
Abstract
Aims/hypothesis
Although clear peripheral nerve pathological abnormalities have been demonstrated in diabetic peripheral neuropathy (DPN), there is little information with regard to brain involvement. Our aim was to use in vivo proton magnetic resonance specroscopy (H-MRS) in patients with DPN in order to assess the neuro-chemical status of the thalamus, which acts as the gateway to the brain for somatosensory information.
Methods
Participants included 18 type 1 diabetic men (eight without DPN, ten with DPN) and six non-diabetic healthy volunteers, who all underwent detailed clinical and neurophysiological assessments yielding a Neuropathy Composite Score (NCS) derived from Neuropathy Impairment Score of the Lower Limbs plus seven tests of nerve function prior to investigation via a single-voxel H-MRS technique, which was used to sample ventral posterior thalamic parenchyma. Spectroscopic resonances including those due to N-acetyl aspartate (NAA) were assessed at both short and long echo-time, providing putative indicators of neuronal function and integrity, respectively.
Results
At long echo-time we observed significantly lower NAA:creatine (p = 0.04) and NAA:choline (p = 0.02) ratios in DPN patients than in the other groups. No group differences were detected at short echo-time. We found a significant positive association between both sural amplitude (ρ = 0.61, p = 0.004) and nerve conduction velocity (r = 0.58, p = 0.006) and NAA:creatine signal among participants with diabetes. Vibration detection threshold (ρ = −0.70, p = 0.004) was significantly related to NAA:choline ratio. Heart rate variability with deep breathing (ρ = −0.46, p = 0.05) and NCS (ρ = −0.53, p = 0.03) were significantly related to NAA:creatine ratio.
Conclusions/interpretation
The significantly lower NAA:creatine ratio in DPN is suggestive of thalamic neuronal dysfunction, while the lack of difference in short echo-time between the groups does not suggest neuronal loss. Taken together with the observed correlations between NAA and neurophysiological assessments, these findings provide evidence for thalamic neuronal involvement in DPN.
Similar content being viewed by others

Introduction
Diabetes is a leading cause of distal symmetric polyneuropathy or diabetic peripheral neuropathy (DPN) [1] with important associated health and economic implications [2]. Research into DPN has focused mainly on the peripheral nervous system with central nervous system involvement being overlooked. However, we have reported that spinal cord ‘atrophy’ is present not only in patients with established DPN, but also in those with early (subclinical) DPN [3]. This made us question whether the brain, too, may be involved.
Ascending sensory pathways of the spinal cord terminate within the ventroposterior lateral (VPL) thalamic subnucleus before high-order sensory projections are sent to the cortex [4]. The thalamus plays a crucial role by modulating information presented to the cortex [4]. Hence, in the presence of the sensory nerve dysfunction accompanying DPN, the specific aim of this study was to investigate whether thalamic function is also impaired.
The investigative technique used was proton magnetic resonance spectroscopy (H-MRS). In-vivo H-MRS offers the unique possibility of non-invasive detection of several cerebral metabolites, in particular, N-acetyl aspartate (NAA), a marker of neuronal and axonal integrity [5], creatine/phosphocreatine (hereafter creatine), which is involved in cellular bioenergetics [6], choline-containing compounds, which are involved in membrane synthesis [6] and myo-inositol, thought to be present mostly [7] in glial cells.
We conducted an H-MRS study, using NAA resonance as a neuronal marker to test the hypothesis that DPN coexists with thalamic neuronal involvement. We also characterised the relationship between thalamic neuronal biochemistry and traditional neurophysiological peripheral nerve assessments reflecting DPN severity.
Methods
Participants
We screened 28 right-handed men with type 1 diabetes from the Royal Hallamshire Hospital Diabetes Register. Exclusion factors were: cerebrovascular disease, non-diabetic neuropathies, history of alcohol consumption of more than 20 units a week, diabetic neuropathies other than DPN, painful DPN, internal diseases potentially affecting cerebral metabolism, diabetic ketoacidosis in the preceeding 6 months, hypoglycaemia in the previous 24 h and factors precluding magnetic resonance (MR) imaging. We also recruited six age-matched male non-diabetic healthy volunteers. All participants gave written, informed consent before participation in the study, which had prior approval by the South Sheffield Regional Ethics Committee.
Neuropathy assessment
Detailed neurological assessment was undertaken to identify the presence and quantify the severity of neuropathy. Neuropathic symptoms were documented by completion of the Neuropathy Symptom Score questionnaire and the outcome of a detailed neurological examination was graded using the Neuropathy Impairment Score questionnaires [8]. All participants underwent: (1) vibration detection thresholds acquired from the right foot using computer-assisted sensory evaluation (CASE IV; W. R. Electronics, Stillwater, MN, USA) [9]; (2) measurement of heart rate variability with deep breathing (O’Brien protocol) [10]; and (3) nerve conduction studies performed at a stable skin temperature of 31°C and a room temperature of 24°C, using an electrophysiological system (Medelec; Synergy Oxford Instruments, Oxford, UK).
Based on these clinical and neurophysiological assessments, diabetic participants were divided into two groups: (1) No-DPN consisting of asymptomatic diabetic patients with normal clinical and neurophysiological assessments; (2) DPN, comprising participants with both clinical and neurophysiological abnormalities (at least two abnormalities in neurophysiological assessment) [11]. In addition, a Neuropathy Composite Score (NCS), which was derived from the assessments above (Neuropathy Impairment Score of the Lower Limbs plus seven tests of nerve function), was calculated [12].
Magnetic resonance spectroscopy
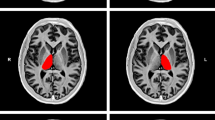
All participants underwent H-MRS examination at 1.5 T (Eclipse; Philips Medical Systems, Cleveland, OH, USA). Single-voxel spectra were obtained from an 8 ml cubic volume of interest placed within the right thalamus to encompass the VPL sub-nucleus (Fig. 1a). Two spectra were acquired: (1) long echo time (TE) (TE = 135 ms, resonance time [TR] = 1600 ms) using a point-resolved technique (Fig. 1b); and (2) short TE (TE = 20 ms, TR = 5000 ms) using a stimulated-echo acquisition mode technique with a mixing time of 12 ms (Fig. 1c).

a Axial section of the brain with voxel positioned to encompass the ventroposterior thalamic subnucleus. b Example spectra obtained at long TE using a point-resolved acquisition technique and c at short TE using a stimulated-echo acquisition mode acquisition technique with a mixing time of 12 ms. Cho Choline, Cr creatine, G + sI scyllo-inositol; Glx, glutamine/glutamate; lac, lactate; lip, lipids; mI, myo-inositol; NAA, N-acetyl aspartate
MR spectra were anonymously analysed by an experienced clinical MR physicist. All post-acquisition processing was performed using fully integrated proprietary software from the manufacturer (Twinstar workstation, Philips Medical Systems, Cleveland, OH, USA). By convention, long TE results are expressed as ratios under the three prominent resonances assigned to choline (at 3.22 ppm), creatine (at 3.02 ppm) and NAA (at 2.02 ppm), i.e. NAA:creatine, NAA:choline and choline:creatine ratios. Short TE results were calculated as the areas under the myo-inositol (3.56 ppm), choline (3.22 ppm), creatine (3.02 ppm) and NAA (2.02 ppm) resonances relative to that of unsuppressed water.
Statistical method
Statistical analyses were performed using the SPSS 11.1 (SPSS Chicago, IL, USA). Baseline characteristics and study endpoints were described as means and standard deviations for normally distributed variables and as medians and range for variables with a skewed distribution.
The appropriate tests for normality were conducted to guide subsequent analysis. Subgroup H-MRS metabolite endpoints were compared using non-parametric tests (Kruskal–Wallis). Any relationships between these endpoints and neurophysiological assessments were analysed in more detail among participants with diabetes, using Spearman’s rank correlation coefficients.
Results
Clinical characteristics of participants
The mean ages, height and weight of the DPN, No-DPN and healthy volunteer groups were not significantly different (ANOVA, p = 0.63, p = 0.29 and p = 0.24 respectively) (Table l). As expected, participants in the DPN group had a longer duration of diabetes (p = 0.02) and higher HbA1c (p = 0.02).
Group spectroscopy findings
The DPN group had lower mean NAA:creatine ratios at long TE (median [range] 1.55 [1.14−1.95]; p = 0.04) than No-DPN participants (1.79 [1.22−2.20]) and healthy volunteers (1.90 [1.65−2.07]) (Table 1, Fig. 2a). NAA:choline ratio was also lowest in participants with DPN (1.55 [1.37−1.93]; p = 0.02) compared with No-DPN (1.80 [1.60−2.05]) and healthy volunteers (1.76 [1.58−1.86]) (Fig. 2b). The choline:creatine ratio obtained at long TE did not vary significantly between the groups (DPN 1.04 [0.84−1.23], No-DPN 0.95 [0.52−1.26], healthy volunteers 1.06 [0.89−1.25]; p = 0.87).

a N-Acetyl aspartate/creatine (NAA/Cr) and (b) N-acetyl aspartate/choline (NAA/Cho) ratios at long echo time in healthy volunteers (HV), diabetic patients with no neuropathy (No-DPN) and neuropathy (DPN). Horizontal lines represent group medians. Subgroup H-MRS metabolite differences (DPN vs No-DPN and HV) p = 0.04 (a), p = 0.02 (b)
Analysis of mean NAA metabolite resonances obtained at short-TE revealed no statistically significant difference between the groups (p = 0.07). No significant differences were observed with any of the other metabolites at short TE between any of the groups.
Spectroscopy and neurophysiological assessments
A significant positive association between both sural amplitude (ρ =0.61, p = 0.004) and nerve conduction velocity (ρ = 0.58, p = 0.006) and long TE NAA:creatine ratio signal was observed among participants with diabetes. In addition, the NAA:creatine ratio signal was significantly associated with peroneal latency (ρ = −0.53, p = 0.01), amplitude (ρ = 0.67, p = 0.001) and velocity (ρ = 0.37, p = 0.06) in this group. Similar correlations among participants with diabetes were noted with tibial nerve latency (ρ = −0.51, p = 0.02), but not with velocity (ρ = 0.23, p = 0.18). In this group, vibration detection threshold was related to NAA:choline ratio at a significant level (ρ = −0.70, p = 0.004), and we also observed a significant correlation between heart rate variability with deep breathing and NAA:creatine ratio (ρ = −0.46, p < 0.05). In addition, the NAA:creatine ratio was also found to relate to overall NCS (ρ = −0.53, p = 0.03) among diabetic participants.
Discussion
The main finding of this preliminary study is the significantly lower long TE thalamic NAA:creatine and NAA:choline ratios in participants with DPN compared with the No-DPN and healthy volunteer groups. The data also demonstrate significant correlations between NAA:creatine ratio and neurophysiological markers (overall NCS and individual nerve function tests) of DPN severity.
NAA is one of the highest concentrations of free amino acids in the brain and is widely used as a spectroscopic neuronal marker of intracranial disease progression. It is believed that a reduction in NAA resonance can be associated with neuronal/axonal loss or loss of neuron viability and dysfunction [13]. In the present study, spectra were acquired at two different TEs (short TE, long TE) from the same voxel. Each spectrum can provide different information regarding NAA. Reduced NAA signal obtained at short TE is thought to reflect decreased neuronal density, which may represent irreversible neuronal loss/shrinkage. Possible reversible neuronal injury/dysfunction can be implicated when the NAA:creatine ratio is reduced at long TE, as this results from changes in the NAA relaxation rates. Just as with standard spin–spin relaxation time (T2)-weighted imaging, variations in T2 imply alterations to the local neurochemical environment of NAA, which in turn affect its relaxation rates [14]. However, unlike reduction in neuronal density, alterations in T2 can be reversible which may account for restoration of metabolite ratio normality concomitantly with improvement in clinical neurological status [15, 16]. We demonstrated a significant difference in NAA:creatine ratio at long TE, but no significant difference in NAA resonance at short TE. In such a scenario, our findings may reflect thalamic neuronal dysfunction in patients with DPN rather than overt neuronal death.
The mechanism of thalamic involvement in DPN is unclear. One explanation may be that loss of afferent input, as a result of peripheral nerve damage, subsequently causes changes to occur at progressively higher levels in the central nervous system (‘dying back’ mechanism). The correlations observed between NAA and neurophysiological markers of neuropathy severity would seem to support this. It is also possible that the observed changes in the thalamus could be occurring concomitantly with changes in the peripheral nervous system. Nonetheless, thalamic neuronal involvement, whether early, late or concomitant, is likely to result in disturbed sensory gating in patients with DPN. This may have consequences for sensory perception and pain modulation. Prospective studies including a subgroup with painful DPN are required to determine at what stage during the course of the disease these abnormalities occur and what impact thalamic dysfunction has on pain perception in patients with diabetes.
Taken together with early spinal cord involvement in DPN [3], the possibility of thalamic sensory neuronal dysfunction suggests that nervous system involvement is not merely confined to the peripheral nerves, but involves the spinal cord and brain. Interestingly, various therapeutic interventions specifically targeted at peripheral nerve damage in DPN patients have thus far been ineffective [17], and it is possible that this may in part be due to inadequate appreciation of the full extent of central nervous system involvement. Recognition that DPN is a disease affecting the whole nervous system should trigger a critical rethinking of this disorder, opening a new direction for further research.
Abbreviations
- DPN:
-
diabetic peripheral neuropathy
- H-MRS:
-
proton magnetic resonance spectroscopy
- MR:
-
magnetic resonance
- NAA:
-
N-acetyl aspartate
- NCS:
-
Neuropathy Composite Score
- T2:
-
spin–spin relaxation time
- TE:
-
echo time
- TR:
-
resonance time
- VPL:
-
ventroposterior lateral
References
Boulton AJM, Malik RA, Arezzo JC, Sosenko JM (2004) Diabetic somatic neuropathies. Diabetes Care 27:1458–1486
Johnson FN, Williams R (1997) Economic aspects of diabetic neuropathy and related diabetic complications. In: Boulton AJM (ed) Diabetic neuropathy. Marius Press, Carnforth, UK, pp 77–95
Selvarajah D, Wilkinson ID, Emery CJ et al (2006) Early involvement of the spinal cord in diabetic peripheral neuropathy. Diabetes Care 29:2664–2669
McCormick DA, Bal T (1994) Sensory gating mechanisms of the thalamus. Curr Opin Neurobiol 4:550–556
Simmons ML, Frondoza CG, Coyle JT (1991) Immunocytochemical localization of N-acetyl-aspartate with monoclonal antibodies. Neuroscience 45:37–45
Miller BL (1991) A review of chemical issues in 1H NMR spectroscopy: N-acetyl-l-aspartate, creatine and choline. NMR Biomed 4:47–52
Brand A, Richter-Landsberg C, Leibfritz D (1993) Multinuclear NMR studies on the energy metabolism of glial and neuronal cells. Dev Neurosci 15:289–298
Grant IA, O’Brien P, Dyck PJ (1999) Neuropathy tests and normative results. In: Dyck PJ, Thomas PK (eds) Diabetic neuropathy. Saunders, Philadelphia, pp 123–141
Dyck PJ, O’Brien PC, Kosanke JL, Gillen DA, Karnes JL (1993) A 4, 2, and 1 stepping algorithm for quick and accurate estimation of cutaneous sensation threshold. Neurology 43:1508–1512
O’Brien IA, O’Hare P, Corrall RJ (1986) Heart rate variability in healthy subjects: effect of age and the derivation of normal ranges for tests of autonomic function. Br Heart J 55:348–354
American Diabetes Association American Academy of Neurology (1988) Consensus statement: report and recommendations of the San Antonio conference on diabetic neuropathy. Diabetes Care 11592–11597
Dyck PJ, Litchy WJ, Daube JR et al (2003) Individual attributes versus composite scores of nerve conduction abnormality: sensitivity, reproducibility, and concordance with impairment. Muscle Nerve 27:202–210
Nakano M, Ueda H, Li JY, Matsumoto M, Yanagihara T (1998) Measurement of regional N-acetylaspartate after transient global ischemia in gerbils with and without ischemic tolerance: an index of neuronal survival. Ann Neurol 44:334–340
Wilkinson ID, Paley MN, Hall-Craggs MA et al (1996) Cerebral magnetic resonance relaxometry in HIV infection. Magn Reson Imaging 14:365–372
Davie CA, Hawkins CP, Barker GJ et al (1994) Serial proton magnetic resonance spectroscopy in acute multiple sclerosis lesions. Brain 117:49–58
Wilkinson ID, Lunn S, Miszkiel KA et al (1997) Proton MRS and quantitative MRI assessment of the short term neurological response to antiretroviral therapy in AIDS. J Neurol Neurosurg Psychiatry 63:477–482
Boulton AJ (2007) Whither clinical research in diabetic sensorimotor peripheral neuropathy? Problems of end point selection for clinical trials. Diabetes Care 30:2752–2753
Acknowledgements
This study was supported by a grant from Diabetes UK. The authors would like to acknowledge the valuable help of the magnetic resonance spectrographers at the University of Sheffield, UK, and to thank all the volunteers, without whose participation this work would not have been possible.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Selvarajah, D., Wilkinson, I.D., Emery, C.J. et al. Thalamic neuronal dysfunction and chronic sensorimotor distal symmetrical polyneuropathy in patients with type 1 diabetes mellitus. Diabetologia 51, 2088–2092 (2008). https://doi.org/10.1007/s00125-008-1139-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-008-1139-0