1857
Bremer JP, Baron M, Peters H et al (2006) Hormonal, subjective and neurocognitive responses to brief hypoglycaemia in postmenopausal woman and age-matched men with type 2 diabetes mellitus. Metabolism 55:331–338
Hoffman RP (2007) Sympathetic mechanisms of hypoglycemic counterregulation. Curr Diabetes Rev 3:185–193
Eliot RS, Buell JC (1985) Role of emotions and stress in the genesis of sudden death. J Am Coll Cardiol 5(6 Suppl):95B–98B
Krell MJ, Kline EM, Bates ER et al (1986) Intermittent, ambulatory dobutamine infusions in patients with severe congestive heart failure. Am Heart J 112:787–791
Zipes DP, Levy MN, Cobb LA et al (1987) Sudden cardiac death. Neural–cardiac interactions. Circulation 76:I202–I207
Veldkamp MW, Verkerk AO, van Ginneken AC et al (2001) Norepinephrine induces action potential prolongation and early afterdepolarizations in ventricular myocytes isolated from human end-stage failing hearts. Eur Heart J 22:995–963
Darbar D, Smith M, Morike K, Roden DM (1996) Epinephrine-induced changes in serum potassium and cardiac repolarization and effects of pretreatment with propranolol and diltiazem. Am J Cardiol 77:1351–1355
Currie CJ, Peters JR, Tynan A et al (2010) Survival as a function of HbA1c in people with type 2 diabetes: a retrospective cohort study. Lancet 375:481–489
Duckworth W, Abraira C, Moritz T, on behalf of the VADT Investigators et al (2009) Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 360:129–139
ADVANCE Collaborative Group, Patel A, MacMahon S, Chalmers J et al (2008) Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 358:2560–2572
Inagaki M, Kawada T, Lie M, Zheng C, Sunagawa K, Sugimachi M (2005) Intravascular parasympathetic cardiac nerve stimulation prevents ventricular arrhythmias during acute myocardial ischemia. Conf Proc IEEE Eng Med Biol Soc 7:7076–7079
Yang B, Lin H, Xu C et al (2005) Choline produces cytoprotective effects against ischemic myocardial injuries: evidence for the role of cardiac M3 subtype muscarinic acetylcholine receptors. Cell Physiol Biochem 16:163–174
Zhao W, Qi H, Liu Y et al (2009) The antiarrhythmic effect and possible ionic mechanisms of pilocarpine on animal models. J Cardiovasc Pharm Ther 14:242–247
Adler GK, Bonyhay I, Failing H, Waring E, Dotson S, Freeman R (2009) Antecedent hypoglycaemia impairs autonomic cardiovascular function: implications for rigorous glycemic control. Diabetes 58:360–366
Cryer PE (2008) The barrier of hypoglycaemia in diabetes. Diabetes 57:3169–3176
Bellavere F, Ferri M, Guarini L et al (1988) Prolonged QT period in diabetic autonomic neuropathy: a possible role in sudden cardiac death? Br Heart J 59:379–383
Pearson EC, Woolsey RL (2005) QT prolongation and torsades de pointes among methadone users: reports to the FDA spontaneous reporting system. Pharmacoepidemiol Drug Saf 14:747–753
No authors listed (2008) ACCORD blood sugar strategy announcement, 6 February 6, 2008. Available from www.nhlbi.nih.gov/health/prof/heart/other/accord, accessed 8 January 2010
Skyler J, Bergenstal R, Bonow RO et al (2009) Intensive glycemic control and the prevention of cardiovascular events: implications of the ACCORD, ADVANCE and VA Diabetes trials. Diabetes Care 32:187–192
Nathan DM, Cleary PA, Backlund JY et al (2005) Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 353:2643–2653
Holman RR, Paul SK, Bethel MA et al (2008) 10-year follow up of intensive glucose control in type 2 diabetes. N Engl J Med 359:1577–1589
Straus SM, Sturkenboom MC, Bleumink GS et al (2005) Non-cardiac QTc-prolonging drugs and the risk of sudden cardiac death. Eur Heart J 26:2007–2012
Ray WA, Chung CP, Murray KT, Hall K, Stein CM (2009) Atypical antipsychotic drugs and the risk of sudden cardiac death. New Engl J Med 360:225–235
Nordin C, Aronson RS (1987) Interactions between oscillatory currents and membrane conductance in isolated cardiac ventricular myocytes: experimental description and preliminary modeling. In: Sideman S, Beyar R (eds) Activation, metabolism and perfusion of the heart. Martinus Nijhoff, Leiden, pp 151–171
Acknowledgements
No outside funding was used for the preparation of this manuscript.
Duality of interest
The author declares that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nordin, C. The case for hypoglycaemia as a proarrhythmic event: basic and clinical evidence. Diabetologia 53, 1552–1561 (2010). https://doi.org/10.1007/s00125-010-1752-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-010-1752-6