
Abstract
Aims/hypothesis
Chromosome 1q21-q24 has been shown to be linked to type 2 diabetes. The International Type 2 Diabetes 1q Consortium showed that one of the nominal associations was located in the NOS1AP gene. Although this association was not replicated in additional samples of European descent, it remains unknown whether NOS1AP plays a role in Chinese individuals.
Methods
In stage 1 analyses, 79 single nucleotide polymorphisms (SNPs) of the NOS1AP gene were successfully genotyped in a group of Shanghai Chinese individuals, comprising 1,691 type 2 diabetes patients and 1,720 control participants. In stage 2 analyses, the SNP showing the strongest association was genotyped in additional Chinese individuals, including 1,663 type 2 diabetes patients and 1,408 control participants.
Results
In stage 1 analyses, 20 SNPs were nominally associated with type 2 diabetes (p < 0.05), with SNP rs12742393 showing the strongest association (OR 1.24 [95% CI 1.11–1.38]; p = 0.0002, empirical p = 0.019). Haplotype analysis also confirmed the association between rs12742393 and type 2 diabetes. In stage 2 analyses, the difference in allele frequency distribution of rs12742393 did not reach statistical significance (p = 0.254). However, the meta-analysis showed a significant association between rs12742393 and type 2 diabetes with an OR of 1.17 (95% CI 1.07–1.26; p = 0.0005).
Conclusions/interpretation
Our data suggest that NOS1AP variants may not play a dominant role in susceptibility to type 2 diabetes, but a minor effect cannot be excluded.
Similar content being viewed by others

Introduction
With its rapidly rising incidence and prevalence in recent years, type 2 diabetes threatens to become a major global epidemic. In addition to environmental factors such as lifestyle and dietary habits, which are known to play a role in the development of this disease, familial studies, including those on twins, as well as migration and admixture studies suggest that genetic factors also contribute to the risk of type 2 diabetes [1]. In our previous genome-wide linkage studies in a Chinese population, we found that chromosome 1q21-q24 was significantly linked to type 2 diabetes [2], a finding similarly reported in other populations [3–9]. To identify susceptibility genes on chromosome 1q, the International Type 2 Diabetes 1q Consortium examined 5,290 single nucleotide polymorphisms (SNPs) across a 23 Mb region of chromosome 1q in over 3,000 individuals from eight populations, including a Chinese population in Shanghai. It found that one of the nominal associations was located in the gene NOS1AP [10]. However, this association was not replicated in additional samples of European descent [10]. In this ‘1q study’, our Shanghai group had examined 80 type 2 diabetic individuals with positive family history and 80 control participants with normal glucose tolerance and without family history of diabetes [10]. Preliminary analysis from the Shanghai 1q samples also showed a significant association between NOS1AP and type 2 diabetes (p = 0.002 for rs4657139). Given the different genetic background between European and Chinese populations, the effects of genetic variants of NOS1AP on risk of type 2 diabetes remained unknown and worthy of further exploration in a large Chinese sample.
Nitric oxide synthase 1 adaptor protein (NOS1AP) regulates the activity of neuronal nitric oxide synthase (nNOS) and consequently affects nitric oxide release mediated by N-methyl-d-aspartate receptors (NMDARs) [11]. Recent studies have shown that dysfunction of nNOS plays an important role in the development of diabetic autonomic neuropathy [12–14], nephropathy and retinopathy [15–18], and may in fact be involved in the action and secretion of insulin [19–21]. A novel mechanism for beta cell dysfunction has been recently described, namely that elevated cholesterol inhibits insulin secretion by modifying nNOS activity [22]. Moreover, two recent reports have shown that the NOS1AP SNP rs10494366 is associated with the incidence of type 2 diabetes in calcium channel blocker users, as well as with poorer prognosis in patients taking sulfonylurea [23, 24].
Based on these previous observations and initial data, we hypothesised that NOS1AP could affect susceptibility to type 2 diabetes through its genetic variations. To test this hypothesis, we genotyped 70 SNPs in a large region spanning the whole NOS1AP gene and analysed their relationship with type 2 diabetes in a large case–control Chinese population.
Methods
Participants
Upon approval by our institutional review board, we recruited 6,771 case–control participants of Han Chinese ancestry in Shanghai using a two-stage approach for this study. Each participant provided informed written consent.
In stage 1, we recruited 1,892 unrelated type 2 diabetic patients from the inpatient database of Shanghai Diabetes Institute and 1,808 unrelated controls who had been recruited from the general population by the Shanghai Diabetes Studies [25, 26]. In stage 2, we additionally recruited 1,663 type 2 diabetic patients from the Shanghai Diabetes Institute inpatient database and 1,408 controls from the general population in Shanghai. The participants recruited in stage 1 underwent screening and analysis for 70 SNPs. The participants recruited in stage 2 were analysed only for the SNP that had been shown in stage 1 to be most significantly associated with type 2 diabetes. After combining participants from the two stages, we performed an overall meta-analysis for this SNP. This two-stage approach enabled us to perform this analysis of the most significantly associated SNP on an expanded large number of study participants.
All cases of diabetes met the 1999 WHO criteria (fasting plasma glucose ≥7.0 mmol/l and/or 2-h post-challenge plasma glucose ≥11.1 mmol/l) and were treated with oral hypoglycaemic agents and/or insulin [27]. Type 1 diabetes and mitochondrial diabetes were excluded by clinical, immunological and genetic criteria. Control participants were recruited from community-based epidemiological studies of diabetes and related metabolic disorders. Blood samples were obtained at 0 and 120 min of the OGTT to measure plasma glucose levels. In the present study, the inclusion criteria for the control participants were: (1) over 40 years old; (2) normal glucose tolerance as assessed by a standard 75 g OGTT (fasting plasma glucose <6.1 mmol/l, 2 h plasma glucose <7.8 mmol/l); and (3) no family history of diabetes indicated in a standard questionnaire.
The characteristics of the study groups are shown in Table 1. Information was available from all participants on age, sex, ethnic background, residency, age at diagnosis of diabetes (for those with known diabetes) and family history of diabetes. Anthropometric measurements such as height and weight (to calculate BMI) were also obtained.
SNP selection and genotyping
In stage 1, we first selected 70 SNPs that span a 340 kb region of NOS1AP, including 68 tagging SNPs selected from the HapMap Phase II Han Chinese database (www.hapmap.org, accessed 2 April 2009) using the threshold of r 2 ≥ 0.7 and two additional synonymous SNPs. All the SNPs had a minor allele frequency over 0.05 as suggested by HapMap Chinese data. These SNPs were genotyped in stage 1 samples using primer extension of multiplex products with detection by matrix-assisted laser desorption ionisation–time of flight mass spectroscopy on an analysing device (MassARRAY Compact; Sequenom, San Diego, CA, USA). Samples were arrayed on ten 384-well plates with ten replicates and four water controls per plate. Case and control samples were distributed line by line across each 384-well plate, with nearly the same number of cases and controls per plate. Genotyping data underwent a series of quality control checks and cleared data were used in further association analyses. The key quality control requirements were: (1) sample call rates ≥70%; (2) SNP call rate ≥90%; (3) concordance rate between replicate samples ≥90%; and (4) Hardy–Weinberg equilibrium test ≥0.05 in controls and ≥0.01 in cases. Overall, 289 individuals were excluded from the sample call rate checks and 17 SNPs failed the SNP call rate check. Nine SNPs were excluded during the Hardy–Weinberg equilibrium test. Then we additionally genotyped 42 SNPs, including six SNPs that had previously failed quality control, in order to increase the coverage of common variants within the NOS1AP region. Three of these 42 failed the call rate check, while four others were excluded because of departure from Hardy–Weinberg equilibrium. In total, 79 SNPs entered the final analysis. These 79 SNPs captured 95% and 92% of common variants in the NOS1AP gene region under the thresholds of r 2 = 0.5 and r 2 = 0.7 respectively. The average call rate for these SNPs was 97.1%, with an average concordance rate based on 100 duplicate comparisons for each SNP of 99.3%. Detailed quality control information on the SNPs is shown in Electronic supplementary material (ESM) Table 1.
In stage 2, only SNP rs12742393, which was shown to be most significantly associated with type 2 diabetes in the stage 1 analysis, was genotyped in the additionally recruited participants by PCR-restriction fragment length polymorphism. The sequences of primers were 5′-GGT GAA TGT GTA CAA AGG AGA AGG-3′ and 5′-CAA ACT GAA ATG GAC CAC AAA GAG-3′. The amplified PCR products were digested by BsrI restriction endonuclease (New England Biolabs, Boston, MA, USA) at 65°C for 2 h, followed by electrophoresis on a 12% polyacrylamide gel and ethidium bromide staining. Direct sequencing, which was performed using a genetic analyser (Prism 3100; Applied Biosystems, Foster City, CA, USA), was used to ensure accuracy of genotyping. No discrepancy was found in 48 randomly selected samples genotyped by all three approaches.
Statistical analyses
The observed genotypes were tested for fit to the expectation of Hardy–Weinberg equilibrium using χ 2 test. Allele and genotype frequencies of SNPs were compared between cases and controls using χ 2 test or Fisher’s exact test. ORs with 95% CI were obtained for each SNP. Pairwise linkage disequilibrium (LD) including |D′| and r 2 were estimated from the combined data of cases and controls using Haploview (version 4.1) [28]. Sliding windows consisting of two or three adjacent SNPs were generated for haplotype analyses using PLINK (version 1.05) [29]. Correction of multiple testing on allele association was performed using Haploview (version 4.1) through 10,000 permutations that randomly permutated the case/control status independently of genotypes. Correction of multiple testing on genotype association after correcting confounders was performed using PLINK through 10,000 permutations. The imputation of un-genotyped SNPs in the region was based on genotypes that had passed quality control procedures in our samples and on haplotype structure of the HapMap Han Chinese in Beijing, China (CHB) and Japanese in Tokyo (JPT) samples using IMPUTE [30]. Imputed SNPs with proper_info <0.4, posterior probability score <0.9 or missing data proportion >0.15 were removed. The associations between imputed SNPs were determined by SNPTEST [30] under an additive genetic model that takes into account the degree of uncertainty of genotype imputation by predicted allele counts based on estimated probabilities. Test for homogeneity was assessed by the Breslow–Day test. Combined ORs from different cohorts were calculated using Comprehensive Meta Analysis (version 2, Englewood, NJ, USA).
Considering a risk allele frequency of 20% and an additive model, the combined cohort had ∼80% power to detect a minimal OR of 1.17 at a significance level of α = 0.001.
Results
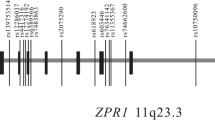
In stage 1, 79 SNPs were successfully genotyped in 1,691 type 2 diabetic patients and 1,720 controls. The LD structure of this region is shown in Fig. 1. The allele frequencies and single SNP associations of all 79 SNPs are shown in ESM Table 2. As shown in Table 2, 20 individual SNPs reached nominal significance by comparing allele frequency distributions in cases and controls (p < 0.05). Among these, the strongest nominal association was observed at rs12742393, which was in low LD (r 2 = 0.08 as suggested by HapMap Chinese data) with and 133 kb away from the SNP reported by the 1q Consortium in European samples, with the C allele showing significant risk for type 2 diabetes (OR 1.24 [95% CI 1.11–1.38], p = 0.0002). The result remained significant after correction for multiple testing by 10,000 permutations (empirical p = 0.019). As age, sex and BMI were significantly different between the case and control participants, adjustment for these confounding factors was performed. The results revealed that diabetes risk association with rs12742393 remained significant (OR 1.22 [95% CI 1.09–1.37], p = 0.0008, empirical p = 0.047).

LD plots for SNPs genotyped in the NOS1AP locus in the stage 1 sample. Location of the exons, including translated and untranslated regions, and introns (line) are shown above the LD plot. Blocks were defined using the CI algorithm. a Shades of grey indicate the strength of pairwise LD based on |D′|. b Shades of grey indicate the strength of pairwise LD based on r 2
For the sliding windows-based haplotype analysis, haplotype TC formed by rs4531272–rs10918974 and TCC formed by rs4531272–rs10918974–rs7522678 were associated with an increased risk of type 2 diabetes after adjustment for multiple testing (p = 5.0E−5 and 5.6E−5, respectively). As these two haplotypes were highly correlated with rs12742393 risk allele C in a manner analogous to LD between individual SNPs (r 2 = 0.944 and 0.974, respectively), the observed association merely confirmed the association between rs12742393 and type 2 diabetes (ESM Fig. 1). As the SNPs rs4531272, rs10918974 and rs7522678, which formed an associated haplotype, resided in a six SNP block together with rs12742393 (Fig. 1), we analysed the association between haplotypes consisting of all these six SNPs and type 2 diabetes. However, no haplotype showed stronger association than rs12742393.
Imputation within the NOS1AP region allowed us to investigate the effects of 137 additional SNPs on type 2 diabetes. We found that multiple SNPs around rs12742393 also showed nominally significant association signals, supporting our finding on the genotyped SNPs (p < 0.05; Fig. 2). However, no SNP showed a stronger association than rs12742393.

Association between both genotyped and imputed SNPs, and type 2 diabetes in the NOS1AP region. Genomic position as shown is based on NCBI Build 37.1 (www.ncbi.nlm.nih.gov/projects/mapview/map_search.cgi?taxid=9606, accessed 3 August 2009). a Genomic location of genes showing intron and exon structure. b Recombination rates, given as cM/Mb. c Plot of −log(p) values for type 2 diabetes under an additive genetic model against chromosome position. Black triangles, SNPs genotyped in the study; white circles, SNPs whose genotypes were imputed. The arrow indicates the position of SNP reported by the International Type 2 Diabetes 1q Consortium
To further investigate the relationship of the SNP rs12742393 with type 2 diabetes, we extended the analysis to an expanded pool of type 2 diabetes case and control participants recruited in stage 2 of the study as described. The allele distribution of rs12742393 was not significantly different between type 2 diabetes patients and controls in stage 2 samples (OR 1.07, 95% CI 0.95–1.21, p = 0.254). We performed a heterogeneity test on the two stage cohorts and found them to be homogenous. We were therefore able to perform a meta-analysis using the fixed effects model. This meta-analysis including 6,469 participants showed a significant association of the SNP rs12742393 with type 2 diabetes, with an OR of 1.17 (95% CI 1.07–1.26, p = 0.0005; Table 3).
Discussion
Considerable data from linkage studies suggest the existence of multiple susceptibility gene(s) for type 2 diabetes at chromosome 1q21-q24 in various populations [2–9]. In fact, several specific genes in this region, such as LMNA, ATF6 and DUSP12, were identified as possibly conferring risk of diabetes in some populations [31–34]. Meta-analysis of the eight populations of the International Type 2 Diabetes 1q Consortium, as well as analysis of the Shanghai Chinese population of this consortium suggested that NOS1AP might also be a susceptibility gene for type 2 diabetes [10]. Although the 1q Consortium failed to replicate this in additional European samples [10], we undertook the present study to explore the relationship between specific genetic variants of the NOS1AP gene and type 2 diabetes in Chinese.
The human NOS1AP gene is ∼300 kb long, with ten exons. NOS1AP is abundant in neuronal tissues, as well as in left ventricular heart tissue and muscle tissues [11, 35, 36]. It has been documented that NOS1AP can compete with postsynaptic density protein 95 by interaction with the C-terminal PDZ domain of nNOS, inhibiting activation of nNOS stimulated by NMDAR and reducing local nitric oxide release [11, 37]. As an adapter protein, NOS1AP can also link nNOS to target proteins, including dexamethasone-induced ras-related protein 1 and synapsins through respective ternary complexes [38, 39]. Certain genetic variants of NOS1AP have been associated with prolonged electrocardiographic QT interval and schizophrenia [35, 40–42]. In particular, a functional study found that rs12742393, which was identified to be associated with type 2 diabetes in the present study, may be a schizophrenia-associated functional variant [42]. Evidence also suggests that nNOS, the target of NOS1AP, might play a role in the development and pathogenesis of diabetes. For example, in nNos knockout mice, insulin sensitivity measured by the clamp technique was reduced [21]. Intracerebroventricular administration of an inhibitor of NOS affected insulin secretion and insulin sensitivity [20]. Another study showed that nNOS dimerisation could affect glucose-stimulated insulin secretion through its effect on glucokinase activity and translocation at high cellular cholesterol levels [22].
Our data do not support a major effect of NOS1AP variants on type 2 diabetes risk. However, our meta-analysis showed evidence that rs12742393 does affect type 2 diabetes susceptibility in Chinese. Recently, International Type 2 Diabetes 1q Consortium reported that they failed to replicate the original association on NOS1AP in additional European samples [10]. We cannot exclude the possibility that the original association on NOS1AP was a false positive result, as two studies failed to replicate it in sufficiently powered samples [10, 43]. It should, however, be noted that both replication studies were done in whites. The effects of NOS1AP variants in other populations remain to be unknown. On the other hand, a functional study suggested that rs12742393 plays a functional role by affecting transcription factor binding and NOS1AP gene expression [42]. We also found that NOS1AP is expressed in pancreatic beta cell line MIN6 (data not shown). But considering that NOS1AP SNP rs10494366 was only shown to affect type 2 diabetes incidence in calcium channel blocker users [24] and the lack of replication of a reported association with type 2 diabetes in additional samples, including the current study and previously reported papers [10, 43], we speculate that NOS1AP may have a small effect on disease susceptibility through interaction with certain, but currently unknown environmental factors.
One limitation of the present study is that the SNPs genotyped cannot tag all common variants in the region and the causal marker(s) may not have been directly genotyped or tagged. We captured over 90% of the common variants and, using imputation, predicted the effects of ungenotyped SNPs. However, such imputed genotypes are based on directly genotyped SNPs and do not indicate independent replication. Thus the possibility still exists that some other important SNPs may have been missed. We also cannot exclude the possibility that other genes in the linkage region are associated with type 2 diabetes in Chinese, even though the International Type 2 Diabetes 1q Consortium failed to identify common variants contributing to the disease in whites [10]. Moreover, we could not entirely exclude that population stratification in the cases and controls was a potential source of bias and incorrect inferences in genotype–disease association. However, the effect of population stratification may be limited in the current study, as the cases and controls were recruited from the same geographic area with the same ancestry.
In summary, we have examined common variants in the NOS1AP gene for their association with type 2 diabetes in a large Chinese cohort. Our data suggest that NOS1AP may not play a major role in susceptibility to type 2 diabetes, but a minor effect cannot be excluded.
Abbreviations
- LD:
-
Linkage disequilibrium
- NMDAR:
-
N-methyl-d-aspartate receptors
- nNOS:
-
Neuronal nitric oxide synthase
- NOS1AP:
-
Nitric oxide synthase 1 adaptor protein
- SNP:
-
Single nucleotide polymorphism
References
Gloyn AL, McCarthy MI (2001) The genetics of type 2 diabetes. Best Pract Res Clin Endocrinol Metab 15:293–308
Xiang K, Wang Y, Zheng T et al (2004) Genome-wide search for type 2 diabetes/impaired glucose homeostasis susceptibility genes in the Chinese: significant linkage to chromosome 6q21-q23 and chromosome 1q21-q24. Diabetes 53:228–234
Elbein SC, Hoffman MD, Teng K, Leppert MF, Hasstedt SJ (1999) A genome-wide search for type 2 diabetes susceptibility genes in Utah Caucasians. Diabetes 48:1175–1182
Hanson RL, Ehm MG, Pettitt DJ et al (1998) An autosomal genomic scan for loci linked to type II diabetes mellitus and body-mass index in Pima Indians. Am J Hum Genet 63:1130–1138
Hsueh WC, St Jean PL, Mitchell BD et al (2003) Genome-wide and fine-mapping linkage studies of type 2 diabetes and glucose traits in the Old Order Amish: evidence for a new diabetes locus on chromosome 14q11 and confirmation of a locus on chromosome 1q21-q24. Diabetes 52:550–557
Langefeld CD, Wagenknecht LE, Rotter JI et al (2004) Linkage of the metabolic syndrome to 1q23-q31 in Hispanic families: the Insulin Resistance Atherosclerosis Study Family Study. Diabetes 53:1170–1174
Ng MC, So WY, Cox NJ et al (2004) Genome-wide scan for type 2 diabetes loci in Hong Kong Chinese and confirmation of a susceptibility locus on chromosome 1q21-q25. Diabetes 53:1609–1613
Vionnet N, Hani EH, Dupont S et al (2000) Genomewide search for type 2 diabetes-susceptibility genes in French whites: evidence for a novel susceptibility locus for early-onset diabetes on chromosome 3q27-qter and independent replication of a type 2-diabetes locus on chromosome 1q21-q24. Am J Hum Genet 67:1470–1480
Wiltshire S, Hattersley AT, Hitman GA et al (2001) A genomewide scan for loci predisposing to type 2 diabetes in a U.K. population (the Diabetes UK Warren 2 Repository): analysis of 573 pedigrees provides independent replication of a susceptibility locus on chromosome 1q. Am J Hum Genet 69:553–569
Prokopenko I, Zeggini E, Hanson RL et al (2009) Linkage disequilibrium mapping of the replicated type 2 diabetes linkage signal on chromosome 1q. Diabetes 58:1704–1709
Jaffrey SR, Snowman AM, Eliasson MJ, Cohen NA, Snyder SH (1998) CAPON: a protein associated with neuronal nitric oxide synthase that regulates its interactions with PSD95. Neuron 20:115–124
Cellek S, Foxwell NA, Moncada S (2003) Two phases of nitrergic neuropathy in streptozotocin-induced diabetic rats. Diabetes 52:2353–2362
Iwasaki H, Kajimura M, Osawa S et al (2006) A deficiency of gastric interstitial cells of Cajal accompanied by decreased expression of neuronal nitric oxide synthase and substance P in patients with type 2 diabetes mellitus. J Gastroenterol 41:1076–1087
Zheng H, Bidasee KR, Mayhan WG, Patel KP (2007) Lack of central nitric oxide triggers erectile dysfunction in diabetes. Am J Physiol Regul Integr Comp Physiol 292:R1158–R1164
Erdely A, Freshour G, Maddox DA, Olson JL, Samsell L, Baylis C (2004) Renal disease in rats with type 2 diabetes is associated with decreased renal nitric oxide production. Diabetologia 47:1672–1676
Koo JR, Vaziri ND (2003) Effects of diabetes, insulin and antioxidants on NO synthase abundance and NO interaction with reactive oxygen species. Kidney Int 63:195–201
Park JW, Park SJ, Park SH et al (2006) Up-regulated expression of neuronal nitric oxide synthase in experimental diabetic retina. Neurobiol Dis 21:43–49
Toda N, Nakanishi-Toda M (2007) Nitric oxide: ocular blood flow, glaucoma, and diabetic retinopathy. Prog Retin Eye Res 26:205–238
Rizzo MA, Piston DW (2003) Regulation of beta cell glucokinase by S-nitrosylation and association with nitric oxide synthase. J Cell Biol 161:243–248
Shankar R, Zhu JS, Ladd B, Henry D, Shen HQ, Baron AD (1998) Central nervous system nitric oxide synthase activity regulates insulin secretion and insulin action. J Clin Invest 102:1403–1412
Shankar RR, Wu Y, Shen HQ, Zhu JS, Baron AD (2000) Mice with gene disruption of both endothelial and neuronal nitric oxide synthase exhibit insulin resistance. Diabetes 49:684–687
Hao M, Head WS, Gunawardana SC, Hasty AH, Piston DW (2007) Direct effect of cholesterol on insulin secretion: a novel mechanism for pancreatic beta-cell dysfunction. Diabetes 56:2328–2338
Becker ML, Aarnoudse AJ, Newton-Cheh C et al (2008) Common variation in the NOS1AP gene is associated with reduced glucose-lowering effect and with increased mortality in users of sulfonylurea. Pharmacogenet Genomics 18:591–597
Becker ML, Visser LE, Newton-Cheh C et al (2008) Genetic variation in the NOS1AP gene is associated with the incidence of diabetes mellitus in users of calcium channel blockers. Diabetologia 51:2138–2140
Hu C, Zhang R, Wang C et al (2009) A genetic variant of G6PC2 is associated with type 2 diabetes and fasting plasma glucose level in the Chinese population. Diabetologia 52:451–456
Jia WP, Pang C, Chen L et al (2007) Epidemiological characteristics of diabetes mellitus and impaired glucose regulation in a Chinese adult population: the Shanghai Diabetes Studies, a cross-sectional 3-year follow-up study in Shanghai urban communities. Diabetologia 50:286–292
Alberti KG, Zimmet PZ (1998) Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med 15:539–553
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–265
Purcell S, Neale B, Todd-Brown K et al (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Marchini J, Howie B, Myers S, McVean G, Donnelly P (2007) A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 39:906–913
Chu WS, Das SK, Wang H et al (2007) Activating transcription factor 6 (ATF6) sequence polymorphisms in type 2 diabetes and pre-diabetic traits. Diabetes 56:856–862
Das SK, Chu WS, Hale TC et al (2006) Polymorphisms in the glucokinase-associated, dual-specificity phosphatase 12 (DUSP12) gene under chromosome 1q21 linkage peak are associated with type 2 diabetes. Diabetes 55:2631–2639
Meex SJ, van Greevenbroek MM, Ayoubi TA et al (2007) Activating transcription factor 6 polymorphisms and haplotypes are associated with impaired glucose homeostasis and type 2 diabetes in Dutch Caucasians. J Clin Endocrinol Metab 92:2720–2725
Wegner L, Andersen G, Sparso T et al (2007) Common variation in LMNA increases susceptibility to type 2 diabetes and associates with elevated fasting glycemia and estimates of body fat and height in the general population: studies of 7, 495 Danish whites. Diabetes 56:694–698
Arking DE, Pfeufer A, Post W et al (2006) A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet 38:644–651
Segalat L, Grisoni K, Archer J, Vargas C, Bertrand A, Anderson JE (2005) CAPON expression in skeletal muscle is regulated by position, repair, NOS activity, and dystrophy. Exp Cell Res 302:170–179
Aarts M, Liu Y, Liu L et al (2002) Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science 298:846–850
Fang M, Jaffrey SR, Sawa A, Ye K, Luo X, Snyder SH (2000) Dexras1: a G protein specifically coupled to neuronal nitric oxide synthase via CAPON. Neuron 28:183–193
Jaffrey SR, Benfenati F, Snowman AM, Czernik AJ, Snyder SH (2002) Neuronal nitric-oxide synthase localization mediated by a ternary complex with synapsin and CAPON. Proc Natl Acad Sci U S A 99:3199–3204
Aarnoudse AJ, Newton-Cheh C, de Bakker PI et al (2007) Common NOS1AP variants are associated with a prolonged QTc interval in the Rotterdam Study. Circulation 116:10–16
Lehtinen AB, Newton-Cheh C, Ziegler JT et al (2008) Association of NOS1AP genetic variants with QT interval duration in families from the Diabetes Heart Study. Diabetes 57:1108–1114
Wratten NS, Memoli H, Huang Y et al (2009) Identification of a schizophrenia-associated functional noncoding variant in NOS1AP. Am J Psychiatry 166:434–441
Andreasen CH, Mogensen MS, Borch-Johnsen K et al (2008) Lack of association between PKLR rs3020781 and NOS1AP rs7538490 and type 2 diabetes, overweight, obesity and related metabolic phenotypes in a Danish large-scale study: case–control studies and analyses of quantitative traits. BMC Med Genet 9:118
Acknowledgements
This work was supported by grants from the Project of National Natural Science Foundation of China (NSFC; 30630061 and 30800617), the Innovation Fund for PhD Students (BXJ0732) from Shanghai Jiao Tong University School of Medicine, National 973 Program (2006CB503901), National 863 Program (2006AA02A409), Shanghai Rising-Star Program (09QA1404400) and Shanghai Key Laboratory of Diabetes Mellitus (08DZ2230200). We would like to thank all the participants of this research. We particularly thank Q. Fang, J. Xu, X. Li, X. Hou, J. Lu, J. Tang, X. Pan and J. Li for their outstanding technical assistance. We also thank all the nursing and medical staff at Shanghai Clinical Center for Diabetes for their dedication and superb professionalism during this study.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
C. Hu and C. Wang contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
(PDF 137 kb)
ESM Table 2
(PDF 136 kb)
ESM Fig. 1
(PDF 91 kb)
Rights and permissions
About this article
Cite this article
Hu, C., Wang, C., Zhang, R. et al. Association of genetic variants of NOS1AP with type 2 diabetes in a Chinese population. Diabetologia 53, 290–298 (2010). https://doi.org/10.1007/s00125-009-1594-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-009-1594-2