
Abstract
Aims/hypothesis
Islet transplantation is a potential cure for diabetes; however, rates of graft failure remain high. The aim of the present study was to determine whether amyloid deposition is associated with reduced beta cell volume in islet grafts and the recurrence of hyperglycaemia following islet transplantation.
Methods
We transplanted a streptozotocin-induced mouse model of diabetes with 100 islets from human IAPP (which encodes islet amyloid polypeptide) transgenic mice that have the propensity to form islet amyloid (n = 8–12) or from non-transgenic mice that do not develop amyloid (n = 6–10) in sets of studies that lasted 1 or 6 weeks.
Results
Plasma glucose levels before and for 1 week after transplantation were similar in mice that received transgenic or non-transgenic islets, and at that time amyloid was detected in all transgenic grafts and, as expected, in none of the non-transgenic grafts. However, over the 6 weeks following transplantation, plasma glucose levels increased in transgenic but remained stable in non-transgenic islet graft recipients (p < 0.05). At 6 weeks, amyloid was present in 92% of the transgenic grafts and in none of the non-transgenic grafts. Beta cell volume was reduced by 30% (p < 0.05), beta cell apoptosis was twofold higher (p < 0.05), and beta cell replication was reduced by 50% (p < 0.001) in transgenic vs non-transgenic grafts. In summary, amyloid deposition in islet grafts occurs prior to the recurrence of hyperglycaemia and its accumulation over time is associated with beta cell loss.
Conclusions/interpretation
Islet amyloid formation may explain, in part, the non-immune loss of beta cells and recurrence of hyperglycaemia following clinical islet transplantation.
Similar content being viewed by others


Introduction
Islet transplantation is a potential treatment for diabetes [1]. Despite the initial promise of a high success rate of insulin independence with the Edmonton protocol of steroid-free immunosuppression [2], a recent follow-up study reported that only 10% of alloislet transplant recipients remained insulin-independent 5 years after transplantation [3]. The decrease in islet graft function cannot be explained solely by immune mechanisms, as a decrease in graft function has been demonstrated in autoislet transplant recipients [4]. Thus, non-immune factors are a critical component of long-term islet graft failure. Islet amyloid may be one of these non-immune factors.
Amyloid has been shown to form in human islets as early as 2 weeks following transplantation into nude mice [5, 6] and has recently been observed in a case in which a sample of transplanted islets was obtained from a human patient [7]. However, these studies using human islets did not determine whether amyloid deposition is associated with beta cell loss in transplanted islets and the recurrence of hyperglycaemia [8]. Thus, the answer to this critical question remains unknown.
Islet amyloid, which is frequently observed in patients with type 2 diabetes but less frequently in non-diabetic individuals [9–11], is associated with the loss of beta cells [12, 13]. The unique amyloidogenic constituent of islet amyloid is the peptide islet amyloid polypeptide (IAPP) or amylin [14, 15], which is a normal secretory product of the beta cell [16]. Human IAPP can aggregate to form fibrils and, eventually, amyloid deposits. While human IAPP-derived amyloid fibrils have been demonstrated to reduce cell viability and induce beta cell death in vitro [17, 18], other studies have suggested human IAPP oligomers to be mediators of cell cytotoxicity [19–21].
While it would be ideal to use human islets to address questions regarding non-immune factors in islet transplantation, their availability is limited. Furthermore, if human islets were used to study the consequences of amyloid formation on transplantation outcomes there would, by definition, be no control islets without the propensity to form amyloid, making it impossible to allow attribution of differences in outcomes to amyloid formation. On the other hand, while rodent islets are plentiful, they never form amyloid, as rodent islet amyloid polypeptide is not amyloidogenic [22]. Thus, we and others have developed human IAPP transgenic mice to study islet amyloid [23–26]. With our mouse model, we have observed in vivo islet amyloid deposits that are morphologically identical to those in humans [27] and result in the loss of beta cells and impaired insulin secretion [28].
We hypothesised that amyloid deposition in transplanted islets contributes to islet graft failure and thus recurrence of hyperglycaemia. To address this hypothesis, we undertook this study in which we transplanted human IAPP transgenic or non-transgenic islets (as controls that lack the propensity to develop amyloid) into a syngeneic mouse model of diabetes induced by streptozotocin and followed them for 1 or 6 weeks to address the following specific questions: Does amyloid form in islet grafts following islet transplantation and, if so, is this associated with the recurrence of hyperglycaemia and a reduction in graft beta cell volume? Does amyloid form prior to the recurrence of hyperglycaemia? If beta cell volume is decreased with amyloid formation, is this associated with an increase in beta cell apoptosis and/or a decrease in beta cell replication?
Methods
Animals
Islet donors were 8–10-week-old hemizygous transgenic mice producing human IAPP in their pancreatic islet beta cells [25] and non-transgenic littermates (F1 C57BL/6 × DBA/2J). Mice for breeding were obtained from a commercial source (Jackson, Bar Harbor, ME, USA) and all breeding was done locally. All islet recipients were syngeneic non-transgenic male mice (F1 C57BL/6 × DBA/2J), rendered diabetic at 7–9 weeks of age by a single intra-peritoneal injection of streptozotocin (Sigma, St Louis, MO, USA; 200 mg/kg body weight) freshly dissolved in citrate buffer (pH 4.5). Mice were fed a moderate-fat diet containing 9% fat (wt/wt; no. 5058; PicoLab, Brentwood, MO, USA), which we have previously shown to be permissive for islet amyloid formation [27]. Mice had free access to food and water. The study was approved by the Institutional Animal Care and Use Committee at the VA Puget Sound Health Care System.
Islet isolation and transplantation
Islets for transplantation were isolated from 8–10-week-old human IAPP transgenic and non-transgenic mice as described previously [29]. Briefly, following anaesthesia with pentobarbital (100 mg/kg i.p.), mice were killed by cervical dislocation. Collagenase P (0.5 mg/ml in RPMI; Roche Applied Sciences, Indianapolis, IN, USA) was injected into the pancreas via the common bile duct. The pancreas was then removed and incubated in a water bath for 15 min at 37°C. Pancreas tissue was disrupted by manual shaking for 1 min. Islets were purified by Histopaque-1077 (Sigma) density centrifugation (400×g for 10 min) and washed twice in RPMI medium before being handpicked. These islets were cultured in RPMI medium containing 11.1 mmol/l glucose at 37°C, 5% CO2 for 90 min, after which they were placed into PE50 polyethylene tubing using a Hamilton syringe (Fisher Scientific, Pittsburgh, PA, USA) for centrifugation (145×g for 3 min) to form a pellet for transplantation.
Mice with plasma glucose levels ≥22.2 mmol/l 5 days after streptozotocin treatment were used as islet transplant recipients. Mice were anaesthetised with sodium pentobarbital (100 mg/kg i.p.), and then the left kidney was exposed by a small lumbar incision. The tip of the PE50 tubing was placed into a hole made in the kidney capsule, the islets were injected under the capsule and the hole sealed by cauterisation. The peritoneum and muscle were sutured in layers; the skin incision was closed with staples and the animals made their post-operative recovery under a warm lamp. Thus, 100 human IAPP transgenic or non-transgenic islets were transplanted under the left kidney capsule through PE50 tubing, and islet loss was <5% in all mice (according to manual counting).
Study design—1 week and 6 week studies
Mice were followed for 1 or 6 weeks following transplantation. Plasma glucose levels and body weight were measured 8 days (the day of streptozotocin injection) before, 2 days before and on the day of islet transplantation in both studies. Following transplantation, plasma glucose levels and body weight were measured every 2–3 days until the mice were killed. Plasma glucose levels were determined using a glucose oxidase method on non-fasting blood samples obtained from the lateral saphenous vein between 12:00 and 17:00 hours.
In the 1 week study, mice were anaesthetised 1 week after transplantation and the graft-bearing kidney removed. These mice were monitored for another week before they were killed. For the 6 week study, mice were anaesthetised on day 42, the graft-bearing kidney was removed and the mice killed immediately.
Characterisation of islet graft morphology: amyloid severity, beta cell volume, beta cell apoptosis and beta cell replication rates
After nephrectomy, the islet graft was fixed in phosphate-buffered paraformaldehyde (4% wt/vol., pH 7.4) and embedded in paraffin. The entire islet graft was cut into 5 μm sections. Sections were examined for islet amyloid following thioflavin S staining (0.5% wt/vol. in water) and for beta cells using insulin immunostaining with an anti-insulin antibody (1:2,000 dilution; Sigma) followed by Cy-3-conjugated secondary antisera (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Graft sections were co-stained with TUNEL (in situ cell death detection kit, Roche Applied Sciences) and insulin or the nuclear proliferative marker Ki-67 (1:50 dilution; Dako, Carpinteria, CA, USA) and insulin to detect apoptotic or replicating beta cells, respectively. All sections were counterstained with Hoechst 33258 (2 μg/ml; Sigma) to identify cell nuclei. Islet graft area, insulin positive area and amyloid area were determined using a computer-based quantitative system [30]. Amyloid severity was calculated as: Σ amyloid area (μm2)/Σ islet graft area (μm2) × 100 (to give values as percentages). Beta cell volume (μm3) was determined as the product of beta cell area and the interval between selected sections (Σ beta cell area [μm2] × 50 [μm]). Apoptotic beta cells were identified by manual counting of insulin-positive cells with TUNEL-positive nuclei; replicating beta cells were similarly identified as insulin-positive cells with Ki-67-positive nuclei. The total number of islet graft cells was determined using the same approach we have previously used to calculate total islet cell number [31]. At least 1,200 cells were counted per graft. We have previously shown that the proportion of apoptotic and replicating beta cells calculated per total number of beta cells are highly correlated with the respective proportions expressed per total number of islet cells in pancreas sections [31]. Therefore, beta cell apoptotic and replication rates are reported as a percentage of total islet graft cell number. All histological assessments were performed in a blinded manner.
Characterisation of islets before transplantation and in control pancreases
A piece of pancreas from each mouse used as an islet donor was excised prior to islet isolation, fixed and analysed to ensure amyloid was not present prior to transplantation. Amyloid was not detected in pancreases from any of the human IAPP transgenic or non-transgenic donor mice. Similarly, no amyloid was detected in pancreases from 14–16-week-old, non-transplanted, non-diabetic, human IAPP transgenic mice that were age-matched to transplant recipients at the end of the study. Finally, the volume of beta cells in samples of 100 isolated islets (transgenic n = 5, non-transgenic n = 5) from 8–10-week-old mice that were age-matched to donors was not different between the two groups (data not shown).
Data analysis and statistical methods
Data are expressed as mean ± SEM. Comparisons between two groups were performed using the Mann–Whitney U test. Correlation analysis was performed to determine Pearson’s correlation coefficient. The presence and severity of amyloid deposition between the two groups were compared using Fisher’s exact probability test. Plasma glucose over time was evaluated by repeated-measures ANOVA. Comparison of graft failure as defined by recurrence of hyperglycaemia was performed by Kaplan–Meier analysis. A p value of less than 0.05 was considered significant.
Results
Plasma glucose levels pre and post islet transplantation
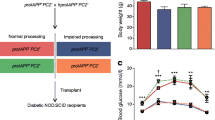
In animals that were followed for 1 week following transplantation of 100 islets, non-fasting plasma glucose levels before streptozotocin injection were not different between mice that received human IAPP transgenic islets (11.3 ± 0.6 mmol/l) and those that received non-transgenic islets (12.5 ± 0.7 mmol/l). Similarly, plasma glucose levels before islet transplantation (transgenic islet recipients 31.8 ± 1.9, non-transgenic islet recipients 34.7 ± 1.3 mmol/l) did not differ between groups (Fig. 1a). Transplantation was associated with a similar decrease in non-fasting plasma glucose levels in both groups of mice by day 2 and they remained similar in both groups 1 week post transplantation (Fig. 1a). At that time, nephrectomy of the graft-bearing kidney resulted in the recurrence of diabetes as measured 1 week post nephrectomy in all islet graft recipients (transgenic 25.4 ± 2.2, non-transgenic 28.7 ± 3.6 mmol/l). Plasma glucose levels did not differ between the two groups at any time point.

Plasma glucose levels (a, b) and body weight (c, d) before and after streptozotocin-induced diabetes and after islet transplantation in mice that received 100 non-transgenic islets or 100 human IAPP transgenic islets and were followed for 1 week (a, c) (non-transgenic, open circles, n = 6; transgenic, solid circles, n = 8) or for 6 weeks following islet transplantation (b, d) (non-transgenic, open squares, n = 10; transgenic, solid squares, n = 12). Plasma glucose was similar in both studies for the first week after transplantation (a, b) and then increased gradually over 6 weeks following islet transplantation in mice that received transgenic islets (b). Body weight was similar between mice receiving transgenic or non-transgenic islets in both studies (c, d). *p<0.05 for mice transplanted with transgenic vs non-transgenic islets
For animals followed for 6 weeks, the patterns of plasma glucose levels during the first week were similar to those in the 1 week study. Non-fasting plasma glucose prior to streptozotocin (transgenic 12.7 ± 0.4; non-transgenic 12.9 ± 0.4 mmol/l), prior to transplantation (transgenic 27.8 ± 0.8, non-transgenic 28.5 ± 1.0 mmol/l) and 2 days after transplantation (transgenic 11.6 ± 0.9, non-transgenic 12.1 ± 1.3 mmol/l) did not differ between the two groups (Fig. 1b). After that, plasma glucose levels in both groups rose slightly and similarly until 12 days after transplantation (transgenic 14.6 ± 1.0, non-transgenic 14.1 ± 1.0 mmol/l). However, thereafter, plasma glucose levels in the group of mice transplanted with transgenic islets continued to rise over time, reaching 16.2 ± 1.7 mmol/l 6 weeks after transplantation. In contrast, in the mice that received non-transgenic islets, plasma glucose levels decreased slightly and remained stable, being 11.9 ± 0.6 mmol/l at 6 weeks post transplantation. These differences in plasma glucose profiles over time meant that this measure was significantly greater in mice transplanted with transgenic islets from 29–40 days after transplantation (p < 0.05).
Body weight pre and post islet transplantation
In the 1 week study, prior to the injection of streptozotocin, body weight was similar in mice that subsequently received human IAPP transgenic islets (34.1 ± 1.4 g) and in those that received non-transgenic islets (32.1 ± 1.2 g; Fig. 1c). Similarly, for animals that were followed for 6 weeks, prior to the induction of diabetes, body weight was not different between groups of mice that received either human IAPP transgenic islets (33.9 ± 0.9 g) or non-transgenic islets (33.5 ± 0.8 g) (Fig. 1d). In the 1 week study, body weight decreased rapidly in both groups after streptozotocin injection and continued to do so up until the end of the first week after islet transplantation (Fig. 1c). In the mice followed for 6 weeks, body weight decreased similarly after streptozotocin injection and for 1 week after transplantation (transgenic islet recipients 29 ± 0.7, non-transgenic islet recipients 29 ± 0.7 g). Thereafter, body weight increased gradually in both groups until the mice were killed 6 weeks after transplantation (transgenic islet recipients 30.1 ± 0.7, non-transgenic islet recipients 30.9 ± 0.7 g). Body weight did not differ between groups at any time (Fig. 1d).
Histological assessment of amyloid formation, beta cell volume and rates of beta cell apoptosis and beta cell replication in islet grafts
Amyloid was present at 1 week after transplantation in all transgenic grafts, with a severity of 0.2 ± 0.06%, while as expected, no amyloid was detected in any of the non-transgenic grafts (Figs 2a, c and 3a). Beta cell volume 1 week post-transplantation did not differ significantly between groups (transgenic 3.7 ± 0.8 × 107, non-transgenic 5.9 ± 1.4 × 107 µm3; p = 0.20; Fig. 3c). At the same time point, there were no differences between transgenic and non-transgenic islet grafts with respect to either beta cell apoptosis rates (transgenic 0.20 ± 0.03%, non-transgenic 0.12 ± 0.04%; p = 0.10; Fig. 3e) or beta cell replication rates (transgenic 0.7 ± 0.11%, non-transgenic 0.9 ± 0.11%; p = 0.22; Fig. 3g).

Representative sections of transplanted islet grafts under the kidney capsule stained for amyloid with thioflavin S (green) and insulin (red; original magnification ×20). As expected, no amyloid was detected in non-transgenic islet grafts 1 week (a) and 6 weeks (b) after transplantation. Amyloid deposits are present in transgenic islet grafts at 1 week (c) and 6 weeks (d) following transplantation

Severity of amyloid deposition, beta cell volume, rate of beta cell apoptosis and rate of beta cell replication in non-transgenic and transgenic islet grafts at 1 week (a, c, e, g) and 6 weeks (b, d, f, h) after islet transplantation. Amyloid deposits were found in all eight transgenic grafts 1 week after islet transplantation (a) and were associated with lower beta cell volume (c), an increased rate of beta cell apoptosis (e) and a decreased rate of beta cell replication (g), although these did not reach statistical significance. Amyloid deposits were present in 11 of 12 transgenic islet grafts 6 weeks after islet transplantation (b) and were associated with a 30% reduction in beta cell volume (d), a twofold higher rate of beta cell apoptosis (f) and a 50% decrease in beta cell replication (h), all of which were statistically significant. As expected, no amyloid was detected in any of the non-transgenic islet grafts at 1 or 6 weeks after islet transplantation (a, b), as mouse IAPP is not amyloidogenic. *p < 0.05, **p < 0.001, ***p < 0.0001 for transgenic vs non-transgenic islet grafts
Six weeks after transplantation, amyloid deposition was detected in 11 of 12 transgenic islet grafts, with a severity of 0.33 ± 0.12% in the transgenic grafts and, again, as expected, in none of the non-transgenic grafts (p < 0.0001; Figs 2b,d and 3b). Beta cell volume was reduced by 30% in transgenic vs non-transgenic grafts (transgenic 6.9 ± 0.7 × 107, non-transgenic 9.9 ± 1.0 × 107 μm3; p<0.05; Fig. 3d). Increasing amyloid severity was associated with decreasing beta cell volume in transgenic grafts (r = −0.65, p < 0.05) and with increasing plasma glucose levels at 40 days after transplantation (r = 0.75, p < 0.005). In islet grafts harvested 6 weeks after transplantation, beta cell apoptosis was twofold higher in transgenic than in non-transgenic grafts (transgenic 0.07 ± 0.01%, non-transgenic 0.03 ± 0.01%; p = 0.02; Fig. 3f), while beta cell replication was reduced by 50% in transgenic vs non-transgenic grafts (transgenic 0.51 ± 0.08%, non-transgenic 1.00 ± 0.09%; p < 0.001; Fig. 3h). Figure 4 illustrates TUNEL- and Ki-67-positive beta cells in non-transgenic and transgenic islet grafts at 6 weeks after islet transplantation.

Staining of apoptotic and replicating beta cells with TUNEL (a, c) and Ki-67 (b, d) in non-transgenic (a, b) and transgenic (c, d) islet grafts 6 weeks post transplantation (original magnification ×40). Arrows indicate apoptotic and replicating beta cells. Nuclear staining is in blue and insulin staining is in red (a, c) or green (b, d)
Effect of amyloid formation on beta cell measures in human IAPP transgenic islet grafts of mice with and without recurrence of hyperglycaemia
Recurrence of hyperglycaemia, as a measure of graft failure, was defined as a plasma glucose level of >13.9 mmol/l on three consecutive occasions during the 6 week follow-up period. The first time point at which this outcome was reached was at 2 weeks, and by 6 weeks, 50% of the mice that had received transgenic islets had a recurrence of hyperglycaemia (Fig. 5). In contrast, none of the ten mice that received non-transgenic islets had a recurrence of hyperglycaemia (p = 0.01 vs transgenic).

Long-term recurrence of hyperglycaemia as defined by plasma glucose levels >13.9 mmol/l on three consecutive occasions occurred in 50% of mice that received transgenic (solid squares, n = 12) but in none of the mice that received non-transgenic (open squares, n = 10) islets over the 6 weeks following transplantation. *p = 0.01 for mice transplanted with transgenic vs non-transgenic islets
To assess whether there were differences between transgenic grafts that were associated with graft failure and those that were not, we compared graft morphology in grafts from mice with (n = 6) and without recurrence of hyperglycaemia (n = 6). Amyloid deposits were detected in all six transgenic grafts with and in five out of six transgenic grafts without recurrence of hyperglycaemia (Table 1). While amyloid severity and the rate of beta cell apoptosis were no different in the two groups, the rate of beta cell replication was lower (p < 0.01) and beta cell volume tended to be lower (p = 0.06) in the grafts from the mice with recurrence of hyperglycaemia. Body weights did not differ between these two subgroups at any time point.
To determine whether amyloid formation affected beta cell viability under euglycaemic conditions, we compared the metabolic and histological graft characteristics of the six mice that received transgenic islets and did not develop hyperglycaemia 6 weeks after transplantation with those of the ten mice that received non-transgenic islets. Plasma glucose levels and body weight did not differ significantly between the two groups at any time following islet transplantation. While beta cell volume did not differ between the transgenic and non-transgenic groups, the rate of beta cell apoptosis was higher (p = 0.04) and the rate of beta cell replication lower (p = 0.03) in the transgenic grafts (Table 1).
Discussion
We have shown that amyloid deposition occurs as early as 1 week after transplantation of human IAPP transgenic islets, and after 6 weeks, is associated with reduced beta cell volume and recurrence of hyperglycaemia. This deleterious effect of amyloid is associated with an increase in beta cell apoptosis and a decrease in beta cell replication.
Transplantation of both human IAPP transgenic and non-transgenic islets initially restored euglycaemia, and plasma glucose concentrations were similar in both groups for the first week. At that time, we found amyloid deposits in all human IAPP transgenic islet grafts. Thus, amyloid deposition is an early event following islet transplantation and it occurs independently from hyperglycaemia. One week following transplantation, beta cell volume, beta cell apoptosis and beta cell replication were not different between human IAPP transgenic and non-transgenic islet grafts. However, the direction of these changes was similar to the transgenic and non-transgenic groups 6 weeks after islet transplantation and is in keeping with a deleterious effect of amyloid on the graft.
Six weeks after transplantation, beta cell volume was significantly reduced in the transgenic grafts and was inversely correlated with amyloid severity, in line with amyloid deposition following islet transplantation being associated with beta cell loss. The mechanism by which amyloid may produce this loss of beta cells appears to be related to an increase in beta cell apoptosis, which was increased twofold, coupled with a 50% reduction in beta cell replication. These findings are in keeping with in vitro studies that have demonstrated that amyloid fibrils and oligomers induce cell death by apoptosis and necrosis [17, 19] and that replicating beta cells have increased susceptibility to hIAPP-induced apoptosis [32]. Our findings strongly suggest, therefore, that there is a dual negative effect of hIAPP-associated amyloid formation on beta cell turnover components, which may contribute to the loss of beta cell volume and the recurrence of hyperglycaemia that occurs following islet transplantation in humans.
Additional support for islet amyloid formation being responsible for beta cell loss and the recurrence of hyperglycaemia comes from the morphological analyses of islet grafts from mice in which hyperglycaemia did and did not reoccur. Six weeks after islet transplantation, half of the mice that received transgenic islets had graft failure defined as a recurrence of hyperglycaemia. Amyloid was detected in human IAPP transgenic islet grafts, regardless of the glucose level, consistent with the 1 week data. However, in mice that redeveloped hyperglycaemia, amyloid formation was associated with a tendency towards decreased beta cell volume and with decreased beta cell replication compared with amyloid formation in mice that received transgenic islets and did not develop a recurrence of hyperglycaemia. Furthermore, human IAPP transgenic grafts from mice that did not redevelop hyperglycaemia exhibited an increased rate of apoptosis and a decreased rate of replication compared with non-transgenic islet grafts, despite the fact that the plasma glucose levels were not different between these two groups. Thus, the cytotoxic effect of amyloid on the beta cell was already evident in human IAPP transgenic islet grafts when the glucose level was still within the euglycaemic range. We anticipate that, with additional time, the effect of these changes would have resulted in the loss of more beta cells and the recurrence of hyperglycaemia in all mice transplanted with transgenic islets.
The rate of beta cell apoptosis differed at 6 weeks by genotype. Compared with values at 6 weeks, at 1 week, beta cell apoptosis was threefold higher in the transgenic grafts and fourfold higher in the non-transgenic grafts. The finding of increased beta cell apoptosis in the first week post transplantation is not unexpected since considerable dynamic changes in islet cell turnover and graft tissue remodelling occur in the early post-transplant period [33]. This may explain why beta cell volume was low in both non-transgenic and transgenic islet grafts at 1 week compared with 6 weeks. Once transplanted islets survive this initial critical period, stressors that lead to long-term graft failure include both immune and non-immune factors.
The role of immunological factors in our study was minimised by using syngeneic donors and recipients, and no immune infiltration in islet grafts was observed at 1 or 6 weeks following transplantation (data not shown). Our experimental design using non-transgenic islets that do not have the propensity to, and did not, develop amyloid, provided controls for other non-immune causes of islet transplantation failure, such as ongoing hypoxia [34, 35] and altered metabolic milieu [36, 37], which are independent of amyloid formation. Furthermore, using light microscopy there was no visible evidence of amyloid in donor pancreases at the time of islet isolation or in pancreases of non-diabetic, non-transplanted human IAPP transgenic mice age-matched to islet transplant recipients 6 weeks post-transplantation. Therefore, amyloid formation over 6 weeks in transplanted islets most likely explains the reduction in beta cell volume and subsequent increase in plasma glucose levels in recipients of human IAPP transgenic islets.
It is possible that insulin release from the endogenous pancreas could have been responsible for differences in glycaemia in the different groups of mice. However, nephrectomy of the graft-bearing kidney at 1 week resulted in a return of plasma glucose levels to pre-transplant values in both transgenic and non-transgenic islet recipients. While we did not perform a nephrectomy on mice at 6 weeks, we have observed a similar recurrence of overt diabetes following nephrectomy 12 weeks after islet transplantation in both genotypes (J. Udayasankar, R. Hull, S. Zraika, K. Aston-Mourney, S.L. Subramanian, M.V. Faulenbach and S.E. Kahn, unpublished observation). This is in keeping with the glycaemic control following islet transplantation being due to a functioning islet graft rather than regeneration of endogenous beta cells in the pancreas of the streptozotocin-diabetic mice under the euglycaemic conditions produced by islet transplantation.
In human islet transplantation, islets are transplanted intraportally, rather than under the renal capsule as in this study. Westermark et al. [6] demonstrated that amyloid deposition occurs in human islets transplanted into the liver and spleen of nude mice, suggesting that it is not likely related to the site of transplantation. Of interest, widespread amyloid has recently been observed in a sample obtained from transplanted islets in a diabetic human [7]. Therefore, the deleterious effects of amyloid deposition on islet graft outcome in the present study are likely relevant to intraportally transplanted islets in human recipients.
In summary, we have observed that amyloid forms in transplanted islets as early as 1 week after transplantation and that this occurs prior to the recurrence of hyperglycaemia. Six weeks post transplantation, amyloid formation in islet grafts is associated with beta cell volume loss and the redevelopment of hyperglycaemia. This effect is likely mediated by an amyloid-associated increase in beta cell apoptosis and suppression of beta cell replication that would be required to prevent net beta cell loss. Thus, islet amyloid formation appears likely to be an important factor contributing to beta cell loss and the recurrence of hyperglycaemia in clinical islet transplantation, and inhibition of amyloid formation may be a strategy that could improve the long-term survival of transplanted human islets.
Abbreviations
- IAPP:
-
islet amyloid polypeptide
References
Robertson RP (2000) Successful islet transplantation for patients with diabetes—fact or fantasy? N Engl J Med 343:289–290
Shapiro AM, Lakey JR, Ryan EA et al (2000) Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med 343:230–238
Ryan EA, Paty BW, Senior PA et al (2005) Five-year follow-up after clinical islet transplantation. Diabetes 54:2060–2069
Clayton HA, Davies JE, Pollard CA, White SA, Musto PP, Dennison AR (2003) Pancreatectomy with islet autotransplantation for the treatment of severe chronic pancreatitis: the first 40 patients at the Leicester General Hospital. Transplantation 76:92–98
Westermark P, Eizirik DL, Pipeleers DG, Hellerstrom C, Andersson A (1995) Rapid deposition of amyloid in human islets transplanted into nude mice. Diabetologia 38:543–549
Westermark GT, Westermark P, Nordin A, Tornelius E, Andersson A (2003) Formation of amyloid in human pancreatic islets transplanted to the liver and spleen of nude mice. Ups J Med Sci 108:193–203
Westermark GT, Westermark P, Berne C, Korsgren O (2008) Widespread amyloid deposition in transplanted human pancreatic islets. N Engl J Med 359:977–979
Westermark P, Andersson A, Westermark GT (2005) Is aggregated IAPP a cause of beta-cell failure in transplanted human pancreatic islets? Curr Diab Rep 5:184–188
Bell ET (1959) Hyalinization of the islets of Langerhans in nondiabetic individuals. Am J Pathol 35:801–805
Clark A, Saad MF, Nezzer T et al (1990) Islet amyloid polypeptide in diabetic and non-diabetic Pima Indians. Diabetologia 33:285–289
Zhao HL, Lai FM, Tong PC et al (2003) Prevalence and clinicopathological characteristics of islet amyloid in Chinese patients with type 2 diabetes. Diabetes 52:2759–2766
Westermark P (1972) Quantitative studies on amyloid in the islets of Langerhans. Ups J Med Sci 77:91–94
Clark A, Wells CA, Buley ID et al (1988) Islet amyloid, increased A-cells, reduced B cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res 9:151–159
Westermark P, Wernstedt C, Wilander E, Hayden DW, O’Brien TD, Johnson KH (1987) Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc Natl Acad Sci U S A 84:3881–3885
Cooper GJ, Willis AC, Clark A, Turner RC, Sim RB, Reid KB (1987) Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc Natl Acad Sci U S A 84:8628–8632
Kahn SE, D’Alessio DA, Schwartz MW et al (1990) Evidence of cosecretion of islet amyloid polypeptide and insulin by beta-cells. Diabetes 39:634–638
Lorenzo A, Razzaboni B, Weir GC, Yankner BA (1994) Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature 368:756–760
Mirzabekov TA, Lin MC, Kagan BL (1996) Pore formation by the cytotoxic islet amyloid peptide amylin. J Biol Chem 271:1988–1992
Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC (1999) The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes 48:491–498
Konarkowska B, Aitken JF, Kistler J, Zhang S, Cooper GJ (2006) The aggregation potential of human amylin determines its cytotoxicity towards islet beta-cells. FEBS J 273:3614–3624
Ritzel RA, Meier JJ, Lin CY, Veldhuis JD, Butler PC (2007) Human islet amyloid polypeptide oligomers disrupt cell coupling, induce apoptosis, and impair insulin secretion in isolated human islets. Diabetes 56:65–71
Westermark P, Engstrom U, Johnson KH, Westermark GT, Betsholtz C (1990) Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc Natl Acad Sci U S A 87:5036–5040
Fox N, Schrementi J, Nishi M et al (1993) Human islet amyloid polypeptide transgenic mice as a model of non-insulin-dependent diabetes mellitus (NIDDM). FEBS Lett 323:40–44
Hoppener JW, Verbeek JS, de Koning EJ et al (1993) Chronic overproduction of islet amyloid polypeptide/amylin in transgenic mice: lysosomal localization of human islet amyloid polypeptide and lack of marked hyperglycaemia or hyperinsulinaemia. Diabetologia 36:1258–1265
D’Alessio DA, Verchere CB, Kahn SE et al (1994) Pancreatic expression and secretion of human islet amyloid polypeptide in a transgenic mouse. Diabetes 43:1457–1461
Butler AE, Janson J, Soeller WC, Butler PC (2003) Increased beta-cell apoptosis prevents adaptive increase in beta-cell mass in mouse model of type 2 diabetes: evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes 52:2304–2314
Verchere CB, D’Alessio DA, Palmiter RD et al (1996) Islet amyloid formation associated with hyperglycemia in transgenic mice with pancreatic beta cell expression of human islet amyloid polypeptide. Proc Natl Acad Sci U S A 93:3492–3496
Hull RL, Andrikopoulos S, Verchere CB et al (2003) Increased dietary fat promotes islet amyloid formation and beta-cell secretory dysfunction in a transgenic mouse model of islet amyloid. Diabetes 52:372–379
Zraika S, Hull RL, Udayasankar J et al (2007) Identification of the amyloid-degrading enzyme neprilysin in mouse islets and potential role in islet amyloidogenesis. Diabetes 56:304–310
Wang F, Hull RL, Vidal J, Cnop M, Kahn SE (2001) Islet amyloid develops diffusely throughout the pancreas before becoming severe and replacing endocrine cells. Diabetes 50:2514–2520
Hull RL, Kodama K, Utzschneider KM, Carr DB, Prigeon RL, Kahn SE (2005) Dietary-fat-induced obesity in mice results in beta cell hyperplasia but not increased insulin release: evidence for specificity of impaired beta cell adaptation. Diabetologia 48:1350–1358
Ritzel RA, Butler PC (2003) Replication increases beta-cell vulnerability to human islet amyloid polypeptide-induced apoptosis. Diabetes 52:1701–1708
Davalli AM, Scaglia L, Zangen DH, Hollister J, Bonner-Weir S, Weir GC (1996) Vulnerability of islets in the immediate posttransplantation period. Dynamic changes in structure and function. Diabetes 45:1161–1167
Carlsson PO, Mattsson G (2002) Oxygen tension and blood flow in relation to revascularization in transplanted adult and fetal rat pancreatic islets. Cell Transplant 11:813–820
Carlsson PO, Palm F, Mattsson G (2002) Low revascularization of experimentally transplanted human pancreatic islets. J Clin Endocrinol Metab 87:5418–5423
Korsgren O, Jansson L, Andersson A (1989) Effects of hyperglycemia on function of isolated mouse pancreatic islets transplanted under kidney capsule. Diabetes 38:510–515
Montana E, Bonner-Weir S, Weir GC (1993) Beta cell mass and growth after syngeneic islet cell transplantation in normal and streptozocin diabetic C57BL/6 mice. J Clin Invest 91:780–787
Acknowledgements
We thank S. Wilbur, M. Watts, R. Bhatti, R. Vogel, R. Hollingworth and R. Koltz for excellent technical support. G. Weir and S. Bonner-Weir from Joslin Diabetes Center, (Boston, MA, USA) are thanked for their valuable advice during the development of the study. This work was financially supported by the Department of Veterans Affairs and National Institutes of Health grant no. DK-17047. K. Kodama was financially supported by the Manpei Suzuki International Diabetes Foundation.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
J. Udayasankar and K. Kodama contributed equally to this study.
Rights and permissions
About this article
Cite this article
Udayasankar, J., Kodama, K., Hull, R.L. et al. Amyloid formation results in recurrence of hyperglycaemia following transplantation of human IAPP transgenic mouse islets. Diabetologia 52, 145–153 (2009). https://doi.org/10.1007/s00125-008-1185-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-008-1185-7