
Abstract
Aims/hypothesis
The aim of this study was to investigate the action of the glucose-lowering compound sodium tungstate on glucose transport in muscle myotubes and to unravel the molecular events underlying the effects observed.
Methods
We studied the effects of tungstate on 2-deoxy-d-glucose uptake, levels and translocation of the glucose transporters GLUT4 and GLUT1, and Glut4 (also known as Slc2a4) promoter activity. We also measured the modifications of individual components of the signalling pathways involved in the effects observed.
Results
Tungstate increased 2-deoxy-d-glucose uptake in differentiated L6 myotubes through an increase in the total amount and translocation of GLUT4 transporter. The effects on glucose uptake were additive to those of insulin. Tungstate activated transcription of the Glut4 promoter, as shown by an increase in Glut4 mRNA, and by a promoter reporter assay. The assay of deletions of the Glut4 promoter indicated that the effect of tungstate is mediated by the myocyte enhancer factor 2 (MEF2)-binding domain. Accordingly, MEF2 levels and DNA binding activities were increased in response to the treatment. Tungstate-induced glucose uptake and GLUT4 transcriptional activation were dependent on the activation of extracellular signal-regulated kinases 1 and 2 (ERK1/2), while no changes were observed in the phosphorylation state of the β subunit of the insulin receptor, in the phosphatidylinositol 3-kinase pathway or in the activation of 5′AMP-activated protein kinase.
Conclusions/interpretation
Tungstate activates glucose uptake in myotubes through a novel ERK1/2-dependent mechanism. This effect is exerted by an increase in the content and translocation of the GLUT4 transporter. This is the first report of a glucose-lowering compound activating Glut4 transcription through an ERK1/2-dependent increase in MEF2 levels.
Similar content being viewed by others


Introduction
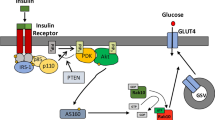
The major metabolic pathways regulated by insulin include the stimulation of glucose transport in muscle. Glucose uptake is mediated in this tissue mainly by the insulin-regulated glucose transporter GLUT4. In the absence of insulin, this transporter slowly recycles between an intracellular storage compartment and the plasma membrane. In response to the hormone or to physical exercise, the amount of GLUT4 located at the plasma membrane increases, which accounts for the augmented transport capacity [1]. Insulin-induced GLUT4 translocation is dependent on the activation of phosphatidylinositol 3-kinase (PI3K) [2, 3], while physical exercise enhances translocation by the activation of 5′AMP-activated protein kinase (AMPK) [4]. A novel mechanism of glucose signalling through a PI3K-independent mechanism, but dependent on the activation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) and phospholipase D, has also been described and may be relevant to GLUT translocation [2, 5].
A second regulatory mechanism of glucose transport is the control of GLUT4 (also known as SLC2A4) gene expression, which is subject to tissue, hormonal and metabolic regulation (as reviewed in [6]). Changes in GLUT4 gene expression levels are observed in physiological states of altered glucose homeostasis and in those of relative insulin deficiency [7]. The GLUT4 gene is subjected to a complex system of tissue-specific and metabolic regulation. Two regulatory domains of the 5′-flanking DNA region are required to direct a complete programme of GLUT4 gene regulation [7–10]. The region referred to as domain II or myocyte enhancer factor 2 (MEF2) domain binds the isoforms of the MEF2 family of transcription factors (MEF2A, MEF2B, MEF2C and MEF2D) [11–13]. A second region, named domain I, binds nuclear factor I (NF1) [14] and a transcriptional activator named GLUT4 enhanced factor [13–15].
Tungstate is an oral glucose-lowering [16–22] and anti-obesity [23] agent. This compound has a low toxicity profile in animals and humans, and is currently undergoing Phase II clinical trials. Tungstate normalises carbohydrate metabolism in liver [16, 18, 19], stimulates insulin secretion and regenerates pancreatic beta cells in neonatally streptozotocin-induced diabetic rats [24]. In streptozotocin-induced diabetic rats, tungstate also increases GLUT4 production and translocation in diaphragm [25]. Here, we dissect the molecular mechanisms that drive the tungstate-induced increase in glucose transport in muscle cells. We found that tungstate increases glucose transport in L6 differentiated myotubes through an ERK1/2-dependent mechanism involving an increase in the production and translocation of GLUT4. This double effect on glucose transport emphasises the glucose-lowering potential of this compound.
Methods
Materials
Sodium tungstate, PD98059 and horseradish peroxidase-conjugated secondary antibodies were from Sigma (St Louis, MO, USA). 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) was from Toronto Research Chemicals (North York, ON, Canada). Tissue culture media and supplements were from Sigma and Invitrogen (Carlsbad, CA, USA). FCS was from Cultek (Madrid, Spain). ERK1/2 and phosphotyrosine (4G10) antibodies were from Upstate (Waltham, MA, USA). Phospho-p44/42 mitogen-activated protein kinase E10 (Thr202/Tyr204), AMPKα2 and phospho-AMPKα2 (Thr172) antibodies were from Cell Signaling (Beverly, MA, USA). Phospho-protein kinase B/Akt (Ser473) antibody was from Nanotools (Teningen, Germany). GLUT1, NF1, MEF2, MEF2C and MEF2D antibodies were from Santa Cruz Biotechnologies (Santa Cruz, CA, USA).
Cell culture
The L6.C11 rat skeletal muscle myoblast line (ECACC No.92102119) was grown in DMEM containing 10% (vol./vol.) FCS. Cells were differentiated by culturing for 5 to 6 days in DMEM containing 2% (vol./vol.) FCS (>50% fusion into multinucleated myotubes). Prior to each experiment, myotubes were incubated in FCS-free medium for 18 to 24 h. For long-term incubations, a tungstate concentration of 100 μmol/l was chosen, while higher concentrations (500 μmol/l to 1 mmol/l) were used for short-term experiments.
2-Deoxy-d-[1-3H]glucose uptake
Differentiated myotubes were incubated in FCS-free medium for 18 to 24 h. Tungstate and insulin treatments were performed in serum-free medium. Triplicate measurements of 2-deoxy-d-[1-3H]glucose (2-DG) uptake were taken after 10 min of incubation as described [26].
Subcellular fractionation
Membrane fractions from myotubes were prepared as described [26, 27]. 5′-Nucleotidase and cytochrome c reductase activities were assayed as marker enzymes for plasma membranes and low-density microsomes, respectively [25].
GLUT1 and GLUT4 protein analyses
L6 myotube membrane fraction proteins were electrophoresed in 10% (wt/vol.) SDS-polyacrylamide gels and processed for western blot with anti-GLUT1 or anti-GLUT4 antibodies (Biogenesis, Poole, UK). Immunoreactive bands were visualised by chemiluminescence and quantified with NIH Image Software [28].
Quantitative RT-PCR
cDNA was generated with a kit (First-strand cDNA Synthesis; GE Health Care, Uppsala, Sweden) using total RNA from the myotubes [29]. Real-time PCR was performed using SYBR Green I dye and the primers: Glut4 5′-GTCATCAACGCCCCACAGAA-3′ and 5′-GCAAGGACAGTGGACGCTCTCTTTC-3′; MyoD (also known as Myod1) 5′-AGGGAAGGGAAGAGCAGAAG-3′ and 5′-TACACCTGTTACACCCGAGAT-3′; and β-actin 5′-GGCCAACCGTGAAAAGATG-3′ and 5′-GGATCTTCATGAGGTAGTCTGTC-3′. Amplifications were carried out using a Stratagene Mx3000P system (Cedar Creek, TX, USA). In each run, standard curves were generated for a primer set by serial dilution of plasmid DNA encoding the relevant cDNA. Melting curves were generated after each run and the fidelity of RT-PCR was also confirmed by electrophoresis (not shown). Glut4 and MyoD mRNA levels were calculated using the real-time PCR standard curve method.
Protein phosphorylation analysis
L6 myotubes were incubated in FCS-free medium for 18 to 24 h and then treated with tungstate (1 mmol/l) or insulin (100 nmol/l) in serum-free medium. After treatment, plates were processed as described in [30]. Proteins were separated by SDS-PAGE and immunoblotted with selected antibodies.
Western blots of 5′AMP-activated protein kinase
L6 myotubes were incubated in FCS-free medium for 24 h and then treated in the absence or presence of insulin (100 nmol/l), sodium tungstate (1 mmol/l) or AICAR (0.5 mmol/l). Plates were flash-frozen in liquid nitrogen and scraped with lysis buffer: 50 mmol/l Tris–HCl (pH 7.4), 150 mmol/l NaCl, 1% (vol./vol.) Triton X-100, 10% (vol./vol.) glycerol, 1 mmol/l EDTA, 1 mmol/l EGTA, 10 mmol/l NaF, 10 mmol/l sodium pyrophosphate, 1 mmol/l sodium orthovanadate, 1 mmol/l dithiothreitol, 4 μg/l leupeptin and 0.5 mmol/l phenylmethylsulfonyl fluoride. The lysates were centrifuged for 30 min at 11,000×g and 4°C. Samples were separated by SDS-PAGE and immunoblotted with anti-AMPKα2 or anti phospho-AMPKα2 (Thr172) antibodies.
Design of reporter gene constructs
The pGL3-ratGLUT4 construct was made by cloning a genomic DNA fragment encompassing positions −2212 to + 164 of the rat Glut4 gene from clone −2212/+147G4CAT (a gift from A. Zorzano [31]), into pGL3-Basic vector (Promega, Madison, WI, USA). pGL3-ratGLUT4Δ1 was obtained by deletion of a SmaI–BstXI fragment of pGL3-ratGLUT4, encompassing positions −502 to −243. pGL3-ratGLUT4Δ2 was generated by removing a SmaI–SacI fragment, which encompasses positions −1042 to −502. The sequence of all constructs was verified by automated DNA sequencing.
DNA transfection assays
Cells were used at 80–90% confluence. Transfection was performed using LipofectAMINE 2000 (Invitrogen). The DNA mixture comprised the various GLUT4-luciferase reporters and the reference plasmid pRL-TK (ratio 1:5). The pRL-TK plasmid codes for Renilla luciferase under the control of the tk promoter as an internal control. Following removal of the transfection reagent, fresh medium was added to the cultures. In the case of cultures set to differentiate into myotubes, differentiation medium was applied 24 h later. Next, where indicated, the cells were incubated for 24 h with insulin (100 nmol/l) or tungstate (0.1–1 mmol/l), and luciferase activity was determined using the dual luciferase method (Promega). Results were standardised for Renilla luciferase activity. All transfections were performed at least three times, in triplicate, using at least two preparations of plasmid DNA. To allow comparison of the expression patterns between myoblasts and myotubes, the data are expressed as relative changes in luciferase activity: luciferase activity of control myoblasts was normalised to a value of 100% and the remaining activities were referred to this value.
Preparation of nuclear extracts
Nuclear extracts from L6 cells were prepared as described [32]. Aliquots of the extracts (40 μg) were resolved by 10% (wt/vol.) SDS-PAGE and analysed by immunoblotting using anti-NF1, anti-MEF2, anti-MEF2C or anti-MEF2D antibodies.
Electrophoretic mobility shift assays
Electrophoretic mobility shift assays (EMSAs) were performed using a double-stranded oligonucleotide probe that contains the functional MEF2 binding site present in the Glut4 promoter: forward, 5′-GATCGCTCTAAAAATAACCCTGTCG-3′; reverse, 5′-CGACAGGGTTATTTTTAGAGCGATC-3′; the recognition sequence for MEF2 is italicised [33]. The double-stranded oligonucleotide was end-labelled using T4 polynucleotide kinase and γ-[32P]ATP (GE Healthcare). Binding reactions of the probes (200 fmol) were performed with 10 μg protein from nuclear extracts, at 4°C for 45 min, in 20 μl of the binding buffer consisting of 20 mmol/l HEPES, pH 7.9, 2.5 mmol/l MgCl2, 10% (vol./vol.) glycerol, 1 mmol/l DTT, 0.1 mg/ml bovine serum albumin and 30 ng/μl of polydeoxyinosinic-deoxycytidylic acid. Competitive assays were conducted under the same conditions, with the addition of 100-fold molar excess of unlabelled oligonucleotides. For supershift studies, extracts were pre-incubated on ice for 30 min with 1 μg of MEF2A, MEF2C or MEF2D antibody before addition of the labelled probe. The DNA–protein complexes were electrophoresed on 5% (wt/vol.) non-denaturing polyacrylamide gels at 4°C in 45 mmol/l Tris, 45 mmol/l borate pH 8 and 1 mmol/l EDTA buffer.
Creatine kinase activity assay
Creatine kinase activity in L6 myotube cultures was assayed in accordance with a previously described method [34]. Cells were lysed with 1% (vol./vol.) Triton X-100 in PBS. After centrifugation (16,000×g), creatine kinase activity of the supernatant fractions was measured using a creatine kinase NAC kit (Chemelex, Barcelona, Spain).
Statistical methods
Results are expressed as means ± SEM for the number of experiments indicated. The statistical significance of variations was evaluated using one-way ANOVA. When a significant effect was found, post hoc comparisons of the means were done using the t adjusted Tukey test. A p value <0.05 was considered significant.
Results
Tungstate stimulates glucose uptake
L6 myotubes were treated for 24 h in serum-free medium and then incubated with increasing concentrations of tungstate (0–500 μmol/l) for 24 h. Tungstate increased 2-DG uptake in a dose-responsive manner (Fig. 1a). To make a comparative study of tungstate- vs insulin-induced increase in glucose transport, myotubes were treated for 24 h in serum-free medium and then incubated with tungstate (100 μmol/l), insulin (100 nmol/l) or a combination for 2 or 24 h. Both tungstate and insulin increased 2-DG uptake in a similar manner (Fig. 1b). Furthermore, when myotubes were incubated for 2 h with both compounds or for 24 h with tungstate and then for an additional 2 h with only insulin, 2-DG uptake was higher than that for either tungstate or insulin alone. Accordingly, the dose–response curve for insulin in the presence or absence of tungstate showed an additive effect on 2-DG uptake when tungstate was present (Fig. 1c).

Effects of tungstate on 2-DG uptake in L6 myotubes. a Cells were incubated for 24 h with increasing concentrations of tungstate (0–500 μmol/l). The relative increments of 2-DG uptake (expressed as percentage increase) compared with basal transport are shown, and the data have been fitted to a hyperbolic saturation curve with a semi-maximal stimulating concentration of tungstate of 75.9 ± 16.1 μmol/l and a maximal increase in 2-DG uptake of 91.8 ± 6.4%. 2-DG uptake was determined in triplicate as described in the ‘Methods’. Results are means ± SEM of four independent experiments. b Cells were incubated in the absence (control [C]) or the presence of insulin (I, 100 nmol/l) or tungstate (W, 100 μmol/l) or both effectors for 2 h (white bars) or 24 h (black bars). The last black bar on right shows the 2-DG uptake of L6 myotubes incubated with W for 24 h and then with I for 2 h (W + I2h). Results are means ± SEM of four independent experiments. a p < 0.05 and b p < 0.001 for difference from control cells; c p < 0.05 for difference from cells treated with W for 2 h; d p < 0.001 for difference from cells treated with W for 24 h. c Cells were incubated for 24 h with increasing concentrations of I (0–100 nmol/l) in the absence (black circles) or presence (white circles) of W (100 μmol/l). The relative increments of 2-DG uptake (expressed as percentage increase) compared with basal transport are shown. Results are the means ± SEM of four independent experiments
Tungstate treatment increases the total amount of GLUT4 and transcription of the gene
We next studied whether the stimulation of 2-DG uptake was correlated with an increase in the total amount of GLUT4 or GLUT1. After incubating myotubes with 100 μmol/l tungstate for 24 h, the amount of these transporters in cell homogenates was quantified using actin as a control. Tungstate-treated myotubes showed a higher content of GLUT4 than control or insulin-treated cells (Fig. 2a). In contrast, the amount of GLUT1 was not modified. Accordingly, a comparative analysis by quantitative RT-PCR showed an increase in Glut4 transcript abundance in tungstate-treated cells (Fig. 2b). To rule out an action of tungstate on myotube differentiation, we measured MyoD mRNA levels, as a marker of the degree of differentiation [6]. No differences were observed in MyoD mRNA abundance in insulin- or tungstate-treated cells compared with untreated cells (Fig. 2b).

Effects of tungstate on GLUT4 and GLUT1 protein levels and Glut4 transcription in L6 myotubes. Cells were incubated in the absence (control [C]) or presence of tungstate (W, 100 μmol/l) or insulin (I, 100 nmol/l) for 24 h. a Representative Western blots showing GLUT4, GLUT1 and β-actin from control, I-treated and W-treated myotubes. The graph shows the results of the densitometric quantification of GLUT1 (white bars) and GLUT4 (black bars) levels, using β-actin as a reference control. Signal densities from control cells were assigned a value of 100% for each transporter. Results are expressed as the means ± SEM for six samples per condition. a p < 0.001 compared with control cells; b p < 0.001 compared with insulin-treated cells. b Quantitative RT-PCR of L6 myotubes. Abundance of Glut4 (black bars) and MyoD (white bars) mRNA was measured by quantitative RT-PCR and expressed relative to the value in myoblast cells. Results are the means ± SEM of four samples per condition. a p < 0.05, b p < 0.001 compared with control myotube cells. ΔΔCt, difference in ΔCt between the test groups and the reference, where ΔCt corresponds to the change in the threshold cycle value. c Effects of insulin and tungstate on Glut4 promoter activity. pGL3-ratGLUT4 construct was transfected in L6 cells. Differentiated (black bars) and non-differentiated (white bars) cells were incubated in the absence (C) or presence of I (100 nmol/l) or W (100 μmol/l and 1 mmol/l) for 24 h. At the end of the treatments, firefly luciferase activity was measured as described in the ‘Methods’. The luciferase activity from non-differentiated control cells was assigned a value of 100%. Values are the means ± SEM (n = 7). a p < 0.05, b p < 0.01, c p < 0.001 compared with non-differentiated or differentiated control cells; d p < 0.001 compared with myotubes treated with 0.1 mmol/l W. d Luciferase activity of cells transfected with pGL3-ratGLUT4, pGL3-ratGLUT4Δ1 and pGL3-ratGLUT4Δ2 constructs. L6 cells were transfected with the three constructs, differentiated to myotubes, deprived of FCS for 24 h and incubated in the absence (C) or presence of I (100 nmol/l) or W (1 mmol/l) for 24 h. For each plasmid, relative luciferase activity in the control cells (pGL3-ratGLUT4: 1784 ± 72 RLU; pGL3-ratGLUT4Δ1: 1927 ± 193 RLU and pGL3-ratGLUT4Δ2: 4940 ± 741 RLU) was normalised to a value of 100%. Values are means ± SEM (n = 6). a p < 0.001 vs control cells. RLU, relative luminescence units
To determine the actions of tungstate on the transcription of Glut4, we assayed the effects of this compound on the Glut4 promoter. For this purpose, a pGL3-ratGLUT4 luciferase construct encompassing positions −2212 to +164 of the rat Glut4 gene that contain the main regulatory elements of the promoter [7] was transfected in L6 cells. Differentiated and non-differentiated cells were pre-incubated for 24 h with FCS-free medium and further incubated (24 h) with insulin (100 nmol/l) or tungstate (100 μmol/l and 1 mmol/l). The luciferase activity in myotubes was approximately twice that in myoblasts (Fig. 2c). Insulin treatment decreased the activity of the Glut4 promoter in myoblasts and myotubes, as previously described in adipocytes [14]. In contrast, tungstate increased luciferase activity up to 1.6-fold at 100 μmol/l and 1 mmol/l. In non-differentiated cells, the compound still had a significant, but lower effect (Fig. 2c).
We next examined the role of domains I and II of the Glut4 promoter [12] on tungstate-stimulated Glut4 transcription. For this purpose, we used two constructs: pGL3-ratGLUT4Δ1, which includes the MEF2 binding site in the rat promoter [11, 31], and pGL3-ratGLUT4Δ2, which includes the putative NF1 binding site [14]. L6 cells were transfected and luciferase activity was assayed in myotubes after the addition of insulin (100 nmol/l) or tungstate (1 mmol/l).
When the pGL3-ratGLUT4Δ2 construct was assayed, transcription levels were higher than with the undeleted promoter. This can probably be attributed to the abolition of the negative effects of NF1 binding on transcriptional activity [14]. When the cells were incubated with insulin or tungstate, the already elevated transcription levels were not further modified (Fig. 3a). Accordingly, tungstate treatment did not change the total amount of NF1 in nuclear extracts, while insulin increased its levels more than fourfold (Fig. 3b).

Effects of tungstate treatment on the protein levels of MEF2 family and NF1 in nuclear extracts from L6 myotubes. Cells were incubated in the absence (control [C]) or presence of insulin (I, 100 nmol/l) or tungstate (W, 1 mmol/l) for 24 h. Nuclear extracts (40 μg) were subjected to 10% (wt/vol.) SDS-PAGE and immunoblotted against MEF2A, MEF2C and MEF2D antibodies (a) or NF1 antibody (b). a Levels of the MEF2 family. A representative western blot of MEF2C and MEF2D is presented together with a graph showing the results of the densitometric quantification of MEF2A (white bars), MEF2C (black bars) and MEF2D (grey bars). Levels (mean ± SEM, n = 4) are expressed as a percentage relative to those in control cells. a p < 0.05, b p < 0.01, c p < 0.001 vs control cells. b A representative western blot of NF1, together with a graph of the results of densitometric analysis. Levels (mean ± SEM, n = 4), are expressed relative to those in control cells. a p < 0.001 vs control cells. c Representative autoradiograph of an MEF2 EMSA. Cells were incubated in the absence or presence of W (100 μmol/l) for 24 h. W-treated nuclear extracts were pre-incubated with a 100-fold excess of unlabelled DNA probe (−). The arrow marks the position of the shifted oligonucleotide band. The intensity in W-treated cells was quantified and is expressed in the graph (mean ± SEM, n = 4) relative to that in control cells. a p < 0.001 vs control cells. d Cells were incubated in the absence (C) or presence of I (I20: 20 nmol/l or I100: 100 nmol/l) or W (100 μmol/l) or a combination of the two I concentrations plus W for 24 h. Creatine kinase activity was measured in the cell lysates as described (‘Methods’). Results are means ± SEM for four samples per condition. a p < 0.05, b p < 0.01 vs control cells; c p < 0.01 vs insulin-treated cells; d p < 0.01 vs W-treated cells
In contrast to the pGL3-ratGLUT4Δ2 construct, basal transcription levels from pGL3-ratGLUT4Δ1 were similar to those with the undeleted construct. The luciferase activity of myotubes transfected with pGL3-ratGLUT4Δ1 was not modified by treatment with tungstate. In contrast, insulin-treated cells still showed a significant reduction in transcription (Fig. 2d). These data indicate that the MEF2 binding site is essential for the promotion of Glut4 transcription induced by tungstate. To further this observation, we first studied whether tungstate treatment increased the amount of individual components of this family of transcription factors. A comparative analysis showed that MEF2A and MEF2C nuclear levels were moderately increased in tungstate-treated myotubes (∼1.25-fold) compared with either control or insulin-treated cells (∼0.85-fold). Strikingly, the nuclear content of MEF2D in tungstate-treated cells was increased more than twofold (Fig. 3a). Second, we analysed by means of an EMSA with a double-stranded oligonucleotide corresponding to the rat GLUT4 MEF2 binding domain whether MEF2 binding activity was modified by tungstate (Fig. 3c). MEF2 binding activity was significantly increased in tungstate-treated myotubes (∼1.65-fold) compared with control cells. A supershift analysis for distinct MEF2 isoforms using anti-MEF2 A, C and D antibodies showed changes in band mobility for MEF2D in the tungstate-treated cells (Electronic supplementary material [ESM] Fig. 1).
Since the transcription of the gene encoding creatine kinase (Ckm) is upregulated by MEF2 [35], we tested the effects of tungstate on total creatine kinase activity in L6-treated myotubes as additional proof of the action of this compound on this family of transcription factors. Creatine kinase activity was increased in cells treated either with insulin (20 or 100 nmol/l) or tungstate (0.1 mmol/l). The combination of both effectors produced an additive effect (Fig. 3d).
Taken together, these results indicate that tungstate treatment increased the total amount of GLUT4 in L6 myotubes through an upregulation of Glut4 transcription driven by MEF2 transcription factors.
Tungstate treatment increases GLUT4 translocation to the plasma membrane
Since GLUT4 translocation to the plasma membrane is the rate-limiting step for glucose uptake in muscle, we tested whether tungstate treatment stimulated GLUT4 translocation in addition to increasing its total cellular content. The analysis of GLUT4 and GLUT1 distribution in plasma membranes and low-density microsomes of myotubes was performed after subcellular fractionation. 5′-Nucleotidase and cytochrome c reductase activities were assayed as marker enzymes of the two fractions and were enriched in the isolated plasma membranes and low-density microsomes respectively (ESM Table 1). Tungstate treatment induced an approximately 60% increase in GLUT4 in plasma membrane-enriched fractions when compared with control counterparts (Fig. 4a). This increase was similar to that observed after insulin treatment. Accordingly, the treatments with insulin or tungstate induced a significant decrease in GLUT4 in low-density microsomes. These results indicate that both insulin and tungstate promote translocation of the transporter from the low-density microsomes to the plasma membrane (Fig. 4a). In contrast, GLUT1 (Fig. 4b) was present mainly in the plasma membrane fractions, without variations in its relative amounts between the experimental groups.

Effects of tungstate on the subcellular distribution of GLUT4 and GLUT1 in L6 myotubes. Cells were incubated in the absence (control [C]) or presence of tungstate (W, 100 μmol/l) or insulin (I 100 nmol/l) for 24 h. Subcellular fractions of the L6 myotubes were prepared and subjected to SDS-PAGE and western blotting using GLUT4 (a) or GLUT1 antibodies (b). Blots show typical results in plasma membranes (PM) and low-density microsomes (LDM). Bar graphs show the results of quantification of GLUT4 (a) or GLUT1 (b) protein levels in PM (white bars) and in LDM (black bars) by densitometry. Levels (means ± SEM, n = 4) are expressed in relation to those of GLUTs in plasma membranes of control cells. a p < 0.05, b p < 0.01 compared with PM from control cells; c p < 0.01 compared with LDM from control cells in a. a p < 0.001 compared with PM from control cells in b
Tungstate-induced GLUT4 translocation is independent of PI3K and AMPK activation
To gain insight into the targets of tungstate action involved in its actions in muscle cells, we studied how the key components of the insulin and AMPK transduction pathways are modulated by this compound. We first analysed the involvement of the insulin receptor in the tungstate-mediated increase in GLUT4 translocation. After incubation (5, 15 and 60 min) of myotubes with tungstate, no significant changes were observed in the phosphorylation of the insulin receptor β subunit. In contrast, insulin induced a rapid phosphorylation of its receptor (not shown).
We next assayed whether tungstate-induced GLUT4 translocation was dependent on PI3K activation. Myotubes were incubated for 2 h with insulin or tungstate in the absence or presence of 100 nmol/l wortmannin, a PI3K inhibitor. Wortmannin treatment reduced (∼40%) the insulin-stimulated 2-DG uptake (Fig. 5a), while it did not modify the tungstate-induced glucose uptake. Therefore, in contrast to insulin, the increase in glucose transport capacity induced by tungstate was not dependent on active PI3K. Accordingly, tungstate treatment did not affect the phosphorylation of protein kinase B (Akt), a pivotal downstream element of the PI3K cascade (Fig. 5b).

Analysis of the effects of tungstate on the signalling pathways involved in GLUT4 translocation. a Analysis of the effects of wortmannin on 2-DG uptake in L6 myotubes treated with insulin (I; 100 nmol/l) or tungstate (W; 0.1 mmol/l) for 2 h. Wortmannin (100 nmol/l) was added 30 min before incubation with the effectors and was essentially not diluted by the addition of the treatments. Results are expressed as means ± SEM of four independent experiments. a p < 0.01 and b p < 0.001 compared with control cells; c p < 0.01 compared with insulin-treated cells. b Blot and quantification of W-induced phosphorylation of protein kinase B (PKB). L6 myotubes were incubated in the absence (control [C]) or presence of I (100 nmol/l) or W (1 mmol/l) for 5, 15 or 60 min. Cells were lysed and total protein was subjected to 10% SDS-PAGE. It was then immunoblotted with antibodies against total (Pan) PKB, and PKB phosphorylated at Ser 473. The results are representative of three independent experiments. c Blot and quantification of W-induced phosphorylation of AMPK. L6 myotubes were incubated for 15 min with I or W as above (b), lysed and analysed for total AMPK or phospho-AMPK content, as described in the ‘Methods’. As a positive control, L6 myotubes were incubated with 0.5 mmol/l AICAR (A) for 15 min
We next assayed whether tungstate-stimulated GLUT4 translocation was dependent on the activation of AMPK [4]. We analysed the key phosphorylation site for AMPK activation, Thr172 from α subunit. Neither tungstate nor insulin treatment modified the phosphorylation of this kinase, in contrast to the AMPK activator, AICAR (Fig. 5c).
Tungstate-stimulated GLUT4 synthesis and translocation is dependent on stimulation of the ERK1/2 signalling pathway
The activation of ERK1/2 has been described as one of the key features of tungstate action in other cell systems. Therefore, we tested whether tungstate modulated these kinases in myotubes. Neither insulin nor tungstate treatments had a significant effect on ERK1/2 levels. In contrast, both insulin and tungstate induced ERK1/2 phosphorylation on Thr202/Tyr204 (Fig. 6a), which was sensitive to PD98059, a specific inhibitor of ERK1/2 (Fig. 6b). Furthermore, incubation of myotubes with increasing concentrations of PD98059 (0–50 μmol/l) reduced the tungstate-induced increase in 2-DG uptake in a dose-dependent way (Fig. 6c). No changes were observed in the increase of 2-DG uptake mediated by insulin in the PD98059-treated cells. Consequently, these results indicate that the increase in glucose uptake induced by tungstate is dependent on the activation of ERK1/2.

Analysis of the effects of tungstate on ERK1/2 activation and its involvement in the tungstate-induced increase of glucose uptake. a Blot and quantification of tungstate-induced phosphorylation of ERK1/2. L6 myotubes were incubated in the absence (control [C]) or presence of insulin (I; 100 nmol/l) or tungstate (W; 1 mmol/l) for 5, 15 or 60 min. Cells were lysed and total protein was subjected to 10% (wt/vol.) SDS-PAGE. It was then immunoblotted with p44/42 mitogen-activated protein kinase (MAPK) and phospho-p44/42 MAPK antibodies. The results are representative of three independent experiments. b Blot and quantification of effects of PD98059 on W-induced ERK1/2 phosphorylation. L6 myotubes were pre-incubated in the absence or presence of PD98059 (30 μmol/l) and then incubated for 15 min with I (100 nmol/l) or W (W0.1: 0.1 mmol/l; W1: 1 mmol/l). PD98059 was added 30 min before incubation with the effectors and was essentially not diluted by the addition of the treatments. Cells were processed as above (a). c L6 myotubes were pre-incubated with increasing concentrations of PD98059 (0–50 μmol/l) and then incubated for 2 h in the absence (white bars) or presence (hatched bars) of I (100 nmol/l) or W (1 mmol/l) (black bars). PD98059 was added 30 min before the effectors and maintained during the treatments. 2-DG uptake was determined as described in the ‘Methods’. Results are means ± SEM of four independent experiments. a p < 0.05, b p < 0.01, c p < 0.001 compared with control cells
In addition, we assayed the dependence of the tungstate-induced Glut4 transcription on the activation of ERK1/2. L6 myotubes transfected with pGL3-ratGLUT4 were incubated in the absence or presence of PD98059 (10 μmol/l) and then in the absence or presence of insulin (100 nmol/l) or tungstate (0.5 mmol/l; Fig. 7a). The tungstate-induced increase in luciferase activity was reduced to control values upon incubation with the specific inhibitor of ERK1/2 phosphorylation.

Involvement of ERK1/2 and phospholipase D in GLUT4 upregulation. a Effects of PD98059 on Glut4 promoter activity. pGL3-ratGLUT4 construct was transfected in L6 cells. L6 myotubes were pre-incubated in the absence (white bars) or presence (black bars) of PD98059 (10 μmol/l), and then incubated for 6 h in the absence or presence of insulin (I, 100 nmol/l) or tungstate (W, 0.5 mmol/l). Cells were pre-treated with PD98059 for 30 min before addition of the effectors and during the treatments. The incubation media were changed every 3 h. At the end of the treatments, firefly luciferase activity was measured. The luciferase activity from control cells was assigned a value of 100. Values are the means ± SEM (n = 4). a p < 0.05, b p < 0.01 compared with control myotubes; c p < 0.01 compared with myotubes treated with 0.5 mmol/l tungstate. b Effect of n-butanol on 2-DG uptake. L6 myotubes were pre-incubated for 30 min in the absence (white bars) or presence (black bars) of 1% n-butanol and then incubated with AICAR (50 μmol/l) or tungstate (W; 1 mmol/l) for 1 h. Results are means ± SEM of four independent experiments. a p < 0.01 compared with control cells
Since our data indicate that tungstate-induced activation of ERK1/2 is independent of PI3K or AMPK activation, we examined whether it was dependent on a transduction pathway variant that is stimulated by glucose [5, 36]. This pathway ultimately involves the ERK1/2-dependent activation of phospholipase D. To address this question, we measured 2-DG uptake in the presence of n-butanol, an inhibitor of phospholipase D [5]. AICAR was used as positive control of 2-DG uptake. The positive effect on 2-DG uptake exerted by tungstate or AICAR (50 μmol/l) was completely blocked by pre-incubation with 1% (vol./vol.) n-butanol (Fig. 7b).
Discussion
The effects of tungstate, a well-characterised glucose-lowering compound, have been studied in several key tissues involved in the maintenance of glucose homeostasis, namely liver, pancreas and adipose tissue [16–19, 23, 24]. In contrast, little is known about the effects of this compound in muscle. A previous report showed that tungstate treatment normalises both the intracellular distribution of GLUT4 and its levels in diaphragms of diabetic animals, thereby suggesting that it increases glucose uptake in muscle [25]. The present study addresses this question at the molecular level. First, our results demonstrate that tungstate treatment increases 2-DG transport in myotubes, through a rise in the total amount of GLUT4 transporter (long-term effect) and its translocation to the plasma membrane (short-term effect). Moreover, the short-term effects of tungstate and insulin on 2-DG uptake were additive, thereby indicating distinct but convergent signalling pathways. This finding may be of relevance for treatment purposes, as by acting through an alternative mechanism, this compound may circumvent the insulin resistance that is characteristic of type 2 diabetic patients. Accordingly, the tungstate-induced increase in 2-DG uptake did not involve activation of the insulin receptor and members of its amplification cascade. In addition, we show that this increase is also independent of the phosphorylation of AMPK. These observations indicate the existence of a PI3K- and AMPK-independent pathway to increase capacity for glucose uptake in muscle cells upon treatment with tungstate. We show that the tungstate-induced effects on GLUT4 (increased translocation and production) are dependent on the activation of ERK1/2. In this context, several authors have associated the activation of ERK1/2 with increased glucose uptake in adipocytes and muscle cells when incubated with elevated concentrations of glucose [2, 5]. They propose that high glucose concentrations increase GLUT4 translocation through the activation of AMPK and initiation of a signalling cascade involving ERK1/2 phosphorylation, and through the activation of phospholipase D as well as atypical protein kinase Cs. Our results suggest that tungstate partly stimulates this pathway, but without the participation of AMPK. This is supported by the observation that an inhibitor of phospholipase D blocks short-term tungstate-mediated 2-DG uptake. Furthermore, tungstate-stimulated glycogen deposition in hepatocytes occurs through an ERK1/2-dependent mechanism [30]. This observation indicates that the activation of these kinases is a key step underlying the glucose-lowering actions of this compound.
One of the key actions of tungstate in myotubes is the stimulation of Glut4 gene transcription. Tungstate-induced Glut4 promoter activation requires the presence of the domain containing the MEF2 binding site. In addition, tungstate-treated myotubes increased the levels and DNA-binding activities of MEF2A, MEF2C and MEF2D. This increase was confirmed after analysis of creatine kinase activity in tungstate-treated myotubes (in muscle, the Ckm promoter contains a canonical MEF2 binding site) [33, 34]. This effect could contribute to the improvement in cardiac performance of tungstate-treated streptozotocin-induced diabetic rats [22]. Taken together, these results indicate that the key point in the tungstate-stimulated transcription of Glut4 is the induced production of members of the MEF2 family. It has been described that Glut4 transcription is significantly upregulated through an increase in the production or activity of MEF2 transcription factors [6]. In fact, an increase in MEF2A and MEF2D levels contributes to the adaptation of skeletal muscle to exercise [37] while, in diabetic animals, MEF2 DNA-binding activity and/or concentration is substantially reduced and correlates with a decrease in Glut4 transcription [12, 38].
In contrast to the effects of tungstate, insulin treatment moderately decreased the GLUT4 reporter gene activity in myotubes. This effect has not been previously described for muscle cells, but is consistent with what has been reported for adipocytes [14] and can be explained by the fourfold increase in NF1 levels induced by the hormone.
It is important to stress that the stimulatory actions of tungstate on Glut4 transcription were dependent on the activation of ERK1/2 through an increase in the content of MEF2, which again places these kinases at the centre of the mechanism of action of this compound.
In conclusion, our results show that tungstate treatment increases the potential for glucose transport in muscle cells by two mechanisms, both of which are ERK1/2-dependent. The first involves an increase in the translocation of GLUT4 and the second a MEF2-dependent increase in transcription of the transporter gene. Compounds like tungstate (currently in phase II clinical trials), which exert their action through the improvement of glucose uptake in skeletal muscle via an increase in MEF2 levels and GLUT4 translocation, are excellent candidates for restoring the impaired muscle glucose metabolism that is characteristic of diabetes mellitus, and through this reducing hyperglycaemia.
Abbreviations
- AICAR:
-
5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside
- AMPK:
-
5′AMP-activated protein kinase
- 2-DG:
-
2-deoxy-d-[1-3H]glucose
- EMSA:
-
electrophoretic mobility shift assay
- ERK1/2:
-
extracellular signal-regulated kinases 1 and 2
- MEF2:
-
myocyte enhancer factor 2
- NF1:
-
nuclear factor 1
- PI3K:
-
phosphatidylinositol 3-kinase
References
Watson RT, Pessin JE (2006) Bridging the GAP between insulin signaling and GLUT4 translocation. Trends Biochem Sci 31:215–222
Sajan MP, Bandyopadhyay G, Kanoh Y et al (2002) Sorbitol activates atypical protein kinase C and GLUT4 glucose transporter translocation/glucose transport through proline-rich tyrosine kinase-2, the extracellular signal-regulated kinase pathway and phospholipase D. Biochem J 362:665–674
Zhou QL, Park JG, Jiang ZY et al (2004) Analysis of insulin signalling by RNAi-based gene silencing. Biochem Soc Trans 32:817–821
Kurth-Kraczek EJ, Hirshman MF, Goodyear LJ, Winder WW (1999) 5′-AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes 48:1666–1671
Bandyopadhyay G, Sajan MP, Kanoh Y et al (2001) Glucose activates protein kinase C-zeta /lambda through proline-rich tyrosine kinase-2, extracellular signal-regulated kinase, and phospholipase D: a novel mechanism for activating glucose transporter translocation. J Biol Chem 276:35537–35545
Zorzano A, Palacín M, Gumà A (2005) Mechanisms regulating GLUT4 glucose transporter expression and glucose transport in skeletal muscle. Acta Physiol Scand 183:43–58
Olson AL, Pessin JE (1995) Transcriptional regulation of the human GLUT4 gene promoter in diabetic transgenic mice. J Biol Chem 270:23491–23495
Oshel KM, Knight JB, Cao KT, Thai MV, Olson AL (2000) Identification of a 30-base pair regulatory element and novel DNA binding protein that regulates the human GLUT4 promoter in transgenic mice. J Biol Chem 275:23666–23673
Knight JB, Eyster CA, Griesel BA, Olson AL (2003) Regulation of the human GLUT4 gene promoter: interaction between a transcriptional activator and myocyte enhancer factor 2A. Proc Nat Acad Sci U S A 100:14725–14730
Liu ML, Olson AL, Edgington NP, Moye-Rowley WS, Pessin JE (1994) Myocyte enhancer factor 2 (MEF2)-binding site is essential for C2C12 myotube-specific expression of the rat GLUT4/muscle-adipose facilitative glucose transporter gene. J Biol Chem 269:28514–28521
Thai MV, Guruswamy S, Cao KT, Pessin JE, Olson AL (1998) Myocyte enhancer factor 2 (MEF2)-binding site is required for GLUT4 gene expression in transgenic mice. J Biol Chem 273:14285–14292
Mora S, Pessin JE (2000) The MEF2A isoform is required for striated muscle-specific expression of the insulin-responsive GLUT4 glucose transporter. J Biol Chem 275:16323–16328
Molkentin JD, Olson EN (1996) Defining the regulatory networks for muscle development. Curr Opin Genet Dev 6:445–453
Cooke DW, Lane MD (1999) The transcription factor nuclear factor I mediates repression of the GLUT4 promoter by insulin. J Biol Chem 274:12917–12924
Tanaka K, Shouguchi-Miyata J, Miyamoto N, Ikeda JE (2004) Novel nuclear shuttle proteins, HDBP1 and HDBP2, bind to neuronal cell-specific cis-regulatory element in the promoter of the Huntington’s disease gene. J Biol Chem 279:7275–7286
Barbera A, Rodriguez-Gil JE, Guinovart JJ (1994) Insulin-like actions of tungstate in diabetic rats. Normalization of hepatic glucose metabolism. J Biol Chem 269:20047–20053
Barbera A, Fernandez-Alvarez J, Truc A, Gomis R, Guinovart JJ (1997) Effects of tungstate in neonatally streptozotocin-induced diabetic rats: mechanism leading to normalization of glycaemia. Diabetologia 40:143–149
Barbera A, Gomis RR, Prats N et al (2001) Tungstate is an effective antidiabetic agent in streptozotocin-induced diabetic rats: a long-term study. Diabetologia 44:507–513
Muñoz MC, Barbera A, Domínguez J, Fernandez-Alvarez J, Gomis R, Guinovart JJ (2001) Effects of tungstate, a new potential oral antidiabetic agent, in Zucker diabetic fatty rats. Diabetes 50:131–138
Palanivel R, Sundravel S, Ravichandran P, Govindasamy S (1998) Insulin-like effect of ammonium para-tungstate on glycoprotein metabolism in streptozotocin diabetic rats. Med Sci Res 26:541–544
Nomiya K, Torii H, Hasegawa T et al (2001) Insulin mimetic effect of a tungstate cluster. Effect of oral administration of homo-polyoxotungstates and vanadium-substituted polyoxotungstates on blood glucose level of STZ mice. J Inorg Biochem 86: 657–667
Nagareddy PR, Vasudevan H, McNeill JH (2005) Oral administration of sodium tungstate improves cardiac performance in streptozotocin-induced diabetic rats. Can J Physiol Pharmacol 83:405–411
Claret M, Corominola H, Canals I et al (2005) Tungstate decreases weight gain and adiposity of obese rats through increased thermogenesis and lipid oxidation. Endocrinology 146:4362–4369
Fernandez-Alvarez J, Barbera A, Nadal B et al (2004) Stable and functional regeneration of pancreatic beta-cell population in nSTZ-rats treated with tungstate. Diabetologia 47:470–477
Girón MD, Caballero JJ, Vargas AM, Suarez MD, Guinovart JJ, Salto R (2003) Modulation of glucose transporters in rat diaphragm by sodium tungstate. FEBS Lett 542:84–88
Yonemitsu S, Nishimura H, Shintani M et al (2001) Troglitazone induces GLUT4 translocation in L6 myotubes. Diabetes 50:1093–1101
Mitsumoto Y, Klip A (1992) Development regulation of the subcellular distribution and glycosylation of GLUT1 and GLUT4 glucose transporters during myogenesis of L6 muscle cells. J Biol Chem 267:4957–4962
Rasband WS, Bright DS (1995) NIH Image: a public domain image processing program for the Macintosh. Microbeam Anal Soc J 4:137–149
Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162:156–159
Domínguez JE, Muñoz MC, Zafra D et al (2003) The antidiabetic agent sodium tungstate activates glycogen synthesis through an insulin receptor-independent pathway. J Biol Chem 278:42785–42794
Santalucía T, Moreno H, Palacín M, Yacoub MH, Brand NJ, Zorzano A (2001) A novel functional co-operation between MyoD, MEF2 and TRa1 is sufficient for the induction of GLUT4 gene transcription. J Mol Biol 314:195–204
Bhasvar PK, Dellow KA, Yacoub MH, Brand NJ, Barton PJR (2000) Identification of cis-acting DNA elements required for expression of the human cardiac troponin I gene promoter. J Mol Cell Cardiol 32:95–108
Ju JS, Smith JL, Oppelt PJ et al (2005) Creatine feeding increases GLUT4 expression in rat skeletal muscle. Am J Physiol Endocrinol Metab 288:E347–E352
Hagiwara Y, Shimo-Oka T, Okamura K et al (1989) Basis for the assay of myogenic cell growth in vitro using creatine kinase activity as an index, with special reference to measurement of power ratio of transferrins in growth promotion. Jpn J Pharmacol 49:53–58
Amacher SL, Buskin JN, Hauschka SD (1993) Multiple regulatory elements contribute differentially to muscle creatine kinase enhancer activity in skeletal and cardiac muscle. Mol Cell Biol 13:2753–2764
Chen HC, Bandyopadhayay G, Sajan MP et al (2002) Activation of the ERK pathway and atypical protein kinase C isoforms in exercise- and aminoimidazole-4-carboxamide-1-β-d-riboside (AICAR)-stimulated glucose transport. J Biol Chem 277:23554–23562
Ojuka EO, Jones TE, Nolte LA et al (2002) Regulation of GLUT4 biogenesis in muscle: evidence for involvement of AMPK and Ca2+. Am J Physiol Endocrinol Metab 282:E1008–E1013
Mora S, Yang C, Ryder JW, Boeglin D, Pessin JE (2001) The MEF2A and MEF2D isoforms are differentially regulated in muscle and adipose tissue during states of insulin deficiency. Endocrinology 142:1999–2004
Acknowledgements
This work was supported by grants ERDF1FD97-0812 (Ministerio de Ciencia y Tecnología, Spain) and PAI-2003-04 (Plan Andaluz de Investigación), to R. Salto; and by grants SAF2004-06962, SAF2007-64722 (Ministerio de Educación y Ciencia, Spain), 2005-SGR-00570 (Generalitat de Catalunya), PI042402 (Instituto de Salud Carlos III [ISCIII], Spain), and a grant from Fundación Marcelino Botín to J. J. Guinovart. N. Sevillano is the recipient of a predoctoral fellowship from Fundación Ramón Areces. We thank T. Yates for assistance in preparing the English manuscript. CIBER de Diabetes y Enfermedades Metabólicas is an initiative of the ISCIII.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
M. D. Girón and N. Sevillano contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
Representative autoradiograph of an MEF2 EMSA. Cells were incubated in the absence or presence of tungstate (100 µmol/l) for 24 h. Lanes corresponding to either control cells (C; far left) or tungstate-treated cells (W; far right) flank the lanes corresponding to samples of tungstate-treated cells that were pre-incubated in the presence of antibodies to MEF2 isoforms A, C and D as described in Methods. The arrows mark the positions of the shifted and supershifted oligonucleotide bands(PDF 85.0 KB)
ESM Table 1
Characterisation of membranes isolated from L6 myotubes (PDF 63.4 KB)
Rights and permissions
About this article
Cite this article
Girón, M.D., Sevillano, N., Vargas, A.M. et al. The glucose-lowering agent sodium tungstate increases the levels and translocation of GLUT4 in L6 myotubes through a mechanism associated with ERK1/2 and MEF2D. Diabetologia 51, 1285–1295 (2008). https://doi.org/10.1007/s00125-008-1003-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-008-1003-2