
Abstract
Aims/hypothesis
Increased glucagon secretion predicts deterioration of glucose tolerance, and high glucagon levels contribute to hyperglycaemia in type 2 diabetes. Inhibition of glucagon action may therefore be a potential novel target to reduce hyperglycaemia. Here, we investigated whether chronic treatment with a glucagon receptor antagonist (GRA) improves islet dysfunction in female mice on a high-fat diet (HFD).
Materials and methods
After 8 weeks of HFD, mice were treated with a small molecule GRA (300 mg/kg, gavage once daily) for up to 30 days. Insulin secretion was studied after oral and intravenous administration of glucose and glucagon secretion after intravenous arginine. Islet morphology was examined and insulin secretion and glucose oxidation were measured in isolated islets.
Results
Fasting plasma glucose levels were reduced by GRA (6.0 ± 0.2 vs 7.4 ± 0.5 mmol/l; p = 0.017). The acute insulin response to intravenous glucose was augmented (1,300 ± 110 vs 790 ± 64 pmol/l; p < 0.001). The early insulin response to oral glucose was reduced in mice on HFD + GRA (1,890 ± 160 vs 3,040 ± 420 pmol/l; p = 0.012), but glucose excursions were improved. Intravenous arginine significantly increased the acute glucagon response (129 ± 12 vs 36 ± 6 ng/l in controls; p < 0.01), notably without affecting plasma glucose. GRA caused a modest increase in alpha cell mass, while beta cell mass was similar to that in mice on HFD + vehicle. Isolated islets displayed improved glucose-stimulated insulin secretion after GRA treatment (0.061 ± 0.007 vs 0.030 ± 0.004 pmol islet−1 h−1 at 16.7 mmol/l glucose; p < 0.001), without affecting islet glucose oxidation.
Conclusions/interpretation
Chronic glucagon receptor antagonism in HFD-fed mice improves islet sensitivity to glucose and increases insulin secretion, suggesting improvement of key defects underlying impaired glucose tolerance and type 2 diabetes.
Similar content being viewed by others

Introduction
Glucagon plays a key role in maintaining circulating glucose mainly through its regulation of hepatic glucose production [1, 2]. The hormone binds to specific glucagon receptors on the hepatocytes and activates the glycogenolytic and gluconeogenetic pathways, thereby increasing hepatic glucose output [3, 4]. The action of glucagon is opposed by insulin; in fact, the ratio between insulin and glucagon is a major determinant of ambient circulating glucose levels [5, 6]. Several studies have suggested that glucagon is implicated in the development of type 2 diabetes [7–10]. Thus, it has been shown that increased secretion of glucagon predicts IGT in humans [11] and that IGT is associated with impaired suppression of glucagon secretion [12–14]. Furthermore, plasma glucagon levels are elevated in subjects with type 2 diabetes [2, 15, 16]. Particularly in the presence of low insulin, hyperglucagonaemia may result in increased glucose production and thereby further contribute to the hyperglycaemia caused by insulin deficiency resulting from beta cell failure and insulin resistance [17]. Together these findings suggest that inhibition of glucagon action may be a novel target for treatment of type 2 diabetes. In fact, both glucagon-neutralising antibodies [18–20] and peptide glucagon receptor antagonists (GRA) [21, 22] efficiently reduce glycaemia in various rodent models. For a potential drug development, several non-peptidyl small molecule GRAs have been tested and proven to inhibit glucagon-mediated metabolic effects [23–25]. Glucagon receptor antagonism reduces hepatic glucose production in healthy humans [26], further indicating that glucagon antagonists may be a new class of agents for treating type 2 diabetes. Recent studies have used the anti-sense technique to reduce the expression of glucagon receptors, which led to improved glycaemia in diabetic rodents [27, 28]. It is also of interest that mice with targeted disruption of the glucagon receptor gene have significantly reduced blood glucose levels and improved glucose tolerance compared with wild-type mice [29–31].
The underlying cause of hyperglycaemia in type 2 diabetes is impaired beta cell function in association with insulin resistance [32–35]. Glucagon stimulates insulin secretion and glucagon receptor signalling has been shown to be required for full glucose competence in beta cells [36]. This would suggest that glucagon receptor antagonism, if developed as a glucose-lowering agent, might deteriorate insulin secretion in type 2 diabetes, thus counterbalancing its beneficial effects. However, a recent study using the antisense technique to reduce glucagon receptor expression demonstrated increased insulin secretion, thereby suggesting improved islet function [28]. It is thus unclear whether islet function is perturbed by inhibition of glucagon action. In this study, therefore, we investigated whether glucagon receptor antagonism affects islet function in a model of type 2 diabetes using mice fed a high-fat diet (HFD). This model has previously been shown to be associated with: (1) insulin resistance, as determined by euglycaemic–hyperinsulinaemic clamp [37]; (2) impaired insulin secretion resulting in glucose intolerance [38]; and (3) hyperglucagonaemia [39]. We thus treated HFD-fed mice chronically with a small molecule that has glucagon receptor antagonistic properties and studied insulin secretion in vivo and in vitro in isolated islets. The GRA has been characterised previously and demonstrated to dose-dependently inhibit glucagon-stimulated hepatic glucose production in live dogs [40]. We also examined whether the glucagon response to intravenous administration of arginine was affected by long-term glucagon receptor antagonism. The rationale for this was that following glucagon receptor antagonism or after genetic ablation of the glucagon receptor, alpha cell hyperplasia can evolve, resulting in exaggerated plasma levels of glucagon [26, 28–30]. It would therefore be of value to establish whether glucagon secretion is also affected after chronic treatment with a GRA.
Materials and methods
Characterisation of the glucagon receptor antagonist
The glucagon receptor antagonist NNC 25-0926 [(2R)-N-[4-({4-(1-cyclohexen-1-yl)[(3,5-dichloroanilino)carbonyl]anilino}methyl)benzoyl]-2-hydroxy-β-alanine] has only 12% bioavailability after oral administration, a short half-life (t 1/2 = 41 min) and is rapidly cleared from the circulation in rats (clearance rate: 32 ml min−1 kg−1). Furthermore, NNC 25-0926 is a potent GRA with an inhibitory concentration of 50% (IC50) value of 12 nmol/l for inhibition of glucagon binding to cloned human glucagon receptors expressed in baby hamster kidney cells (E. Nishimura and U. G. Sidelmann, unpublished observations). It is also specific for the glucagon receptor in that it binds poorly to related receptors (IC50: human glucose-dependent insulinotropic polypeptide receptor: 625 nmol/l; human vasoactive intestinal polypeptide receptor: 429 nmol/l; and human glucagon-like peptide 1 [GLP-1] receptor: 21,000 nmol/l). Based on these characteristics, and particularly the low oral bioavailability, a dose of 300 mg/kg was selected for this study.
Animals and study design
Female C57BL/6JBomTac mice (8 weeks old; Taconic, Skensved, Denmark) were maintained in a temperature-controlled room (22°C) on a 12 h light–dark cycle. The mice were divided into two groups and fed either a normal diet (ND; 11% energy as fat, 16% as protein, 73% as carbohydrate; Research Diets, New Brunswick, NJ, USA) or an HFD (58% fat, 16% protein, 26% carbohydrate; Research Diets). Body weight was measured once a week. Blood samples were taken at indicated time points from the intraorbital, retrobulbar plexus from overnight fasted (16 h), anaesthetised mice (20 mg/kg fluanisone–0.8 mg/kg fentanyl; Janssen, Beerse, Belgium; 10 mg/kg midazolam; Hoffman-LaRoche, Basel, Switzerland). After feeding mice with the ND or HFD for 8 weeks, the HFD-fed mice were divided into two groups receiving either 300 mg/kg GRA dissolved in vehicle (VEH; 0.13 mg/ml vitamin E as tocopheryl polyethylene glycol 1,000 succinate (TPGS), 0.015 mg/ml Kollidon 12 PF, 65 mmol/l sodium phosphate buffer, pH 7.0) (HFD-GRA) or VEH only (HFD-VEH), by gavage once daily (08.00 h, 0.5 ml per mouse). ND-fed mice were treated with VEH (ND-VEH). The in vivo experiments were performed in overnight-fasted, anaesthetised mice. The mice were given their daily dose of GRA or VEH 2 h before the experiments. The study was approved by the Animal Ethics Committee in Lund/Malmö, Sweden.
Glucose tolerance tests and insulin release
After 17–18 days of GRA treatment, IVGTTs were performed. Mice were anaesthetised as described 30 min prior to the IVGTT. Thereafter a blood sample (50 μl) was taken from the retrobulbar, intraorbital capillary plexus, after which d-glucose (1 g/kg [5.6 mmol/kg]; Sigma, St Louis, MO, USA) was injected intravenously in a tail vein (volume load 10 μl/g). Additional blood samples were taken at 1, 5, 10, 20, 50 and 75 min after injection. For OGTTs, anaesthetised mice were given 75 mg [0.42 mmol] d-glucose by intragastric gavage. Blood samples were collected 0, 15, 30, 60 and 120 min after glucose administration. Following immediate centrifugation (10,000 g) at 4°C, plasma was separated and stored at −20°C until analysis of glucose and insulin.
Glucagon secretion
Glucagon secretion was measured after 17–21 days of GRA treatment after intravenous injections of l-arginine (0.25 g/kg). Blood samples (60 μl) were collected in 10 μl aprotinin (1.4 mg/ml; Bayer Health Care, Leverkusen, Germany) at 0, 1, 5, 20 and 50 min after the arginine injection and handled as described above. Samples were stored at −20°C until analysis of glucose and glucagon.
Euglycaemic–hyperinsulinaemic clamp
HFD-fed mice were subjected to a euglycaemic–hyperinsulinaemic clamp after 21–24 days of GRA treatment, as described [37]. Two hours before the clamp study, non-fasted mice were given their daily treatment. Mice were anaesthetised (as above) and the right jugular vein and left carotid artery were catheterised for infusion of glucose and insulin (Actrapid; Novo Nordisk, Bagsværd, Denmark) and for blood sampling, respectively. Thirty minutes after insertion of the catheters, insulin was infused at a rate of 40 mU kg−1 min−1 for 1 min, followed by constant infusion of 20 mU kg−1 min−1. Blood glucose levels were determined every 5 min for 90 min using a glucose analyser (Hemocue B; Hemocue, Ängelholm, Sweden). A glucose solution (20%, by weight) was infused at a variable rate to maintain blood glucose levels at the target of 6.5 mmol/l. Blood samples were taken at 0 and 90 min to determine insulin levels. Insulin sensitivity was calculated as the mean of the glucose infusion rates (GIR) from 60 to 90 min.
Insulin and glucose measurements
Glucose was measured with the glucose oxidase method using 2,2′-azino-bis(3-ethyl-benzothialozine-6-sulfonate) as substrate, absorbance being measured at 420 nm on a microtitre plate reader (Fluostar/Polarstar Galaxy; BMG Labtechnologies, Offenburg, Germany). Insulin was determined radioimmunochemically using a guinea pig anti-rat insulin antibody, 125I-labelled human insulin as tracer and rat insulin as standard (Linco Research, St Charles, MO, USA). Glucagon was analysed using a radioimmunoassay with a guinea pig anti-glucagon antibody, 125I-labelled glucagon as tracer and glucagon standard (Linco Research). For both assays free and bound radioactivity were separated by use of an anti-IgG (goat anti-guinea pig) antibody. Radioactivity was measured on a gamma counter (Wallac Wizard 1470; Perkin Elmer, Turku, Finland).
Islet isolation and insulin secretion
After mice had been treated with the GRA for 24–30 days, islets were isolated from the pancreas by collagenase digestion and handpicked under microscope. Batches of islets were preincubated in HEPES balanced salt solution (HBSS; 125 mmol/l NaCl, 5.9 mmol/l KCl, 1.28 mmol/l CaCl2, 1.2 mmol/l MgCl2, 25 mmol/l HEPES [pH 7.4], 5.6 mmol/l glucose and 0.1% fatty-acid-free bovine albumin) (Boehringer Mannheim, Mannheim, Germany) for 60 min. Thereafter, islets in groups of three were incubated in 200 μl of the HBSS with varying concentrations of glucose and incubated for 60 min at 37°C. After incubation, aliquots of 25 μl in duplicates were collected and stored at −20°C until analysis of insulin as above.
Islet insulin and glucagon content
Batches of four islets were frozen, thawed and then sonicated in acidic ethanol (0.2 mol/l HCl in 87.5% ethanol, by volume). The procedure was repeated twice. The samples were then centrifuged (10,000 g) and total insulin and glucagon contents were measured in the supernatant fractions.
Caspase 3/7 activity
Islets were solubilised in 20 mmol/l Tris-HCl, 150 mmol/l NaCl, 2 mmol/l EDTA and 1% Triton X-100, pH 7.5. Caspase 3/7 activity was measured in the supernatant, using CaspaseGlo (Promega, Madison, WI, USA) according to the manufacturer’s protocol. Luminescence was measured on a microtitre plate reader (Victor 2 Wallac; Perkin-Elmer, Turku, Finland).
Islet glucose oxidation
Glucose oxidation was measured in isolated islets. Batches of 30 islets in quadruplicates were incubated in a reaction mixture containing 3.7 or 26 kBq [14C]glucose (specific activity 11.5 GBq/mmol; NEN, Boston, MA, USA) as tracer, at final concentrations of 2.8 or 16.7 mmol/l glucose. The reaction was terminated after incubation of the samples for 2 h at 37°C, and the amount of released 14CO2 trapped with benzetonium hydroxide was determined by liquid scintillation counting.
Immunocytochemistry and morphometry
The islets were examined using indirect immunofluorescence as described [41]. Sections were incubated with primary antibodies (rabbit anti-porcine-glucagon, code 7811, dilution 1:5,120; guinea pig anti-human-proinsulin, code 9003, dilution 1:2,560; EuroDiagnostica, Malmo, Sweden) and with secondary antibodies with specificity for rabbit- or guinea pig-IgG coupled to fluorescein isothiocyanate (Jackson, West Grove, PA, USA) or Texas Red (Sigma). The specificity of the immunostaining was tested by pre-absorption with homologous peptide (100 μg/ml diluted antiserum). Islet alpha and beta cell areas were determined as previously reported [42]. In brief, serial sections (nine for each animal) on coded slides from five HFD-VEH and three HFD-GRA mice, double-stained for glucagon and insulin, were analysed using Image Pro Plus software (Media Cybernetics, Bethesda, MD, USA). We randomly selected ten islets in each section and calculated the immunostained area.
Statistical analysis
All data are presented as mean ± SEM. In the IVGTT, the acute insulin response to intravenous glucose was calculated as the mean of suprabasal 1 and 5 min values, and the glucose elimination was quantified using the glucose elimination constant, K G, calculated as the slope of the logarithmic transformation of circulating glucose between 1 and 20 min after the glucose bolus. The suprabasal AUC was calculated from 0 to 60 min by the trapezoid rule. The early insulin response in OGTT was defined as the 15-min peak value over basal secretion. The acute glucagon response to arginine was calculated as the mean of suprabasal 1 and 5 min values. Multiple comparisons between the different groups were performed by one-way ANOVA and Tukey’s post hoc test to calculate statistical differences between the groups. Significant statistical difference was considered to be present at p < 0.05.
Results
Body weight and fasting blood parameters
Body weight was significantly higher in mice fed HFD for 8 weeks than in ND-fed mice (p < 0.001; Fig. 1). During treatment with the GRA or VEH, body weight decreased slightly in HFD-fed mice, although with no significant difference between the two treatment groups (Fig. 1).

Body weight gain in mice fed normal diet (open triangles) or high-fat diet (open squares, closed squares) for 12 weeks. The mice were 8 weeks old at the start of the experiment and were fed the different diets for 8 weeks before starting the once-daily gavage treatment with the glucagon receptor antagonist (closed squares) or with vehicle (open squares). Data are presented as mean ± SEM, with n = 10 in each group. Arrow device, vertical: beginning of treatment; horizontal: duration of treatment
Treatment with the GRA for 17–18 days resulted in significant reduction in overnight fasting plasma glucose levels compared with HFD-VEH (Table 1). There was a trend towards lower plasma glucose levels in HFD-GRA than in ND-VEH, although this difference was not statistically significant. In contrast, there was no difference in fasting insulin levels between the treatment groups. The HFD-VEH group had elevated glucagon levels compared with the ND-VEH group, an increase that was even more marked and significantly so in HFD-GRA mice compared with both VEH groups (Table 1).
Intravenous and oral glucose tolerance test
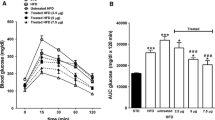
Glucose tolerance was impaired in HFD-fed mice compared with ND-fed mice (Fig. 2a). Following GRA treatment for 17–18 days, glucose elimination was improved and did not differ from that in ND-VEH mice (Fig. 2a, Table 1). The acute insulin response was augmented in HFD-VEH mice compared with ND-VEH and significantly augmented in HFD-GRA compared with both VEH groups (Table 1). Hence, as evident from the results of the IVGTT, GRA improved glucose tolerance and the insulin response to glucose in mice with HFD-induced glucose intolerance (Fig. 2).

IVGTT (1 g/kg [5.6 mmol/kg] glucose) was performed in overnight-fasted, anaesthetised mice after 17–18 days of treatment with the GRA or with VEH and fed ND or HFD. Plasma levels of glucose (a, b) and insulin (c) were measured at the indicated time-points in ND-VEH (open triangles; n = 10), HFD-VEH (open squares; n = 17) or HFD-GRA (closed squares; n = 16). Data are presented as mean ± SEM
There were only small differences in glucose tolerance between HFD- and ND-fed mice, which is different from previous data demonstrating clear glucose intolerance in the former [38]. This difference is explained by the experimental conditions. Here, the IVGTT was performed in mice fasted overnight, conditions in which glucose intolerance is less pronounced in HFD-fed mice. In mice fasted for 4 h, basal glucose and insulin levels were elevated compared with ND-fed mice. After an overnight fast, basal blood glucose and insulin levels were not different between the two feeding groups (Table 1). However, logarithmic transformation of the glucose levels in the IVGTT revealed that mice fed the HFD were glucose intolerant (Fig. 2b) despite significantly elevated insulin levels (Fig. 2c). The results thus show that GRA improved glucose tolerance by increasing the insulin secretion.
The OGTT demonstrated that the increase in plasma glucose was reduced in HFD-GRA compared with HFD-VEH mice (Fig. 3a), which was illustrated by a reduced AUCglu0-60 (Table 1). The improved glucose excursions observed in HFD-GRA mice were accompanied by reduced insulin secretion (Fig. 3b, Table 1), with significantly reduced early insulin response (3,040 ± 420 vs 1,890 ± 160 pmol/l, p = 0.012) and AUCins0-60 (Table 1).

OGTT (75 mg [0.42 mmol] glucose per mouse) was performed in overnight-fasted, anaesthetised mice after 24 days of treatment with the GRA or with VEH and fed the HFD. Plasma levels of glucose (a) and insulin (b) were measured at the indicated time-points in HFD-VEH (open squares; n = 16) or HFD-GRA (closed squares; n = 16). Data are presented as mean ± SEM
Glucagon secretion
After treatment with the GRA for 17–21 days, the glucagon response to intravenous l-arginine was examined. Again, basal glucose levels were significantly reduced in HFD-GRA compared with HFD-VEH mice (Fig. 4a), while basal glucagon levels were significantly elevated (p < 0.05 compared with ND and HFD-VEH; Fig. 4b, Table 1). Comparing the acute glucagon response to arginine, we found it significantly higher in HFD-GRA than in HFD-VEH mice (p < 0.01; Table 1). Notably, no effect on glucose levels was seen in the HFD-GRA treatment group despite elevated glucagon levels, suggesting that inhibition of the glucagon signal had occurred after GRA administration, because the acute rise in glucose after arginine administration is largely dependent on glucagon.

Arginine was injected intravenously into overnight-fasted, anaesthetised mice treated with the GRA or VEH for 17–21 days. Plasma levels of glucose (a) and glucagon (b) were measured at the indicated time points. Data are presented as mean ± SEM. ND-VEH (open triangles; n = 10); HFD-VEH (open squares; n = 17); HFD-GRA (closed squares; n = 17)
Euglycaemic–hyperinsulinaemic clamp
After 21–25 days of GRA treatment, HFD-fed mice were subjected to euglycaemic–hyperinsulinaemic clamp experiments (Fig. 5a). During the 60–90 min clamp, plasma glucose levels were 6.5 ± 0.2 mmol/l in HFD-GRA, 6.3 ± 0.2 mmol/l in HFD-VEH and 6.3 ± 0.06 mmol/l in ND mice. The corresponding insulin levels were 14,500 ± 2,600, 13,900 ± 2,000 and 11,000 ± 1,200 pmol/l, respectively. The mean GIR needed to maintain a stable glucose level between 60 and 90 min, was significantly higher in the HFD-GRA than in the HFD-VEH group (0.059 ± 0.007 vs 0.042 ± 0.008 mmol kg−1 min−1 in HFD-VEH; p = 0.021) (Fig. 5b). This suggests that insulin action is improved after glucagon receptor antagonism in HFD-fed mice. In the ND-fed control mice GIR was 0.105 ± 0.013 mmol kg−1 min−1.

Glucose levels (a) and GIR (b) in the euglycaemic–hyperinsulinaemic clamp study performed on ND (open triangles; n = 7), HFD-VEH (open squares; n = 7) and HFD-GRA (closed squares; n = 7) mice at 21–25 days after beginning of GRA treatment. Glucose was infused to maintain plasma levels at 6.5 mmol/l. Blood samples were taken every 5 min during the 90 min experiment
In vitro islet effects of glucagon receptor antagonist treatment
After 24–30 days of GRA treatment, insulin secretion in freshly isolated islets from the HFD-GRA group was augmented in response to 11.1 and 16.7 mmol/l glucose compared with islets from HFD-VEH mice (p < 0.001 at both concentrations) and with those from ND-VEH mice at 16.7 mmol/l glucose (p = 0.036; Fig. 6a). The glucose concentration at which half-maximal insulin response was observed was significantly lower in islets from HFD-GRA mice (11.3 mmol/l) than in those from the HFD-VEH group (13.7 mmol/l) and similar to that in islets from ND-VEH mice (11.0 mmol/l). This shows that both the insulin response to glucose and the glucose sensitivity of the islet were augmented by GRA treatment.

a Glucose-stimulated insulin secretion in isolated islets from ND-VEH (open triangles), HFD-VEH (open squares) and HFD-GRA (closed squares) mice after once-daily gavage treatment for 24–30 days with the GRA or VEH. Data are from three independent experiments. Islets were isolated from three mice in each treatment group and pooled. Batches of three islets in eight replicates were analysed for insulin. The results are presented as mean ± SEM. **p < 0.01, ***p < 0.001. b Islet glucose oxidation (ox) measured in islets isolated from ND-VEH (white bars), HFD-VEH (black bars) and HFD-GRA (grey bars) mice after once daily gavage treatment for 24–30 days. Data are presented as mean ± SEM from three independent experiments where each condition was run in quadruplicate
To evaluate whether the increased glucose stimulated insulin secretion (GSIS) observed in HFD-GRA mice was due to increased glucose metabolism, glucose oxidation was measured at 2.8 and 16.7 mmol/l glucose in freshly isolated islets. However, no difference in islet glucose oxidation was seen between the different treatment groups (Fig. 6b), suggesting that other mechanisms are involved in potentiating GSIS in HFD-GRA mice.
Islet insulin content was increased in response to the HFD. There was a slight reduction in islet insulin content in islets from HFD-GRA mice, though not statistically different from the two VEH-treated groups (Table 1). Islet glucagon content was similar in the three treatment groups (Table 1).
To evaluate whether GRA treatment may potentially damage islets and cause apoptosis, caspase 3/7 activity was measured in isolated islets. There were no differences in caspase 3/7 activity in the various groups (Table 1).
Immunocytochemistry
Immunocytochemical analysis illustrated increased alpha cell mass with increased intensity in glucagon immunostaining in HFD-GRA mice but without overt differences in islet morphology or insulin staining (Fig. 7). Morphometric analysis demonstrated a significantly increased alpha cell mass after GRA treatment (3,000 ± 600 in HFD vs 6,600 ± 1,200 μm2 in HFD-GRA; p = 0.017). In contrast, beta cell mass did not differ between the groups (11,800 ± 2,300 in HFD vs 16,500 ± 3,400 μm2 in HFD-GRA; p > 0.05). This resulted in a tendency towards a lower ratio of beta cell to alpha cell mass in HFD-GRA mice (3.9 ± 0.9 in HFD, 2.3 ± 0.2 in HFD-GRA).

Glucagon immunostaining in representative islets from ND-VEH (a), HFD-VEH (b) and HFD-GRA (c) mice after once daily gavage treatment with the GRA or VEH for 30 days. Pancreases from three to five mice in each treatment group were examined. Bar = 20 μm
Discussion
This study demonstrates that glucagon receptor antagonism using a specific GRA, NNC 25-0926, improves glycaemia and increases insulin secretion, resulting in improved glucose tolerance in female mice with HFD-induced insulin resistance. Previous data have shown that this compound specifically inhibits glucagon binding to cloned human glucagon receptors expressed in baby hamster kidney cells with an IC50 value of 12 nmol/l (E. Nishimura and U. G. Sidelmann, unpublished observations) and blocks glucagon-stimulated hepatic glucose output in dogs [40].
Our study demonstrates that glucagon receptor antagonism improves GSIS, which was evident under in vivo conditions in IVGTT and also in vitro in freshly isolated islets incubated with glucose, with improved islet sensitivity to glucose. Interestingly, this effect was not due to increased islet glucose oxidation. However, it could not be ruled out that glucose metabolism might be altered at other levels resulting in increased insulin secretion. Another possible explanation for the improved GSIS is increased expression of GLP-1 in islets. Others have suggested that increased expression of proglucagon, which is apparent after inhibition of glucagon signalling, may result in increased islet production of GLP-1 [28, 29]. Proglucagon is cleaved by prohormone convertases in the alpha cells to produce mainly glucagon and, to a much smaller extent, GLP-1 [43, 44]. The results demonstrate that long-term treatment with GRA results in improved islet glucose sensitivity, indicating that inhibition of the glucagon signal, directly or indirectly, has significant and beneficial effects on beta cell function.
GRA treatment increased alpha cell mass as well as glucagon secretion in response to arginine. Mice with a null mutation in the glucagon receptor have elevated glucagon levels, with pancreas and islets significantly enlarged, and massive alpha cell hyperplasia [29, 30]. These findings are similar to those in mice treated with glucagon receptor antisense oligonucleotide inhibitors, which also resulted in alpha cell hypertrophy, hyperplasia and hyperglucagonaemia, although to a less pronounced degree than in glucagon receptor knockout mice [28]. Hyperglucagonaemia has been observed in other studies using other GRAs [26]. The reason for this compensatory increase in the levels of glucagon after inhibition of glucagon signalling is currently not known. One possibility is that inhibition of glucagon signalling in the alpha cell may disrupt an autocrine negative feedback signal on glucagon secretion, which may augment glucagon secretion in response to secretagogues. In contrast, insulin expression was maintained during glucagon receptor antagonism, with similar immunostaining and with no difference in islets content compared with ND-fed mice or HFD-fed control mice. Although inhibition of the glucagon signal results in increased alpha cell mass, there has been no evidence of neoplastic formations in the pancreas after disruption of the glucagon signal [28, 29]. Also in the present study, up to 30 days of GRA treatment did not cause any apparent damage to islets, with no apparent apoptotic effect of the compound, as evidenced by the fact that caspase 3/7 activity, which is increased when apoptotic pathways are activated [45], was similar in the three treatment groups.
The reduction of glucose levels by GRA is consistent with other studies where glucagon signalling was inhibited, either through administration of GRA [25, 46], neutralising glucagon antibodies [18–20], or disruption of glucagon receptor expression [27–30]. In our study, improved glycaemia was not accompanied by elevated basal insulin levels. Furthermore, the results from the OGTT suggest that the demand for insulin was reduced in HFD-GRA-treated mice, since glucose excursions were improved despite lower insulin levels. Together with the results from the clamp study, this suggests that GRA treatment leads to improved insulin sensitivity. It remains to be established whether the improved insulin sensitivity depends on hepatic or extra-hepatic effects. Glucagon receptor antagonists may also reduce glucose through inhibition of hepatic glucose production [25, 40].
The use of glucagon receptor antagonism as a possible treatment of type 2 diabetes is tempting due to glucagon’s role in maintaining plasma glucose levels and the fact that patients with type 2 diabetes exhibit elevated fasting glucagon levels, which contribute to their hyperglycaemia [8, 9, 11]. The current treatment modalities for type 2 diabetes do not take increased glucagon levels into account and new drugs need to be developed to tackle this issue. Inhibition of glucagon signalling has, in several studies using several different techniques, been demonstrated to potently reduce glycaemia [18, 26, 28, 29]. One concern with this strategy is the risk of hypoglycaemia. However, no studies have observed this phenomenon, except in overnight-fasted mice lacking glucagon receptors, which is an extreme animal model with complete block of the glucagon signal [29]. However, the use of GRAs with long duration of action might not be feasible, whereas antagonists whose action is of shorter duration may possibly minimise the risk of hypoglycaemia. Another concern with glucagon receptor antagonism is the alpha cell hyperplasia that has been observed in all models where glucagon signalling was inhibited. It is, however, possible that one effect of alpha cell hyperplasia is increased islet GLP-1 production, which then would be an important mechanism contributing to increased insulin secretion [6].
In conclusion, this study demonstrates that glucagon receptor antagonism, through a small glucagon receptor-binding molecule, efficiently increases GSIS in mice with HFD-induced insulin resistance in association with increased glucose sensitivity in the islet beta cells. Glucagon receptor antagonism also increases glucagon secretion and results in increased alpha cell mass. Overall glucagon receptor antagonism reduces basal glucose and normalises glucose tolerance. The results encourage further exploration of the feasibility of glucagon antagonism as a potential treatment for type 2 diabetes.
Abbreviations
- GIR:
-
glucose infusion rate
- GLP-1:
-
glucagon-like peptide-1
- GRA:
-
glucagon receptor antagonist
- GSIS:
-
glucose stimulated insulin secretion
- HBSS:
-
HEPES balanced salt solution
- HFD:
-
high-fat diet
- IC50 :
-
inhibitor concentration of 50%
- ND:
-
normal diet
- VEH:
-
vehicle
References
Cherrington AD, Chiasson JL, Liljenquist JE, Lacy WW, Park CR (1978) Control of hepatic glucose output by glucagon and insulin in the intact dog. Biochem Soc Symp 43:31–45
Jiang G, Zhang BB (2003) Glucagon and regulation of glucose metabolism. Am J Physiol Endocrinol Metab 284:E671–E678
Ikeda T, Hoshino T, Honda M et al (1989) Effect of glucagon on glucose output from bivascularly perfused rat liver. Exp Clin Endocrinol 94:383–386
Beuers U, Jungermann K (1990) Relative contribution of glycogenolysis and gluconeogenesis to basal, glucagon- and nerve stimulation-dependent glucose output in the perfused liver from fed and fasted rats. Biochem Int 21:405–415
Shah P, Basu A, Basu R, Rizza R (1999) Impact of lack of suppression of glucagon on glucose tolerance in humans. Am J Physiol 277:E283–E290
Sloop KW, Michael MD, Moyers JS (2005) Glucagon as a target for the treatment of type 2 diabetes. Expert Opin Ther Targets 9:593–600
Dobbs R, Sakurai H, Sasaki H et al (1975) Glucagon: role in the hyperglycemia of diabetes mellitus. Science 187:544–547
Unger RH, Orci L (1975) The essential role of glucagon in the pathogenesis of diabetes mellitus. Lancet 1:14–16
Gerich JE, Lorenzi M, Karam JH, Schneider V, Forsham PH (1975) Abnormal pancreatic glucagon secretion and postprandial hyperglycemia in diabetes mellitus. JAMA 234:155–159
Unger RH (1978) Role of glucagon in the pathogenesis of diabetes: the status of the controversy. Metabolism 27:1691–1709
Larsson H, Ahren B (2000) Glucose intolerance is predicted by low insulin secretion and high glucagon secretion: outcome of a prospective study in postmenopausal Caucasian women. Diabetologia 43:194–202
Muller WA, Faloona GR, Aguilar-Parada E, Unger RH (1970) Abnormal alpha-cell function in diabetes. Response to carbohydrate and protein ingestion. N Engl J Med 283:109–115
Aronoff SL, Bennett PH, Unger RH (1977) Immunoreactive glucagon (IRG) responses to intravenous glucose in prediabetes and diabetes among Pima Indians and normal Caucasians. J Clin Endocrinol Metab 44:968–972
Ohneda A, Watanabe K, Horigome K, Sakai T, Kai Y, Oikawa S (1978) Abnormal response of pancreatic glucagon to glycemic changes in diabetes mellitus. J Clin Endocrinol Metab 46:504–510
Reaven GM, Chen YD, Golay A, Swislocki AL, Jaspan JB (1987) Documentation of hyperglucagonemia throughout the day in nonobese and obese patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab 64:106–110
Baron AD, Schaeffer L, Shragg P, Kolterman OG (1987) Role of hyperglucagonemia in maintenance of increased rates of hepatic glucose output in type II diabetics. Diabetes 36:274–283
Basu R, Chandramouli V, Dicke B, Landau B, Rizza R (2005) Obesity and type 2 diabetes impair insulin-induced suppression of glycogenolysis as well as gluconeogenesis. Diabetes 54:1942–1948
Brand CL, Jorgensen PN, Knigge U et al (1995) Role of glucagon in maintenance of euglycemia in fed and fasted rats. Am J Physiol 269:E469–E477
Brand CL, Rolin B, Jorgensen PN, Svendsen I, Kristensen JS, Holst JJ (1994) Immunoneutralization of endogenous glucagon with monoclonal glucagon antibody normalizes hyperglycaemia in moderately streptozotocin-diabetic rats. Diabetologia 37:985–993
Brand CL, Jorgensen PN, Svendsen I, Holst JJ (1996) Evidence for a major role for glucagon in regulation of plasma glucose in conscious, nondiabetic, and alloxan-induced diabetic rabbits. Diabetes 45:1076–1083
Johnson DG, Goebel CU, Hruby VJ, Bregman MD, Trivedi D (1982) Hyperglycemia of diabetic rats decreased by a glucagon receptor antagonist. Science 215:1115–1116
Van Tine BA, Azizeh BY, Trivedi D et al (1996) Low level cyclic adenosine 3′,5′-monophosphate accumulation analysis of [des-His1, des-Phe6, Glu9] glucagon-NH2 identifies glucagon antagonists from weak partial agonists/antagonists. Endocrinology 137:3316–3322
Ling A, Plewe M, Gonzalez J et al (2002) Human glucagon receptor antagonists based on alkylidene hydrazides. Bioorg Med Chem Lett 12:663–666
Shen DM, Zhang F, Brady EJ et al (2005) Discovery of novel, potent, and orally active spiro-urea human glucagon receptor antagonists. Bioorg Med Chem Lett 15:4564–4569
Qureshi SA, Rios Candelore M, Xie D et al (2004) A novel glucagon receptor antagonist inhibits glucagon-mediated biological effects. Diabetes 53:3267–3273
Petersen KF, Sullivan JT (2001) Effects of a novel glucagon receptor antagonist (Bay 27-9955) on glucagon-stimulated glucose production in humans. Diabetologia 44:2018–2024
Liang Y, Osborne MC, Monia BP et al (2004) Reduction in glucagon receptor expression by an antisense oligonucleotide ameliorates diabetic syndrome in db/db mice. Diabetes 53:410–417
Sloop KW, Cao JX, Siesky AM et al (2004) Hepatic and glucagon-like peptide-1-mediated reversal of diabetes by glucagon receptor antisense oligonucleotide inhibitors. J Clin Invest 113:1571–1581
Gelling RW, Du XQ, Dichmann DS et al (2003) Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci U S A 100:1438–1443
Parker JC, Andrews KM, Allen MR, Stock JL, McNeish JD (2002) Glycemic control in mice with targeted disruption of the glucagon receptor gene. Biochem Biophys Res Commun 290:839–843
Sorensen H, Winzell MS, Brand CL et al (2006) Glucagon receptor knockout mice display increased insulin sensitivity and impaired beta-cell function. Diabetes 55:3463–3469
Bergman RN, Phillips LS, Cobelli C (1981) Physiologic evaluation of factors controlling glucose tolerance in man: measurement of insulin sensitivity and beta-cell glucose sensitivity from the response to intravenous glucose. J Clin Invest 68:1456–1467
Kahn SE, Prigeon RL, McCulloch DK et al (1994) The contribution of insulin-dependent and insulin-independent glucose uptake to intravenous glucose tolerance in healthy human subjects. Diabetes 43:587–592
Larsson H, Ahren B (1996) Failure to adequately adapt reduced insulin sensitivity with increased insulin secretion in women with impaired glucose tolerance. Diabetologia 39:1099–1107
Porte D, Jr, Kahn SE (2001) Beta-cell dysfunction and failure in type 2 diabetes: potential mechanisms. Diabetes 50(Suppl 1):S160–S163
Huypens P, Ling Z, Pipeleers D, Schuit F (2000) Glucagon receptors on human islet cells contribute to glucose competence of insulin release. Diabetologia 43:1012–1019
Pacini G, Thomaseth K, Ahren B (2001) Contribution to glucose tolerance of insulin-independent vs insulin-dependent mechanisms in mice. Am J Physiol Endocrinol Metab 281:E693–E703
Winzell MS, Ahren B (2004) The high-fat diet-fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes 53(Suppl 3):S215–S219
Ahren B, Simonsson E, Scheurink AJ, Mulder H, Myrsen U, Sundler F (1997) Dissociated insulinotropic sensitivity to glucose and carbachol in high-fat diet-induced insulin resistance in C57BL/6J mice. Metabolism 46:97–106
Rivera N, Everett CA, Edgerton DS et al (2007) A novel glucagon receptor antagonist, NNC 25-0926, blunts hepatic glucose production in the conscious dog. J Pharmacol Exp Ther DOI 10.1124/jept.106.115717
Wierup N, Kuhar M, Nilsson BO, Mulder H, Ekblad E, Sundler F (2004) Cocaine- and amphetamine-regulated transcript (CART) is expressed in several islet cell types during rat development. J Histochem Cytochem 52:169–177
Harndahl L, Wierup N, Enerback S et al (2004) Beta-cell-targeted overexpression of phosphodiesterase 3B in mice causes impaired insulin secretion, glucose intolerance, and deranged islet morphology. J Biol Chem 279:15214–15222
Rouille Y, Bianchi M, Irminger JC, Halban PA (1997) Role of the prohormone convertase PC2 in the processing of proglucagon to glucagon. FEBS Lett 413:119–123
Rouille Y, Kantengwa S, Irminger JC, Halban PA (1997) Role of the prohormone convertase PC3 in the processing of proglucagon to glucagon-like peptide 1. J Biol Chem 272:32810–32816
Hui H, Dotta F, Di Mario U, Perfetti R (2004) Role of caspases in the regulation of apoptotic pancreatic islet beta-cells death. J Cell Physiol 200:177–200
Dallas-Yang Q, Shen X, Strowski M et al (2004) Hepatic glucagon receptor binding and glucose-lowering in vivo by peptidyl and non-peptidyl glucagon receptor antagonists. Eur J Pharmacol 501:225–234
Acknowledgements
The authors would like to thank L. Kvist, K. Andersson, L. Bengtsson and D. Persson for excellent technical assistance and J. Ahrén for morphometric analysis of islets. This work was supported by the Swedish Research Council (grant numbers 6834 to B. Ahrén and 4499 to F. Sundler), the Novo Nordisk Foundation, the Albert Påhlsson Foundation, the Crafoord Foundation, the Swedish Diabetes Foundation, Region Skåne and the Medical Faculty of Lund University.
Duality of interest
E. Nishimura, C. L. Brand and U. G. Sidelmann are employed by Novo Nordisk, Denmark and are shareholders in the company. B. Ahrén has received grants for research from Novo Nordisk, Denmark.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Winzell, M.S., Brand, C.L., Wierup, N. et al. Glucagon receptor antagonism improves islet function in mice with insulin resistance induced by a high-fat diet. Diabetologia 50, 1453–1462 (2007). https://doi.org/10.1007/s00125-007-0675-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-007-0675-3