
Abstract
Aims/hypothesis
Controversy surrounds whether the ratio of apolipoprotein B (ApoB) to apolipoprotein A-I (ApoA-I) is the best lipoprotein discriminator of CHD risk in non-diabetic populations, but the issue has never been investigated in type 2 diabetes.
Methods
In 2,627 participants without known vascular disease in the Collaborative Atorvastatin Diabetes Study, ApoB, ApoA-I, LDL-cholesterol (LDLC) and HDL-cholesterol (HDLC) were assayed at baseline.
Results
There were 108 CHD and 59 stroke endpoints over 3.9 years. The ApoB:A-I ratio at baseline was the lipoprotein variable most closely predicting CHD risk both by comparison of the hazard ratio for a 1 SD change or tertiles of frequency distribution. The areas under the receiver–operator curve for the ApoB:ApoA-I and the LDLC to HDL-HDLC ratios, although not significantly different from each other, were greater (p = 0.0005 and p = 0.0125 respectively) than that of non-HDLC:HDLC. The 27% decrease in the ApoB:ApoA-I ratio on atorvastatin predicted a 32% (95% CI 5.4–51.2%) risk reduction in CHD, close to the 36% decrease observed. Neither the ApoB:ApoA-I nor any other lipoprotein concentration or ratio predicted the stroke outcome.
Conclusions/interpretation
Overall, the ApoB:ApoA-I ratio improved on the non-HDLC:HDLC ratio in predicting CHD, but, depending on the assessment chosen, its superiority over LDLC:HDLC may be marginal. The statin-induced decrease in stroke risk may not be lipoprotein mediated.
Trial registration: ClinicalTrials.gov NCT00327418
Funding: The study was supported by unrestricted grants from Diabetes UK, the Department of Health and Pfizer to the University of Manchester and to University College, London.
Similar content being viewed by others


Introduction
Apolipoprotein A-I (ApoA-I) and B-100 (ApoB) are the major components of the protein moieties of LDL and HDL respectively [1]. ApoB is also present in VLDL and lipoprotein(a) [Lp(a)]. There is one molecule of ApoB in each ApoB-containing lipoprotein particle [2, 3]. Thus it is a measure of the particle concentration of VLDL, LDL and Lp(a). ApoA-I does not reflect the particle concentration of lipoproteins to the same extent, because ApoA-I-containing lipoproteins generally have one to four molecules of ApoA-I per particle. However, both ApoB and ApoA-I, unlike LDL-cholesterol (LDLC) and HDL-cholesterol (HDLC), may reveal the presence of LDL and HDL particles that contain little cholesterol, such as small dense LDL [4] and pre-beta HDL [5]. Furthermore HDLC methods vary in the proportion of cholesterol in HDL which they detect [6]. The possibilities that apolipoproteins might provide the clinician with a more accurate assessment of cardiovascular risk and a better target for statin therapy have thus been raised [3].
It has been recognised for nearly 30 years that even when LDLC is in the normal range, particularly in hypertriacyglycerolaemia, LDL-ApoB can be raised [7]. Sniderman and colleagues [8] first proposed that this was because under such circumstances a cholesterol-depleted LDL was present. They termed this condition hyperapobetalipoproteinaemia and reported that it was associated with CHD. It was subsequently revealed that this phenomenon was due to the presence of a small dense LDL particle [3, 4, 9]. After the initial report of a case–control study that the ApoB:A-I ratio provided the closest association between lipoprotein variables and CHD [10], numerous others followed confirming this to a greater or lesser extent [3]. A transnational study revealed that ApoB:ApoA-I ratio explained more of the variation in CHD risk than any other risk factor [11]. The most influential evidence was, however, the large prospective study by Walldius and colleagues [12] that reported that the ApoB:ApoA-I ratio related more closely to CHD than other lipoprotein variables or ratios and a large meta-analysis of prospective studies [13]. The possible use of the ApoB:ApoA-I ratio as a target of lipid-lowering therapy has also been widely discussed [3]; two recent studies—one with a Framingham cohort not treated with statins [14] and another in a secondary study in patients treated with statins [15]—both cast doubt on the clinical need for this. All authors agree, however, that there is a dearth of information about the predictive power of ApoB:ApoA-I for cardiovascular disease in type 2 diabetes.
In the present report, we have examined a wide range of cardiovascular outcomes, including stroke, in a group of type 2 diabetic patients participating in the Collaborative Atorvastatin Diabetes Study (CARDS) [16–18].
Methods
Study population
CARDS was a double-blind, randomised, placebo-controlled, multicentre trial of atorvastatin 10 mg daily in the primary prevention of cardiovascular disease in type 2 diabetes [16–18]. It received ethical approval both centrally and at each participating institution and each patient gave written informed consent. There were 2,838 patients (68% men) aged between 40 and 75 years randomised either to placebo (n = 1,410) or atorvastatin (n = 1,428) [17]. The primary endpoint of the trial was the first acute CHD event (myocardial infarction, hospitalised unstable angina, acute CHD death), coronary revascularisation procedure or stroke. In addition, information about all causes of death was collected. To enter the trials, patients had to be free of macrovascular disease, to have serum LDLC levels of 4.14 mmol/l or less and to have fasting serum triacylglycerol levels of 6.77 mmol/l or less. In addition, they were required to have at least one of the following cardiovascular risk factors: hypertension on treatment, or systolic BP ≥ 140 mm Hg and/or diastolic BP ≥ 90 mm Hg on two successive occasions, any retinopathy, proteinuria including microalbuminuria or current smoking. The trial was terminated 2 years earlier than planned at the request of the Safety Committee because of the clear benefit of active treatment (two-tailed p < 0.001). The median time each patient had spent in the trial was 3.9 years. Complete apolipoprotein data were available at baseline (pre-randomisation) for 2,627 patients. They yielded 196 primary endpoints, 108 fatal and non-fatal myocardial infarctions, 131 deaths and 59 strokes. These were adjudicated by an independent endpoint committee as described in detail elsewhere [16, 17].
Laboratory methods
All participants were asked to fast from 20:00 hours the previous day. Cholesterol in serum and lipoproteins was measured using the CHOD/PAP method (Roche, Basel, Switzerland) on a Cobas Mira analyser. Triacylglycerols were measured by the GPO/PAP method (Roche) and serum ApoB and ApoA-I were measured using immunoturbidimetry (Roche) on the same instrument with calibration traceable to the International Federation of Clinical Chemistry primary standards [19]. HDLC was isolated by precipitation of other serum lipoproteins using heparin–manganese [20]. When serum triacylglycerol exceeded 4.00 mmol/l, VLDL-cholesterol was removed by ultracentrifugation for 18 h at 144,000 g (Beckman L8-55 M) at 1.006 g/ml before the heparin–manganese procedure. The laboratory participated in the UK RIQAS quality control scheme (Randox Laboratories, Belfast, UK). The HDLC method was aligned with the results of the Centers for Disease Control and Prevention (CDC) laboratory participating in this scheme.
LDLC was calculated using the Friedewald formula [21] when serum triacylglycerol levels were 4.00 mmol/l or less and by subtraction of HDLC from that in the D1.006 g/ml ultracentrifuge infranatant obtained by tube slicing when serum triacylglycerol exceeded 4.00 mmol/l [6].
Statistical methods
This study pools 2,627 (93%) of the 2,838 participants in CARDS with complete baseline lipid data. The main analysis in this investigation was based on a Cox proportional hazard regression model, which assessed whether each apolipoprotein variable (as a continuous variable) was a significant predictor of cardiovascular risk in univariate analysis or adjusted by treatment in multivariate analysis. The HR and 95% CI were calculated for a 1 SD increment for each lipoprotein variable. The strength of associations among all apolipoproteins was ranked based on the χ 2 statistics and the corresponding p values estimated from the regression coefficient based on the Cox proportional hazard model. In addition, the relationships between tertiles of baseline ApoB:ApoA-I ratio and HR for cardiovascular risks were assessed by the Cox proportional hazard model using the first tertile of ApoB:ApoA-I ratio as the control group.
The degrees of predictability of cardiovascular risk for the apolipoproteins and lipoproteins were also compared by receiver operating characteristic curve (ROC) analysis based on a logistic regression model. The logistic regression model included the individual study cardiovascular endpoint (binary data) as the dependent variable with individual lipoprotein variables (continuous) as the predictor variable. By applying various cut points to the predicted probability obtained from the logistic regression model, each participant was classified as case or control. For each cut point, a 2 × 2 cross-classification table (observed vs expected based on the model) was constructed and the corresponding sensitivity (true positive rate) and 1−specificity (false positive rate) were calculated. The ROC curve was then constructed by plotting the sensitivity against 1−specificity for all cut points. The AUC was calculated by integrating the area between the ROC curve and the diagonal line (where sensitivity is equal to 1−specificity) based on the trapezoidal rule [22].
In addition, the effect of atorvastatin on lipid concentrations at each annual visit and throughout the trial was assessed by repeated measurement analysis of variance analysis based on linear mixed models. These models included treatment, time and treatment by time interaction as the major fix factors and individual participants nested in the treatment as the cluster unit.
All analyses are performed at a two-sided significance level of 0.05 using SAS statistical software (version 8.12; SAS Institute, Cary, NC, USA).
Results
Cardiovascular outcomes
As previously reported, the primary endpoint was decreased by 37% (95% CI 17–53%) on active treatment compared with placebo, acute coronary events by 36% (9–55%) and stroke by 48% (11–69%; all p < 0.05) [17]. All-cause mortality declined by 27% (−1.48%), which was of borderline significance p = 0.0592) overall, but significant in the final year [17].
Change in lipids, lipoproteins and apolipoproteins
At baseline, patients allocated to active atorvastatin or placebo were closely matched to their lipid, lipoprotein and apolipoprotein levels as previously reported [16]. Thus, in the 2,627 patients in whom complete laboratory data were available at baseline, mean serum cholesterol was 5.36 ± 0.82 mmol/l (mean ± SD), LDLC 3.04 ± 0.71 mmol/l, non-HDLC 3.95 ± 0.83 mmol/l, HDLC 1.40 ± 0.33 mmol/l, ApoA-I 1.53 ± 0.29 g/l and ApoB 1.16 ± 0.24 g/l. The mean difference in LDLC concentration on atorvastatin compared with placebo throughout the trial was 40.9% (mean 95% CI 40.1–41.6%), whereas ApoB decreased by 24.3% (23.4–25.2%; both p < 0.0001). The decrease on atorvastatin compared with placebo in ApoB:ApoA-I was 27.2% (−28.5, −25.9%; p < 0.0001) and, in LDLC:HDLC, −44.6% (−45.6, −43.5%; p < 0.001). HDLC was significantly increased on active treatment compared with placebo by 1.6% (1.0–2.1%; p < 0.05), but ApoA-I was not significantly changed by atorvastatin.
Baseline lipoproteins and cardiovascular outcomes
Table 1 shows the change in HR adjusted by treatment for a primary endpoint in the combined treatment groups predicted by a 1 SD difference in various lipoprotein variables and their ratios. These are ranked according to p values by χ 2. These are inversely related to the strength of the association. The most statistically significant increments in HR were predicted by the ApoB:ApoA-I ratio. Although the less significant increment in HR for a 1 SD change in LDLC:HDLC or LDLC was greater than for ApoB:ApoA-I, this was less statistically significant because the SD was smaller for ApoB:ApoA-I. The level of statistical significance which is related to the Cox regression coefficient (β) and its standard error reveals the true strength of the relationships.
Table 2 shows the subgroup analysis by treatments comparing the predictability of the risk of primary endpoints among studied lipids and lipoproteins. For the placebo group, ApoB:ApoA-I ratio remained the strongest significant risk factor for primary endpoint. Compared with both treatment groups combined, the rank orders among the first four lipid/lipoproteins remained the same with only slight differences in the last four lipids. For the atorvastatin group, all studied lipid/lipoproteins variables were insignificant.
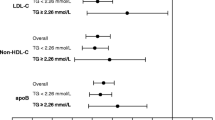
Table 3 shows the intertertiles analysis of ApoB:ApoA-I ratio at baseline and HR, both unadjusted for treatment effect (model 1) and adjusted for treatment effect (model 2). The results based on ‘adjusted for treatment effect’ and HR for primary endpoints, CHD endpoints, total mortality and stroke were very similar to those from an unadjusted analysis. The relationship between both cardiovascular risk and all-cause mortality and tertiles of the ApoB:ApoA-I ratio was stronger compared with other lipoprotein variables (Fig. 1a,b). Furthermore, it was strongest for the CHD component of cardiovascular risk (Fig. 1c) and weakest for stroke for which it had no statistical significance (Fig. 1d). Both the ApoB:ApoA-I and the LDLC:HDLC ratios also identified patients at greater risk of CHD than non-HDLC:HDLC as judged by their ROCs (p = 0.0005 and p = 0.0125 respectively; Fig. 2). However, the apparently greater AUC for ApoB:ApoA-I as opposed to LDL:HDLC did not achieve statistical significance.

The HR for (a) primary endpoints (CHD and stroke events; n = 196), (b) CHD death and non-fatal myocardial infarction (n = 108), (c) all-cause mortality (n = 131) and (d) strokes (n = 59) in the three tertiles (T1, T2 and T3) of lipid, lipoprotein and apolipoprotein risk factors and their ratios. TG, triacylglycerol. Black inverted triangle, ApoB:ApoA-I; white diamond, LDLC:HDLC; white triangle, ApoB; black circle, LDLC; black square, non-HDLC; white square, TG; white circle, HDLC, black triangle, ApoA-I

ROCs for the prediction of the primary endpoint by baseline ApoB:ApoA-I (0.5925 ± 0.0210) (AUC ± SE), LDLC:HDLC (0.5700 ± 0.0210) and non-HDLC:HDLC (0.5456 ± 0.0209). The AUC for ApoB:ApoA-I and LDLC:HDLC were statistically significantly greater than that for non-HDLC:LDLC (p = 0.0005 and 0.0125 respectively). For ApoB:ApoA-I vs LDLC:HDLC, p = 0.1286. Red line, ApoB:ApoA-I; blue line, LDLC:HDLC; green line, non-HDLC:HDLC
Lipoprotein levels on treatment and cardiovascular outcome
The first time apolipoproteins were measured per protocol after baseline in the participants as a whole was at 1 year after commencing treatment. By then, 60 primary endpoints had occurred. Attention to this early benefit has previously been drawn [18]. It meant that many of the total number events in the trial had occurred before the changes in apolipoproteins with the treatment were measured for the first time. Thus, it was not possible to relate individual treatment changes to outcomes, because of insufficient subsequent events. However, the time-dependent Cox proportional hazard regression model, which included percentage change in ApoB:ApoA-I adjusted for treatment effect as a covariate, predicted that the 27.2% decrease in ApoB:ApoA-I on active treatment would produce a 30.2% (95% CI 4.1–49.2%) decrease in the primary endpoints, which is similar to the 37% decrease observed in the full study. The reduction in primary endpoints predicted by the decrease of ApoB:ApoA-I ratio was almost exclusively due to CHD events. Thus, based on a similar time-dependent Cox proportional hazard regression model, it was estimated that the same 27.2% decrease in ApoB:ApoA-I ratio on active treatment would produce a 32% (5.4–51.2%) reduction in CHD risk which was close to the 36% decrease observed. None of the lipid, lipoprotein or apolipoprotein variables, or their ratios, forecast the 48% decrease in the incidence of strokes.
Discussion
This study provides additional evidence that ApoA-I and ApoB are more closely associated with atherosclerotic cardiovascular disease risk, particularly CHD, than other lipid and lipoprotein variables available in the clinic and is the first to show this in diabetes. Furthermore, while the statin benefit in CHD prevention was largely explicable in terms of changes in ApoB, there was the suggestion that the decrease in stroke risk may have been at least partly independent of statin-induced changes in lipids, lipoproteins and apolipoproteins.
Several studies have indicated that ApoA-I and ApoB provide better discrimination of CHD risk than serum LDLC, HDLC and non-HDLC which are the basis of current guidance for statin therapy in cardiovascular disease and CHD prevention [23–25]. A similar conclusion was reached in a randomised primary prevention trial of lovastatin [26], in a non-diabetic population. Ours is the first to do so in type 2 diabetes. The ApoB:ApoA-I ratio was more closely associated with cardiovascular and CHD risk than other lipoprotein variables as judged by the Cox proportional hazard model. Furthermore, most of the observed reduction in cardiovascular and CHD events was explained by the statin-induced change in the ApoB:ApoA-I ratio and the predictive model derived from pre-treatment (baseline) values. However, by ROC analysis, although not clearly better than non-HDLC:HDLC, the ApoB:ApoA-I ratio did not significantly improve on LDLC:HDLC. Although ROC methods have their limitations [27], we would nonetheless concede the argument that a more accurate prediction of cardiovascular risk with ApoA-I and ApoB, while it may be of theoretical interest, might be of little quantitative clinical value over measurement of the LDLC:HDLC ratio. Non-HDLC and indices depending on non-HDLC were, however, significantly less accurate in predicting cardiovascular risk than either the ApoB:ApoA-I or the LDLC:HDLC ratios.
Relevant to the issue over whether ApoB should replace measurement of LDLC in clinical practice is also the accuracy with which it can be routinely measured. It is likely that LDLC and non-HDLC were shown to their best advantage in CARDS, not least because in patients in whom triacylglycerol levels were too high for the use of the Friedewald formula, ultracentrifugation was used to remove triacylglycerol before HDLC was measured [6]. Such a procedure cannot be automated, is impracticable and too costly for routine laboratory use. Commercially available third-generation HDLC methods used in routine laboratories may introduce considerable bias compared with CDC standards [6]. Future studies should, therefore, concentrate on the relative accuracy and cost of automated ApoB and ApoA-I assays and/or direct, automated LDLC compared with LDLC calculated from results of routinely used HDLC methods.
Neither the ApoB:ApoA-I ratio nor any of the other variables studied here explained the 48% decrease in stroke in CARDS. Results of other trials support our finding that statins can decrease ischaemic stroke risk [28–30]. However, this favourable effect of atorvastatin on stroke risk may be through a mechanism not directly mediated by the changes in lipoprotein levels. Although it must be conceded that stroke incidence was lower than that of CHD in CARDS and that there may have been too few strokes to detect a statistically significant association with lipoproteins, the relationship between the mean decreases in LDLC achieved in other trials and the decrease in stroke risk is also generally less strong than for CHD [28]. In epidemiological studies, stroke incidence is also strongly associated with lipoproteins that are associated with CHD [31, 32]. A further study is underway to examine whether in CARDS the stroke outcome relates to pleiotropic effects of atorvastatin, such as the decrease in C-reactive protein [33].
In conclusion, ApoB and ApoB:ApoA-I ratio were only marginally better than LDLC:HDLC in predicting cardiovascular risk in type 2 diabetes. Understanding the mechanism by which statins prevent stroke may lead to the development of more effective therapy.
Abbreviations
- ApoA-I:
-
apolipoprotein A-I
- ApoB:
-
apolipoprotein B100
- CARDS:
-
Collaborative Atorvastatin Diabetes Study
- LDLC:
-
LDL-cholesterol
- Lp(a):
-
lipoprotein(a)
- HDLC:
-
HDL-cholesterol
- ROC:
-
receiver operator curve
References
Havel RJ, Goldstein JL, Brown MS (1980) Lipoproteins and lipid transport. In: Bondy PK, Rosenberg LE (eds) Metabolic control and disease. Saunders, Philadelphia, pp 393–494
Fredrickson DS (1974) Plasma lipoproteins and apolipoproteins. In: The Harvey Lectures 1972–73, Series 68. Academic, London, pp 185–237
Barter PJ, Ballantyne CM, Carmena R et al (2006) Apo B versus cholesterol to estimate cardiovascular risk and to guide therapy: report of the Thirty Person/Ten Country Panel. J Int Med 259:247–258
Austin MA, Breslow JL, Hennekens CH, Buring JE, Willett WC, Krauss RM (1988) Low density lipoprotein subclass patterns and risk of myocardial infarction. J Am Med Assoc 260:1917–1921
Neary R, Bhatnagar D, Durrington PN, Ishola M, Mackness M, Arrol S (1991) An investigation of the role of lecithin: cholesterol acyl transferase and triglyceride-rich lipoproteins in the metabolism of pre-beta high density lipoproteins. Atherosclerosis 89:35–48
Warnick GR, Nauck M, Rifai N (2001) Evolution of methods for measurement of HDL-cholesterol: from ultracentrifugation to homogeneous assays. Clin Chem 47:1579–1596
Durrington PN, Bolton CH, Hartog M (1978) Serum and lipoprotein apolipoprotein B levels in normal subjects and patients with hyperlipoproteinaemia. Clin Chim Acta 82:151–160
Sniderman A, Shapiro S, Marpole D, Skinner B, Teng B, Kwiterovich POJ (1980) Association of coronary atherosclerosis with hyperapobetalipoproteinemia (increased protein but normal cholesterol levels in human plasma low density (beta) lipoproteins). Proc Natl Acad Sci U S A 77:604–608
Griffin BA, Minihane AM, Furlonger N et al (1999) Inter-relationships between small, dense low-density lipoprotein (LDL), plasma triacylglycerol and LDL apoprotein B in an atherogenic lipoprotein phenotype in free-living subjects. Clin Sci 97:267–276
Avogaro P, Bittolo-Bon G, Avogaro G, Cazzolato G, Quinci GB (1979) Are apolipoproteins better discriminators than lipids for atherosclerosis. Lancet 1:901–903
Yusuf S, Hawken S, Öunpuu S et al (2004) INTERHEART Study Investigators. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case–control study. Lancet 364:937–952
Walldius G, Jungner I, Holme I, Aastveit AH, Kolar W, Steiner E (2001) High apolipoprotein B, low apolipoprotein A-1, and improvement in the prediction of fatal myocardial infarction (AMORIS study): a prospective study. Lancet 358:2026–2033
Thompson A, Danesh J (2006) Associations between apolipoprotein B, apolipoprotein AI, the apolipoprotein B/AI ratio and coronary heart disease: a literature-based meta-analysis of prospective studies. J Intern Med 259:481–492
Ingelsson E, Schaefer EJ, Contois JH et al (2007) Clinical utility of different lipid measures for prediction of coronary heart disease in men and women. JAMA 298:776–785
Kastelein JJP, Van der Steeg WA, Holme I et al (2008) Lipids, apolipoproteins, and their ratios in relation to cardiovascular events with statin treatment. Circulation 117:3002–3009
Colhoun HM, Thomason MJ, Mackness MI et al (2002) Collaborative AtoRvastatin Diabetes Study (CARDS). Design of the Collaborative AtoRvastatin Diabetes Study (CARDS) in patients with type 2 diabetes. Diabet Med 19:201–211
Colhoun HM, Betteridge DJ, Durrington PN et al (2004) Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): a multicentre randomized placebo-controlled trial. Lancet 364:685–696
Colhoun H, Betteridge DJ, Durrington PN et al (2005) Rapid emergence of effect of atorvastatin on cardiovascular outcomes in the Collaborative Atorvastatin Diabetes Study (CARDS). Diabetologia 48:2482–2485
Albers JJ, Marcovina SM, Kennedy H (1992) International Federation of Clinical Chemistry standardization project for measurements of apolipoproteins A-I and B. II. Evaluation and selection of candidate reference materials. Clin Chem 38:658–662
Warnick GR, Albers JJ (1978) A comprehensive evaluation of the heparin–manganese precipitation procedure for estimating high density lipoprotein cholesterol. J Lipid Res 19:65–76
Friedewald WT, Levy RI, Frederickson DS (1972) Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem 18:499–502
De Long ER, De Long DM, Clarke-Pearson DL (1988) Comparing the areas under two or more correlated receiver operating characteristic corves: a nonparametric approach. Biometrics 44:837–845
Grundy SM, Cleeman JI, Bairey Merz CN et al (2004) For the Co-ordinating Committee of the National Cholesterol Education Program. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines. Circulation 110:227–239
Graham I, Fourth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (Constituted by representatives of nine societies and by invited experts) (2007) European guidelines on cardiovascular disease prevention in clinical practice: executive summary. Atherosclerosis 194:1–45
McElduff P, Jaefarnezhad M, Durrington P (2006) American, British and European recommendations for statins in the primary prevention of cardiovascular disease applied to British men studied prospectively. Heart 92:1213–1218
Gotto A Jr, Whitney E, Stein EA et al (2000) Relation between baseline and on-treatment lipid parameters and first acute coronary events in the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS). Circulation 101:477–484
Pepe MS, Janes H, Longton G, Leisenring W, Newcomb P (2004) Limitations of the odds ratio in gauging the performance of a diagnostic, prognostic, or screening marker. Am J Epidemiol 159:882–890
Cholesterol Treatment Trialists’ (CTT) Collaborators (2005) Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 366:1267–1278
Amarenco P, Labreuche J, Lavallee P, Touboul PJ (2004) Statins in stroke prevention and carotid atherosclerosis: systematic review and up-to-date meta-analysis. Stroke 35:2902–2909
The Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) Investigators (2006) High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med 355:549–559
Kurth T, Everett BM, Buring JE, Kase CS, Ridker PM, Gaziano JM (2007) Lipid levels and the risk of ischemic stroke in women. Neurology 68:556–562
Lewington S, Whitlock G, Clarke R et al (2007) Prospective Studies Collaboration: blood cholesterol and vascular mortality by age, sex and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet 370:1829–1839
Schaefer EJ, McNamara JR, Asztalos BF et al (2005) Effects of atorvastatin versus other statins on fasting and postprandial C-reactive protein and lipoprotein-associated phospholipase A2 in patients with coronary heart disease versus control subjects. Am J Cardiol 95:1025–1032
Acknowledgement
We are grateful to C. Price, Cardiovascular Research Group, University of Manchester, for expert preparation of the manuscript without compensation.
Duality of interest
C. N. Newman, M. Szarek and D. A. DeMicco are currently or previously were salaried employees of Pfizer. Universities employing P. N. Durrington, H. Colhoun, J. Fuller and V. Charlton-Menys have received research grants from Pfizer. P. N. Durrington, D. J. Betteridge, H. Colhoun, G. A. Hitman, J. Fuller and H. A. W. Neil have received fees for lectures and consultancy work from Pfizer. S. J. Livingstone and M. France have no conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s00125-008-1235-1
Rights and permissions
About this article
Cite this article
Charlton-Menys, V., Betteridge, D.J., Colhoun, H. et al. Apolipoproteins, cardiovascular risk and statin response in type 2 diabetes: the Collaborative Atorvastatin Diabetes Study (CARDS). Diabetologia 52, 218–225 (2009). https://doi.org/10.1007/s00125-008-1176-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-008-1176-8