
Abstract
Aims/hypothesis
AMP-activated protein kinase (AMPK) is a heterotrimeric enzyme that acts as an intracellular fuel sensor, directing multiple metabolic pathways in a catabolic direction in times of nutrient shortage. In humans, three different γ-subunits (γ1, γ2, γ3) have been identified as AMPK regulators. The AMPKγ3 (protein kinase, AMP-activated, gamma 3 non-catalytic subunit, PRKAG3) isoform plays a role in gene regulation in glucose/lipid metabolism and skeletal muscle glycogen content. We investigated whether PRKAG3, in addition to being expressed in skeletal muscle, is also expressed in human liver. We also investigated whether genetic variance in PRKAG3 is associated with glucose and/or lipid metabolism in non-diabetic whites.
Materials and methods
After sequencing a screening cohort (n = 50) in the PRKAG3 locus, we genotyped 1061 participants for frequently found single nucleotide polymorphisms (SNPs). Association analyses between genotypes/haplotypes and metabolic traits were carried out.
Results
We detected PRKAG3 expression in human liver and skeletal muscle. Two SNPs (rs692243, rs6436094) with minor allele frequencies of 0.16 and 0.26 respectively and in moderate linkage disequilibrium (D′ = 0.92; r 2 = 0.47) were found. rs692243 (C/G) confers a Pro71Ala mutation, while rs6436094 (A/G) is located in the 3′ untranslated region. No associations with prediabetic traits such as body fat distribution, insulin resistance or insulin secretion were found (p > 0.15 for all). However, the minor alleles of both SNPs were significantly associated with higher serum LDL-cholesterol and apolipoprotein (Apo) B-100 levels (rs692243: CG:LDL 4.3%, ApoB-100 3.4%; GG:LDL 7.6%, ApoB-100 5.4%; p = 0.008 and p = 0.01 respectively; rs6436094: AG:LDL 3.3%, ApoB-100 1.7%; GG:LDL 11.3%, ApoB-100 11.1%; p = 0.009 and p = 0.05 respectively; dominant model). The GG/GG diplotype homozygous for both minor SNP alleles displayed the highest LDL-cholesterol among all frequent diplotypes (p = 0.059).
Conclusions/interpretation
While genetic variability in PRKAG3 does not seem to have a major effect on glucose metabolism, it may play an important role in lipoprotein metabolism in humans.
Similar content being viewed by others


Introduction
The AMP-activated serine/threonine protein kinase (AMPK) is of pivotal importance for cellular responses to stresses causing depletion of ATP, which is the major intracellular source of energy. Upon increased AMP/ATP ratio, AMP binds and activates AMPK, which subsequently acts on a broad range of signalling pathways involved in metabolic regulation, e.g. cholesterol synthesis, fatty acid transport and oxidation, insulin secretion, glucose uptake, lipogenesis or lipolysis [1, 2]. AMPK not only acts directly on various enzymes involved in these metabolic pathways, but also on the transcriptional level of many genes acting in glucose and lipid metabolism [3]. In this way, AMPK controls cellular energy homeostasis, switching off ATP-consuming (e.g. protein synthesis, cholesterol synthesis) and initiating ATP- and energy-producing processes (e.g. lipid oxidation, glucose uptake) in times of nutrient shortage [4]. Type 2 diabetes emerges when insulin resistance cannot be counterbalanced by increased insulin secretion due to a gradual loss of pancreatic beta cell function [5]. Each of the above-named AMPK-controlled metabolic pathways that are influenced by AMPK activity may contribute to final onset of type 2 diabetes under pathological conditions. Therefore, the gene encoding AMPK is an interesting candidate for type 2 diabetes pathogenesis and a potential therapeutical target, especially as AMPK is sensitive to oral glucose-lowering agents such as metformin and thiazolidinediones [6].
AMPK is a heterotrimeric enzyme, comprising one catalytic α-subunit (two isoforms: α1 and α2), one regulatory β-subunit (β1 and β2) and one regulatory γ-subunit (γ1, γ2 and γ3). The γ-subunits of the AMPK trimeric enzyme are responsible for AMP binding and therefore important for subsequent activation of the AMPK enzyme complex [7]. The different γ-isoforms of each subunit are encoded by different gene loci, and transcription of these genes varies among different tissues. While the γ1 and γ2 isoforms are produced throughout a broad range of different tissues, transcription of the gene encoding the γ3 isoform AMPKγ3 (protein kinase, AMP-activated, gamma 3 non-catalytic subunit, PRKAG3) seems to be more restricted and is mainly ascribed to white skeletal muscle in animals and humans [8]. However, weaker PRKAG3 expression has been reported for heart and pancreas; moreover PRKAG3-associated AMPK activity has also been found in rat liver lysates [9]. In Prkag3 knockout (Prkag3 −/−) mice, post-exercise glycogen resynthesis, as well as gene expression of glucose transporter 4 (also known as solute carrier family 2 [facilitated glucose transporter], member 4 [Slc2a4]) and of several lipid-oxidative enzymes, was impaired in skeletal muscle [10]. The opposite phenotype was found in transgenic animals expressing a constitutively active PRKAG3 mutant (PRKAG3R225Q) under the control of a muscle-specific promoter [11]. These animals show increased exercise-induced muscular glycogen resynthesis and opposing effects on gene expression, resulting in prevention of dietary-induced insulin resistance through increased fat oxidation. Taken together, PRKAG3-associated AMPK activity is thought to be important for the development of insulin resistance in skeletal muscle, while data on a physiological role of PRKAG3 in liver or other insulin-sensitive organs are still lacking.
Several genetic human studies were conducted to investigate the potential association of genetic variance of AMPK subunits with type 2 diabetes. While large-scale testing and haplotype analysis of the genes encoding protein kinase, AMP-activated, alpha 2 non-catalytic subunit (PRKAA2), protein kinase, AMP-activated, beta 1 non-catalytic subunit (PRKAB1) and protein kinase, AMP-activated, beta 2 non-catalytic subunit (PRKAB2) did not reveal any significant association with type 2 diabetes or insulin sensitivity in a Canadian and Scandinavian population [12], PRKAA2 variance was shown to correlate with insulin resistance and susceptibility to type 2 diabetes in a Japanese study [13] and with serum lipids in healthy white women [14]. In a Chinese population, two polymorphisms in the protein kinase, AMP-activated, gamma 2 non-catalytic subunit gene (PRKAG2) were associated with higher concentrations of serum triacylglycerol and total cholesterol, with risk of type 2 diabetes tending to be higher in single nucleotide polymorphism (SNP) carriers [15]. The only mutations at the protein level of human AMPK subunit isoforms that are known to confer functional effects on AMPK activity are localised in the C-terminus of PRKAG2. Most of these polymorphisms impair or even abolish AMPK activation due to a hampered binding affinity of the regulatory PRKAG2 subunit to AMP or ATP [16, 17]. Carriers of these PRKAG2 variants predispose to hypertrophic cardiomyopathy owing to cardiac glycogenosis [18] and often suffer from the Wolff–Parkinson–White syndrome [19]. By contrast, no reports exist so far on PRKAG3 genetic variance and its possible association with prediabetic traits in humans. We, therefore, investigated in this study whether polymorphisms in PRKAG3 correlate with glucose and lipid metabolism in a carefully phenotyped population of non-diabetic whites.
Methods
RT-PCR for PRKAG3 To investigate whether PRKAG3 is expressed in human liver, human liver cDNA cloned into the pACT2 vector was purchased (Clontech, Mountain View, CA, USA) and amplified in competent bacteria before analysis. The primers PRKAG3 forward 5′-CCAGAGGCACTAGGATTACC-3′ and PRKAG3 reverse 5′-TCTCTTGCCACAAGTTCAGG-3′ were used to amplify a 501 bp fragment localised in the 3′ untranslated region (UTR) of the PRKAG3 transcript. Specificity of the PCR fragments obtained was verified by sequencing. As a positive control, cDNA obtained by RT-PCR from skeletal muscle was used as a template. RNA was isolated from a human skeletal muscle biopsy with Trizol (Invitrogen, Carlsbad, CA, USA) and purified by RNAse-free DNAse from a blood RNA kit (PAXGene Blood RNA Kit, PreAnalytiX; Qiagen, Zurich, Switzerland). Synthesis of cDNA was done using a kit (1st Strand cDNA Synthesis KIT for RT-PCR AMV; Roche Diagnostics, Basel, Switzerland). cDNA libraries of human adipose tissue and brain (CLONTECH) were used as negative controls.
Participants
Data from 1,061 non-diabetic whites from southern Germany with and without a family history of type 2 diabetes were analysed for this study. The participants were not taking any medication known to affect glucose tolerance, insulin sensitivity or serum lipids. For the test period, strict abstention from smoking was required. Informed written consent was obtained from all participants and the local medical ethics committee approved the protocol. From this study cohort, 206 participants underwent a hyperinsulinaemic–euglycaemic clamp, 200 were characterised for intramyocellular lipids (IMCL) and 155 characterised for liver fat.
Body composition and body fat distribution
Body composition was measured as percentage of body fat by bioelectrical impedance (RJL, Detroit, MI, USA). BMI was calculated as weight divided by the square of height (kg/m2). Waist and hip circumferences were measured in the upright position and waist-to-hip ratio was calculated as an indicator of body fat distribution.
Determination of intramyocellular and intrahepatic lipids
Intramyocellular lipid content and liver fat were determined by proton magnetic resonance spectroscopy. Local image-guided proton spectra of the mixed tibialis anterior muscle (containing mainly type IIb, anaerobic, glycolytic, fast-twitch fibres) were obtained with a 1.5 T whole-body imager (Magnetom Sonata; Siemens Medical Solutions, Erlangen, Germany). For volume selection, a single-voxel stimulated echo acquisition mode (STEAM) technique was applied (echo time = 10 ms, repetition time = 2 s, volume of interest 11 × 11 × 20 mm3, 40 acquisitions). IMCL was quantified in fixed frequency regions in relation to methylene signal of creatine as described elsewhere [20]. For determination of liver fat content, modified STEAM parameters were used (repetition time = 4 s, echo time = 10 ms, 32 acquisitions) for a voxel of 3 × 3 × 2 cm3 placed in the posterior part of the seventh segment of the liver. Liver fat content was then calculated as described before [21].
Oral glucose tolerance test
OGTT was performed as recommended by the World Health Organization [22]. In addition to blood glucose, we determined plasma insulin, C-peptide and serum NEFA levels at 0, 30, 60, 90 and 120 min.
Calculations of insulin sensitivity and insulin secretion
Insulin sensitivity was calculated for all study participants from OGTT glucose and insulin values as described by Matsuda and deFronzo [23]. Insulin secretion was estimated from C-peptide concentrations obtained at 30 min of the OGTT.
Hyperinsulinaemic–euglycaemic clamp
A subgroup of 206 individuals underwent a hyperinsulinaemic–euglycaemic clamp to determine insulin sensitivity. A bolus-primed insulin infusion rate of 40 mU m−2 min−1 was administered over a 2 h period. Blood glucose was determined every 5 min to adjust the parallel infusion rate of exogenous glucose in order to maintain the baseline glucose level. Plasma insulin was measured at baseline and during the steady state of the clamp. Clamp-derived insulin sensitivity was calculated as the glucose infusion rate necessary for maintaining euglycaemia during the steady-state period (last 40 min) of the clamp divided by the steady-state insulin concentration.
Analytical procedures
Blood glucose was determined using a bedside glucose analyser (Yellow Springs Instruments, Yellow Springs, CO, USA). Plasma insulin was determined by microparticle enzyme immunoassay (Abbott Laboratories, Tokyo, Japan). A colorimetric assay was used for determination of serum cholesterols (Bayer HealthCare, Leverkusen, Germany) and triacylglycerol (Roche Diagnostics, Mannheim, Germany). NEFA were measured with an enzymatic method (WAKO Chemicals, Neuss, Germany), plasma C-peptide by radioimmunoassay (Byk-Sangtec, Dietzenbach, Germany) and leptin and adiponectin by ELISA (Linco Research, St Charles, MO, USA).
Genotyping
DNA was obtained from the Buffy coat of EDTA blood samples after erythrocyte lysis (TRIS–HCl 0.1 mol/l, MgCl2 0.1 mol/l, NaCl 0.5 mol/l, pH = 7.6; 3 × 45 min, room temperature), white blood cell lysis (TRIS–HCl 10 mmol/l, EDTA 10 mmol/l, NaCl 50 mmol/l, SDS 0.2%, proteinase K 200 μg/ml, pH 7.6; 6 h at 42°C) and protein precipitation (Na–Acetat). The DNA obtained was washed with isopropanol (100%, 30 min, room temperature) and ethanol (70% vol./vol., room temperature). PRKAG3 (NM_017431) was sequenced (ABI Prism 310 genetic analyser; Applied Biosystems, Foster City, CA, USA) in a screen of 50 participants, including ∼1,500 bp of the promoter (−1,479 bp referring to the start codon), all exons and exon–intron boundaries, and the whole 3′UTR. Detected SNPs with a minor allele frequency (MAF) >0.05 were selected for further genotyping by the TaqMan assay (Eurogentec, Liege, Belgium) and an ABI Prism 7500 sequence detection system (Applied Biosystems). SNPs are indicated according to the dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP/).
Statistical analyses
Unless otherwise stated, data are given as means ± SEM. The JMP software package (SAS Institute, Cary, NC, USA) was used for statistical analyses and comparison between different genotype groups. Analysis was performed using logarithmically transformed data for non-normally distributed parameters. To adjust the effects of relevant covariates (age, sex, body fat), multivariate linear regression analyses were performed. A p value ≤ 0.05 was considered to be statistically significant. The HPlus v2.5 software package, available from the Fred Hutchinson Cancer Research Center (http://qge.fhcrc.org/hplus/), was used for estimation of inferred diplotypes [24]. SNP linkage disequilibrium (LD) analysis was done by means of the JLIN program provided by the Western Australian Institute for Medical Research [25]. Hardy–Weinberg equilibrium was tested with the χ 2 test. The Tag SNP Picker program, provided by the HapMap project (http://www.hapmap.org/), was used to estimate whether the SNPs detected cover the genetic variance of the entire PRKAG3 locus of whites. A software analysis package purchased from HUSAR Bioinformatics Lab (http://genius.embnet.dkfz-heidelberg.de) was used for protein secondary structure and domain analysis.
Results
Expression of PRKAG3 in human skeletal muscle and liver As previously published reports on PRKAG3 gene expression provide conflicting data concerning hepatic expression of PRKAG3 [8, 9], we tested PRKAG3 expression in cDNA obtained from human brain, adipose tissue (negative controls), skeletal muscle (positive control) and liver. We were able to amplify PRKAG3 in the hepatic cDNA preparation tested (Fig. 1) and conclude that PRKAG3 is not restricted to skeletal muscle in humans, but also expressed in human liver. We, therefore, extended our analyses of PRKAG3 genetic variance to liver-specific prediabetic traits beyond characteristics that are related to skeletal muscle.

Human adipose tissue (AdipT), brain, skeletal muscle (SkM) and liver cDNA were subjected to PCR analysis with primers specific for the PRKAG3 subunit. Expected size of the PCR fragment was 501 bp. Specificity for PRKAG3 was confirmed by additional sequencing
Baseline characteristics of the study cohort The 1,061 non-diabetic participants (362 men/699 women) had a mean (±SEM) age of 37.1 ± 0.4 years, a mean BMI of 26.7 ± 0.2 kg/m2 and a mean body fat of 28.8 ± 0.3%. In the fasting state, mean plasma glucose was 5.03 ± 0.02 mmol/l and plasma insulin was 55.8 ± 1.5 pmol/l. A family history of at least one second-degree relative with diabetes mellitus was confirmed by 71.4% of participants.
Genetic analysis of the PRKAG3 locus The screening analysis revealed two SNPs with a MAF > 0.05 in our population, namely rs692243 (C/G) in exon 3 (MAF = 0.164) and rs6436094 (A/G) in the 3′UTR (MAF = 0.261). rs692243 causes a mutation at the protein level (Pro71Ala) of PRKAG3. Both SNPs were distributed according to Hardy–Weinberg equilibrium (χ 2 test: p = .76 for rs692243; p = 0.98 for 6436094). The two SNPs were in moderate LD (D′ = 0.92, r 2 = 0.47). Interestingly, MAF and LD values of our study cohort were very similar to the CEU population (Utah residents with ancestry from northern and western Europe: D′ = 0.92, r 2 = 0.53; phase II data) of the HapMap Project [26]. The two SNPs investigated for this study were also selected by the Tag SNP Picker program of the HapMap Project; in the PRKAG3 locus (including 5,000 bp of the promoter region) no additional SNPs with a MAF > 0.05 were present in the CEU.
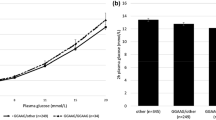
Correlation of PRKAG3 genotype with anthropometrics, blood glucose and parameters of insulin sensitivity and insulin secretion No correlation was found between rs692243 and rs6436094 genotype and anthropometric data of our study population, e.g. age, sex, BMI, waist-to-hip ratio, body fat or family history of diabetes. There were also no associations with fasting plasma glucose or insulin or with the 2 h glucose or insulin levels during the OGTT. Insulin sensitivity was not altered according to the two SNPs, either in the estimation model of Matsuda and DeFronzo [23], or in the group of 206 participants who underwent a hyperinsulinaemic–euglycaemic clamp. Differences in insulin secretion were also not found among PRKAG3 genotypes (Table 1).
Analysis of intramyocellular lipids in association with PRKAG3 genotype Both mice (PRKAG3R225Q) and pigs (PRKAG3R200Q) with a mutated PRKAG3 exhibit a phenotype with increased glycogen content in white, non-oxidative fast-twitch muscle [27, 28]. In diabetic human patients, skeletal muscle glycogen content is inversely correlated with IMCLs [29]. On the other hand, increased IMCLs are associated with insulin resistance in non-diabetic whites [20]. For this reason, we investigated whether the PRKAG3 SNPs are associated with IMCLs of the primarily non-oxidative, fast-twitch, glycolytic tibialis anterior muscle. In 200 participants, spectroscopically determined IMCL were not associated with the different alleles of rs692243 and rs6436094 (Table 1).
Analysis of lipid parameters and association with PRKAG3 genotype and inferred diplotypes After adjustment for age, sex and body fat, LDL-cholesterol was significantly increased in carriers of the minor alleles of both rs692243 (CG, 4.3%; GG, 7.6% vs the common allele CC; p = 0.008, dominant model) and rs6436094 (AG, 3.3%; GG, 11.3% vs the common allele AA; p = 0.009; Table 2). This increase was paralleled by higher serum levels of apolipoprotein (Apo) B-100 (rs692243: CG, 3.4%; GG, 5.4%, p = 0.01; rs6436094: AG, 1.7%; GG, 11.1%; p = 0.05) and total cholesterol (p = 0.02 for rs692243; p = 0.08 for rs6436094). There were no significant associations with fasting and 2 h serum NEFA, HDL-cholesterol or triacylglycerol. We next tested whether serum adiponectin and leptin levels differed among PRKAG3 genotypes, as both adipokines can activate AMPK in different tissues [4] and therefore may interfere with the genetic analysis. However, neither adiponectin nor leptin showed a significant variability across different PRKAG3 genotype groups. In addition, no statistically significant association between spectroscopically determined liver fat content and PRKAG3 variance was found (Table 2).
To test which allele combinations are associated with the highest increase in LDL-cholesterol, we conducted a haplotype analysis with generation of inferred diplotypes. Eight inferred diplotypes were found in our population, five of which had a frequency of >0.02. Adjusted LDL-cholesterol levels were highest in the GG/GG diplotype, which is homozygous for both SNPs. Analysis of variance comprising all five frequent diplotypes reached an almost significant level (p = 0.059), possibly due to the relatively small sample size for the diplotypes containing both minor alleles (Fig. 2).

The five most common (>0.02) diplotypes (1st allele/2nd allele) are presented in the order of appearance of rs692243 (exon 3) and rs6436094 (3′UTR) following their distance from the PRKAG3 start codon. LDL-cholesterol measurements were logarithmically transformed and adjusted for age, sex and body fat. The difference Δlog LDL-cholesterol of all frequent (MAF > 0.02) diplotypes is presented as means + SEM relatively to the mean log LDL-cholesterol measurements of the most common diplotype CA/CA, containing only wild-type alleles for both SNPs (dashed line). CA/CA: n = 568, frequency 0.54; CG/CA: n = 163, frequency 0.15; GG/CA: n = 239, frequency 0.22; GG/CG: n = 33, frequency 0.03; GG/GG: n = 27, frequency 0.03
Discussion
This is the first study to have investigated the associations of genetic variance of PRKAG3 with carefully phenotyped prediabetic traits in a non-diabetic study cohort. We found two frequent SNPs in our population, which reflected very well the CEU population of the HapMap Project in respect to minor allele frequencies and LD of detected SNPs. The rs692243 SNP causes a mutation (Pro71Ala) in the N-terminus of PRKAG3, while the 3′UTR-located rs6436094 may play a role in mRNA stability. Although PRKAG3 has been suggested to play a major role in white skeletal muscle [8], we did not find associations with prediabetic traits reflecting skeletal muscle insulin sensitivity. There was also no significant correlation with other parameters of glucose metabolism, e.g. fasting and 2 h (OGTT) blood glucose, plasma insulin levels or insulin secretion. We additionally tested whether spectroscopically analysed IMCL in fast-twitch, glycolytic type IIb fibres containing tibialis anterior muscle is associated with the PRKAG3 genotypes, as animals in which a mutation in PRKAG3 causes enhanced AMPK activity have a higher glycogen content in white skeletal muscle [27, 28] and IMCLs have been shown to inversely correlate with intramuscular glycogen content in obese diabetic patients [29]. We found no significant association of IMCL with PRKAG3 genotype in our study.
In contrast, we detected significant associations of LDL-cholesterol and ApoB-100 serum levels with the genotypes of both PRKAG3 SNPs. Associations of the genes encoding AMPK with serum lipoproteins have been reported for different populations, namely for the AMPK catalytic subunit PRKAA2 in a British cohort [14] and the regulatory subunit PRKAG2 in a Chinese population [15]. Interestingly, in these two studies, parameters of insulin sensitivity were also not altered between different genotypes. LDL particles and their core protein ApoB-100 are synthesised by the liver in order to transport esterified cholesterol to peripheral tissues [30]. Elevated serum LDL-levels are commonly a result of impaired LDL-receptor function in the liver, a frequently found abnormality in familial hypercholesterolaemia [31]. Therefore, our finding of PRKAG3 in a human liver cDNA library and the detection of a 4.4 kb transcript with a γ3-specific probe by northern blot in an even broader range of human tissues by Cheung et al. [9] support the hypothesis that PRKAG3 activity, though generally reported to be strongest in skeletal muscle, may not be as specific to skeletal muscle as previously thought. This hypothesis is also supported by AMPK enzyme activities found in rat liver lysates that had undergone immunoprecipitation with an antibody directed against the γ3-subunit [9]. Changes in PRKAG3-dependent AMPK activity in liver could explain the observed phenotype in minor allele carriers, as AMPK is able to inhibit the key enzyme of cholesterol synthesis, 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, by phosphorylation on serine 872 [32–34]. In addition, increased LDL-cholesterol levels could also be a consequence of altered sterol regulatory element binding transcription factor 1 (SREBP1) function. SREBP1 positively regulates expression of the genes encoding HMG-CoA reductase and HMG-CoA synthetase; it also induces transcription of the LDL receptor gene in many different tissues [35]. Interestingly, SREBP1 (also known as sterol regulatory element binding protein-1 [SREBF1]) transcription is significantly upregulated in Prkag3 −/− knockout mice and downregulated in PRKAG3R225Q mice [36].
On the other hand, PRKAG3-associated activity in skeletal muscle may also contribute to changes in LDL-cholesterol serum levels. Beyond SREBP1 transcription [36], PRKAG3 regulates gene expression of various glucose- and lipid-metabolic key enzymes in white skeletal muscle [37] and therefore may have an effect on endogenous skeletal muscle cholesterol synthesis and LDL receptor expression. Such changes in cholesterol synthesis or LDL receptor expression of myocytes could alter their LDL-cholesterol ingestion, subsequently also altering serum LDL-cholesterol levels, as skeletal muscle tissue accounts for ∼5% of total peripheral LDL-cholesterol uptake [38, 39].
Based on the assumption that PRKAG3-mediated downregulation of HMG-CoA reductase or PRKAG3-mediated transcriptional regulation of SREBP1 in liver or skeletal muscle accounts for the observed phenotype in our study, it may be speculated that the SNPs in human PRKAG3 reported here confer a negative effect on PRKAG3-associated function. This raises the question of whether the Pro71Ala mutation caused by rs692243 could be relevant for PRKAG3 function. PRKAG33 contains four cystathionine β-synthase domains in its N terminus, while no domains or classical secondary structures have been predicted that would be affected by the Pro71Ala mutation. In this regard, it is interesting that all reported functional polymorphisms in the PRKAG2 isoform have only been found in its C terminus [16]. Therefore, a functional relevance of Pro71Ala in PRKAG3 seems unlikely, and rs692243 (Pro71Ala) may rather tag another functional SNP, e.g. in the PRKAG3 promoter. Indeed, HapMap analysis reveals seven other SNPs in the 25 kb promoter region between PRKAG3 and its neighbouring gene, six of which are in high LD (all D′ = 1.0) with rs692243 and may influence PRKAG3 transcriptional rate.
Some limitations of our study have to be considered. Our analysis is based on cross-sectional data derived from a non-diabetic healthy population. This study design does not allow any assumptions on later onset of clinically relevant hypercholesterolaemia or arteriosclerosis. In addition, the findings have to be reproduced in other populations before final conclusions on the role of PRKAG3 genetics in human glucose and lipid metabolism can be drawn.
In summary, genetic variance of the PRKAG3 locus is significantly associated with LDL–cholesterol and ApoB-100 serum levels in a non-diabetic white population, but not with prediabetic traits. It would therefore be worthwhile to test whether the SNPs in PRKAG3 are determinants of lipid metabolism in populations with familial hypercholesterolaemia. Such findings would strongly support the hypothesis that PRKAG3 may be an interesting therapeutical target for metabolic diseases such as hypercholesterolaemia or atherosclerosis.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- Apo:
-
apolipoprotein
- CEU:
-
Utah residents with ancestry from northern and western Europe
- HMG-CoA:
-
3-hydroxy-3-methylglutaryl coenzyme A
- IMCL:
-
intramyocellular lipid
- LD:
-
linkage disequilibrium
- MAF:
-
minor allele frequency
- PRKAA2:
-
protein kinase, AMP-activated, alpha 2 non-catalytic subunit
- PRKAG2:
-
protein kinase, AMP-activated, gamma 2 non-catalytic subunit
- PRKAG3:
-
protein kinase, AMP-activated, gamma 3 non-catalytic subunit
- SNP:
-
single nucleotide polymorphism
- SREBP1:
-
sterol regulatory element binding transcription factor 1
- UTR:
-
untranslated region
References
Winder WW, Hardie DG (1999) AMP-activated protein kinase, a metabolic master switch: possible roles in type 2 diabetes. Am J Physiol 277:E1–E10
Steinberg GR, Macaulay SL, Febbraio MA, Kemp BE (2006) AMP-activated protein kinase—the fat controller of the energy railroad. Can J Physiol Pharmacol 84:655–665
Leff T (2003) AMP-activated protein kinase regulates gene expression by direct phosphorylation of nuclear proteins. Biochem Soc Trans 31:224–227
Long YC, Zierath JR (2006) AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest 116:1776–1783
Weyer C, Bogardus C, Mott DM, Pratley RE (1999) The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest 104:787–794
Schimmack G, Defronzo RA, Musi N (2006) AMP-activated protein kinase: role in metabolism and therapeutic implications. Diabetes Obes Metab 8:591–602
Wong KA, Lodish HF (2006) A revised model for AMP-activated protein kinase structure: The alpha-subunit binds to both the beta- and gamma-subunits although there is no direct binding between the beta- and gamma-subunits. J Biol Chem 281:36434–36442
Mahlapuu M, Johansson C, Lindgren K et al (2004) Expression profiling of the gamma-subunit isoforms of AMP-activated protein kinase suggests a major role for gamma3 in white skeletal muscle. Am J Physiol Endocrinol Metab 286:E194–E200
Cheung PC, Salt IP, Davies SP, Hardie DG, Carling D (2000) Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem J 346:659–669
Barnes BR, Long YC, Steiler TL et al (2005) Changes in exercise-induced gene expression in 5′-AMP-activated protein kinase gamma3-null and gamma3 R225Q transgenic mice. Diabetes 54:3484–3489
Barnes BR, Marklund S, Steiler TL et al (2004) The 5′-AMP-activated protein kinase gamma3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle. J Biol Chem 279:38441–38447
Sun MW, Lee JY, de Bakker PI et al (2006) Haplotype structures and large-scale association testing of the 5′ AMP-activated protein kinase genes PRKAA2, PRKAB1, and PRKAB2 with type 2 diabetes. Diabetes 55:849–855
Horikoshi M, Hara K, Ohashi J et al (2006) A polymorphism in the AMPKalpha2 subunit gene is associated with insulin resistance and type 2 diabetes in the Japanese population. Diabetes 55:919–923
Spencer-Jones NJ, Ge D, Snieder H et al (2006) AMP-kinase alpha2 subunit gene PRKAA2 variants are associated with total cholesterol, low-density lipoprotein-cholesterol and high-density lipoprotein-cholesterol in normal women. J Med Genet 43:936–942
Xu M, Li X, Wang JG et al (2005) Glucose and lipid metabolism in relation to novel polymorphisms in the 5′-AMP-activated protein kinase gamma2 gene in Chinese. Mol Genet Metab 86:372–378
Daniel T, Carling D (2002) Functional analysis of mutations in the gamma 2 subunit of AMP-activated protein kinase associated with cardiac hypertrophy and Wolff–Parkinson–White syndrome. J Biol Chem 277:51017–51024
Burwinkel B, Scott JW, Buhrer C et al (2005) Fatal congenital heart glycogenosis caused by a recurrent activating R531Q mutation in the gamma 2-subunit of AMP-activated protein kinase (PRKAG2), not by phosphorylase kinase deficiency. Am J Hum Genet 76:1034–1049
Arad M, Maron BJ, Gorham JM et al (2005) Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med 352:362–372
Gollob MH, Green MS, Tang AS et al (2001) Identification of a gene responsible for familial Wolff–Parkinson–White syndrome. N Engl J Med 344:1823–1831
Jacob S, Machann J, Rett K et al (1999) Association of increased intramyocellular lipid content with insulin resistance in lean nondiabetic offspring of type 2 diabetic subjects. Diabetes 48:1113–1119
Stefan N, Schafer S, Machicao F et al (2005) Liver fat and insulin resistance are independently associated with the −514C>T polymorphism of the hepatic lipase gene. J Clin Endocrinol Metab 90:4238–4243
Alberti KG, Zimmet PZ (1998) Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus. Provisional report of a WHO consultation. Diabet Med 15:539–553
Matsuda M, DeFronzo RA (1999) Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 22:1462–1470
Li SS, Khalid N, Carlson C, Zhao LP (2003) Estimating haplotype frequencies and standard errors for multiple single nucleotide polymorphisms. Biostatistics 4:513–522
Carter KW, McCaskie PA, Palmer LJ (2006) JLIN: a java based linkage disequilibrium plotter. BMC Bioinformatics 7:60
The International HapMap Consortium (2005) A haplotype map of the human genome. Nature 437:1299–1320
Milan D, Jeon JT, Looft C et al (2000) A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science 288:1248–1251
Barnes BR, Glund S, Long YC, Hjalm G, Andersson L, Zierath JR (2005) 5′-AMP-activated protein kinase regulates skeletal muscle glycogen content and ergogenics. FASEB J 19:773–779
Levin K, Daa Schroeder H, Alford FP, Beck-Nielsen H (2001) Morphometric documentation of abnormal intramyocellular fat storage and reduced glycogen in obese patients with type II diabetes. Diabetologia 44:824–833
Olofsson SO, Boren J (2005) Apolipoprotein B: a clinically important apolipoprotein which assembles atherogenic lipoproteins and promotes the development of atherosclerosis. J Intern Med 258:395–410
Soutar AK, Naoumova RP (2007) Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med 4:214–225
Istvan ES, Palnitkar M, Buchanan SK, Deisenhofer J (2000) Crystal structure of the catalytic portion of human HMG-CoA reductase: insights into regulation of activity and catalysis. EMBO J 19:819–830
Hardie DG (1992) Regulation of fatty acid and cholesterol metabolism by the AMP-activated protein kinase. Biochim Biophys Acta 1123:231–238
Omkumar RV, Darnay BG, Rodwell VW (1994) Modulation of Syrian hamster 3-hydroxy-3-methylglutaryl-CoA reductase activity by phosphorylation. Role of serine 871. J Biol Chem 269:6810–6814
Sakai J, Rawson RB (2001) The sterol regulatory element-binding protein pathway: control of lipid homeostasis through regulated intracellular transport. Curr Opin Lipidol 12:261–266
Nilsson EC, Long YC, Martinsson S et al (2006) Opposite transcriptional regulation in skeletal muscle of AMP-activated protein kinase gamma3 R225Q transgenic versus knock-out mice. J Biol Chem 281:7244–7252
Long YC, Barnes BR, Mahlapuu M et al (2005) Role of AMP-activated protein kinase in the coordinated expression of genes controlling glucose and lipid metabolism in mouse white skeletal muscle. Diabetologia 48:2354–2364
Pittman RC, Attie AD, Carew TE, Steinberg D (1979) Tissue sites of degradation of low density lipoprotein: application of a method for determining the fate of plasma proteins. Proc Natl Acad Sci USA 76:5345–5349
Carew TE, Pittman RC, Steinberg D (1982) Tissue sites of degradation of native and reductively methylated [14C]sucrose-labeled low density lipoprotein in rats. Contribution of receptor-dependent and receptor-independent pathways. J Biol Chem 257:8001–8008
Acknowledgement
We thank all the research volunteers for their participation. We gratefully acknowledge the technical assistance of M. Weisser, R. Werner and H. Luz. The authors would like to thank all participants for their cooperation. This study was supported by a grant from the Deutsche Forschungsgemeinschaft (KFO 114/1–2) and the European Community’s FP6 EUGENE 2 (LSHM-CT-2004-512013).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Weyrich, P., Machicao, F., Staiger, H. et al. Role of AMP-activated protein kinase gamma 3 genetic variability in glucose and lipid metabolism in non-diabetic whites. Diabetologia 50, 2097–2106 (2007). https://doi.org/10.1007/s00125-007-0788-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-007-0788-8