
Abstract
Aims/hypothesis
Hyperglycaemia, one of the main features of diabetes, results in non-enzymatic glycation of plasma proteins, including apolipoprotein A-I (apoA-I), the most abundant apolipoprotein in HDL. The aim of this study was to determine how glycation affects the structure of apoA-I and its ability to activate lecithin:cholesterol acyltransferase (LCAT), a key enzyme in reverse cholesterol transport.
Materials and methods
Discoidal reconstituted HDL (rHDL) containing phosphatidylcholine and apoA-I ([A-I]rHDL) were prepared by the cholate dialysis method and glycated by incubation with methylglyoxal. Glycation of apoA-I was quantified as the reduction in detectable arginine, lysine and tryptophan residues. Methylglyoxal-AGE adduct formation in apoA-I was assessed by immunoblotting. (A-I)rHDL size and surface charge were determined by non-denaturing gradient gel electrophoresis and agarose gel electrophoresis, respectively. The kinetics of the LCAT reaction was investigated by incubating varying concentrations of discoidal (A-I)rHDL with a constant amount of purified enzyme. The conformation of apoA-I was assessed by surface plasmon resonance.
Results
Methylglyoxal-mediated modifications of the arginine, lysine and tryptophan residues in lipid-free and lipid-associated apoA-I were time- and concentration-dependent. These modifications altered the conformation of apoA-I in regions critical for LCAT activation and lipid binding. They also decreased (A-I)rHDL size and surface charge. The rate of LCAT-mediated cholesterol esterification in (A-I)rHDL varied according to the level of apoA-I glycation and progressively decreased as the extent of apoA-I glycation increased.
Conclusions/interpretation
It is concluded that glycation of apoA-I may adversely affect reverse cholesterol transport in subjects with diabetes.
Similar content being viewed by others

Introduction
Macrovascular complications are responsible for up to 80% of the deaths in subjects with diabetes [1]. There is a large body of evidence to suggest that this is partly due to non-enzymatic protein glycation and AGE formation. AGE have been implicated in the development of premature atherosclerosis in diabetic subjects by virtue of their ability to increase endothelial dysfunction and decrease endothelial cell viability and smooth muscle cell proliferation [2, 3].
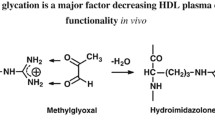
Chronic hyperglycaemia, a feature of both type 1 and type 2 diabetes, generates highly reactive and toxic α-oxoaldehydes, such as methylglyoxal (MG), glyoxal and 3-deoxyglucosone [4]. Covalent binding of these oxoaldehydes to reactive lysine, arginine and cysteine residues as well as to N-terminal amino groups in plasma proteins and apolipoproteins, including apolipoprotein A-I (apoA-I), the major apolipoprotein of HDL, generates early glycation adducts and AGE [5–8]. These modifications are associated with protein cross-linking and aggregation [9]. It has been suggested that MG-derived, as well as other AGE, not only accelerate the development of coronary artery disease in humans [10], but are also responsible for the macro- and microvascular complications frequently observed in diabetes.
The impact of apoA-I glycation on HDL function is poorly understood. Some evidence suggests that glycation adversely affects HDL metabolism by diminishing the binding of HDL to fibroblasts and macrophages [11, 12] and enhancing HDL catabolism [13]. Glycation also increases cholesteryl ester (CE) transfer protein-mediated transfers of CEs from HDL to apoB-containing lipoproteins [14] and alters the ability of lecithin:cholesterol acyltransferase (LCAT) to esterify cholesterol [15].
LCAT is the enzyme that generates almost all of the CEs in plasma. It plays a key role in reverse cholesterol transport and is activated by the apoA-I in HDL [16]. The impact of diabetes on the ability of HDL to act as a substrate for LCAT is unclear, with reports suggesting that it either diminishes [17], does not alter [18] or increases the rate of LCAT-mediated cholesterol esterification [19]. These discrepancies are probably due to variations in the study populations and the methods used to measure cholesterol esterification [15]. Although there is evidence suggesting that glycation of apoA-I in HDL impairs the LCAT reaction in vivo and in vitro [20–22], the underlying mechanism of these observations are not understood. These issues are addressed in the present study using preparations of discoidal reconstituted HDL (rHDL) containing apoA-I and phosphatidylcholine ([A-I]rHDL). These preparations are structurally and functionally indistinguishable from the discoidal HDL that are the main LCAT substrates in plasma.
Materials and methods
Isolation of apolipoprotein A-I
xHDL were isolated from pooled samples of autologously donated human plasma (Gribbles Pathology, Adelaide, SA, Australia) by sequential ultracentrifugation (1.063 < d < 1.21 g/ml). The HDL were delipidated and apoA-I was isolated by anion exchange chromatography on a Q sepharose fast flow column (GE Healthcare, Uppsala, Sweden) attached to a fast protein liquid chromatography system [23–25]. The apoA-I appeared as a single band when subjected to SDS-Phastgel electrophoresis (GE Healthcare) and Coomassie staining.
Preparation of discoidal (A-I)rHDL labelled with unesterified [3H]cholesterol
Discoidal (A-I)rHDL containing 1-palmitoyl-2-oleoyl phosphatidylcholine (POPC) (Avanti Polar Lipids, Alabaster, AL, USA), unesterified cholesterol (UC) (Sigma-Aldrich, St Louis, MO, USA), a trace amount of 1α,2α-3H-labelled UC ([3H]UC; GE Healthcare) and apoA-I (initial POPC:UC:apoA-I molar ratio 100:10:1) were prepared by the cholate dialysis method [26]. The (A-I)rHDL were dialysed against PBS (pH 7.4) containing 0.006% (w/v) NaN3 and 0.005% (w/v) EDTA-Na2 before use.
Isolation of LCAT
LCAT was isolated from pooled samples of human plasma as described [23] and appeared as a single band following SDS-Phastgel electrophoresis and silver staining. LCAT activity was assessed using [3H]UC-labelled discoidal (A-I)rHDL as the substrate [27]. The LCAT preparations used in this study generated 346–1,600 nmol CE ml LCAT−1 h−1.
Glycation of lipid-free apoA-I by glucose
Lipid-free apoA-I (final concentration 1 mg/ml) was incubated at 37°C under 5% CO2 for 3 days to 3 weeks with 5, 25 or 100 mmol/l glucose (Sigma). Unreacted glucose was removed by dialysis against PBS.
Glycation of lipid-free apoA-I and discoidal (A-I)rHDL by methylglyoxal
Lipid-free apoA-I and discoidal (A-I)rHDL (final apoA-I concentration 1 mg/ml) were incubated individually at 37°C under 5% CO2 for 0.5–24 h with either 0, 0.5, 1.5, 3, 15 or 30 mmol/l MG (Sigma). When the incubations were complete, unreacted MG was removed by dialysis against PBS.
ApoA-I arginine, lysine and tryptophan modification
MG-mediated modification of apoA-I arginine, lysine and tryptophan residues was assessed fluorometrically and expressed as a percentage of the values obtained for unmodified samples incubated with PBS [28]. Rate constants for MG-mediated modification of apoA-I arginine (k arg), lysine (k lys) and tryptophan (k trp) residues in discoidal (A-I)rHDL were derived by non-linear regression analysis (GraphPad Prism version 4.0; GraphPadSoftware, San Diego, CA, USA) using the equation:
where t is the duration of the incubation with MG. The results are expressed as h−1.
Determination of discoidal (A-I)rHDL surface charge
Discoidal (A-I)rHDL containing non-enzymatically glycated apoA-I, (A-I)rHDLglyc, were electrophoresed at 90 V for 45 min on 1% agarose gels (Helena Laboratories, Mt Waverly, VIC, Australia), fixed in methanol, stained with Coomassie Blue R and destained with water::acetic acid::isopropanol (77.5::10::12.5, v::v::v). Electrophoretic mobilities were calculated as described [29]
and corrected for pI-dependent retardation effects according to the following relationship:
Conformation of unmodified and glycated apoA-I in the lipid-free form and in discoidal (A-I)rHDL
A Biacore 2000 biosensor (GE Healthcare, Uppsala, Sweden) was used to measure the association rate constant (k a) of seven unique apoA-I-specific monoclonal antibodies (mAbs) for rHDL. Saturating amounts of rabbit anti-mouse Fc were immobilised on all four flow cells of a CM5 chip using amine coupling [30]. Injected isotype control or specific mAbs were captured by the rabbit anti-mouse Fc at dilutions pre-determined to give 400 response units. This was followed by injection of the rHDL (analyte). The rHDL injections were made on the basis of apoA-I molarity.
Data were collected at a high (5 Hz) data collection rate. Evaluation of the data began by synchronising the injection time and zeroing the baselines of the sensorgram. Control antibody sensorgrams were subtracted from each specific antibody sensorgram. Each antibody and rHDL combination was evaluated with a 1:1 (Langmuir) model.
Kinetic studies
Various concentrations of discoidal (A-I)rHDL and discoidal (A-I)rHDLglyc containing unlabelled UC (final UC concentration 0.5–14.0 μmol/l) and a tracer amount of [3H]UC were incubated at 37°C for 1 h in a shaking water-bath with a constant amount of LCAT, fatty acid-free BSA (final concentration 3.7 mg/ml), β-mercaptoethanol (final concentration 5.3 mmol/l) and Tris-buffered saline (pH 7.4). The final volume of the incubation mixtures was 135 μl. The incubations were carried out in triplicate on three separate occasions. Cholesterol esterification was quantitated as described [27]. The kinetic parameters, K m(app) and V max, were determined by non-linear regression analysis (GraphPad Prism) using the equation
where V is the rate of cholesterol esterification. Results are expressed as μmol CE formed ml LCAT−1 h−1.
Competition studies
[3H]UC-labelled discoidal (A-I)rHDL (final UC concentration 0.5–5 μmol/l) was mixed with varying amounts of unlabelled discoidal (A-I)rHDLglyc (final UC concentration 0, 1.5 or 3 μmol/l) and incubated with a fixed amount of LCAT. The rate of cholesterol esterification in the [3H]UC-labelled discoidal (A-I)rHDL was determined according to equation 5 [31].
where: (1) v is the predicted rate of cholesterol esterification (μmol CE formed ml LCAT−1 h−1); (2) V max and K m(app) are the kinetic parameters for [3H]UC-labelled discoidal (A-I)rHDL incubated with LCAT in absence of unlabelled (A-I)rHDLglyc; and (3) [3H]UC-labelled (A-I)rHDL] and [(A-I)rHDLgly] represent the UC concentrations of the unmodified (A-I)rHDL and (A-I)rHDLglyc, respectively. K i is the inhibitor constant for [3H]UC-labelled discoidal (A-I)rHDL in presence of unlabelled (A-I)rHDLglyc, and was calculated as follows:
where V max(app) is the V max for the unmodified (A-I)rHDL incubated with LCAT in presence of different concentrations of (A-I)rHDLglyc.
Other techniques
All chemical analyses were performed in triplicate on an automatic analyser (Hitachi 902; Roche Diagnostics, Mannheim, Germany). UC and POPC concentrations were measured enzymatically [32, 33]. ApoA-I concentrations were determined by the bicinchoninic assay using BSA as a standard [34].
Protein oxidation was determined by HPLC as the formation of the protein-bound tyrosine and phenylalanine oxidation products 3,4-dihydroxy phenylalanine (DOPA), o-o′-di-tyrosine and o-tyrosine [28]. Lipid hydroperoxides were quantified using a lipid hydroperoxide assay kit (Cayman Chemicals, Ann Arbor, MI, USA). Protein cross-linking was assessed by SDS-PAGE. Autofluorescence (excitation 370 nm, emission 440 nm) of cross-linked proteins was determined with a Cytofluor II fluorimeter (Perspective Biosystems, Framingham, MA, USA). Results are expressed in arbitrary fluorescent units (FU) after blank subtraction.
Discoidal (A-I)rHDL diameters were determined by 3–40% non-denaturating polyacrylamide gradient gel electrophoresis and staining with Coomassie Blue R [35]. The stained gels were scanned with a laser densitometer. Particle sizes were determined by reference to high-molecular mass standards of known diameter.
MG-AGE adducts were detected by incubating apoA-I with MG, removing unreacted MG by dialysis against PBS, electrophoresing the incubation mixture on a 20% homogeneous SDS Phastgel (GE Healthcare) and immunoblotting with a 1:1,000 dilution of an anti-MG-AGE mAb (Biologo, Kronshagen, Germany). The blots were developed by electrochemoluminescence (GE Healthcare). Carboxyethyllysine and carboxymethyllysine were quantified by gas chromatography/mass spectrometry and pentosidine levels were quantitated by HPLC as described [36–38].
Statistical analyses
Significant differences in apoA-I residue modifications were determined by one-way ANOVA with Tukey’s multiple comparison post-test analysis. Two-way ANOVA was used to assess differences in cholesterol esterification rates. Surface plasmon resonance data were compared with a two-tailed, unpaired t test. Graphpad Prism (GraphPad Prism Software Inc., San Diego, CA, USA) was used for all analyses. Significance was set at p < 0.05.
Results
Modification of lipid-free apoA-I arginine, lysine and tryptophan residues by glucose
Compared with lipid-free apoA-I incubated with a physiological, non-pathological concentration of glucose (5 mmol/l), 1 week of incubation with either 25 or 100 mmol/l glucose did not modify apoA-I significantly. After 3 weeks of incubation with 25 mmol/l glucose, 6% of the apoA-I lysine residues were modified, but the arginine and tryptophan residues were not affected (data not shown). After 2 weeks of incubation with 100 mmol/l glucose, 10% of the apoA-I lysine residues were modified, while 30% of the lysine residues and approximately 8% of the arginine and 6% of the tryptophan residues were modified at 3 weeks. Given that the half-life of apoA-I in plasma is 3–4 days [39], it was concluded that these modifications are unlikely to be of physiological or pathological significance.
Modification of lipid-free apoA-I and the apoA-I in discoidal (A-I)rHDL by MG: time and concentration dependence
Additional experiments were carried out to determine if the reactive dicarbonyl derivative of glucose, MG, modifies apoA-I arginine, lysine and tryptophan residues more rapidly than glucose. When lipid-free apoA-I was incubated for 24 h with 1.5 mmol/l MG, the arginine, lysine and tryptophan residues were significantly modified (p < 0.001) (Table 1). More extensive modifications were apparent when lipid-free apoA-I was incubated with 15 mmol/l MG (p < 0.001 compared with 1.5 mmol/l MG). The apoA-I lysine and tryptophan, but not the arginine residues, were modified even further by incubation with 30 mmol/l MG (p < 0.001 compared with 15 mmol/l MG). As these modifications impaired the ability of lipid-free apoA-I to associate with phospholipids, these preparations could not be used to generate discoidal rHDL.
To investigate the effects of apoA-I glycation on HDL structure and function, it was therefore necessary to incorporate apoA-I into discoidal rHDL prior to incubation with MG. When discoidal (A-I)rHDL were incubated for 1 h with 3 mmol/l MG, the apoA-I arginine, lysine and tryptophan residues were extensively modified (p < 0.05 for lysine, p < 0.01 for arginine and p < 0.001 for tryptophan compared with [A-I]rHDL incubated for 1 h in the absence of MG) (Table 2). Further lysine and tryptophan, but not arginine, modifications were observed in incubations carried out for 1 h with 30 mmol/l MG.
The apoA-I arginine, lysine and tryptophan residues were not significantly modified when discoidal (A-I)rHDL were incubated with 0.5 mmol/l MG for 24 h (Table 2). By contrast, significant apoA-I modifications were apparent when the rHDL were incubated for 24 h with 1.5 mmol/l MG (p < 0.01 for the arginine, lysine and tryptophan residues in [A-I]rHDL), 3 mmol/l MG (p < 0.01 for the lysine residues in [A-I]rHDL; p < 0.001 for the arginine and tryptophan residues in [A-I]rHDL) or 30 mmol/l MG (p < 0.001 for the arginine, lysine and tryptophan residues in [A-I]rHDL). Comparison of the results in Tables 1 and 2 shows that, during a 24-h incubation, twice as much MG was required to achieve comparable modification of the arginine, lysine and tryptophan residues in the apoA-I in discoidal (A-I)rHDL as in lipid-free apoA-I.
The rate of modification of apoA-I arginine, lysine and tryptophan residues was determined by incubating discoidal rHDL in the absence or presence of 3 mmol/l MG. The arginine residues were modified more rapidly (k arg = 0.34 ± 0.05 h−1) than the lysine (k lys = 0.11 ± 0.02 h−1) (p < 0.001) and tryptophan residues residues (k trp = 0.045 ± 0.02 h−1) (p < 0.01) (Fig. 1).

Methylglyoxal-mediated modification of discoidal (A-I)rHDL: time dependence. Discoidal (A-I)rHDL (final apoA-I concentration 1 mg/ml) were incubated at 37°C for 0.5–24 h with 3 mmol/l MG (final volume 4 ml). The percentage of unmodified arginine (square), lysine (triangle) and tryptophan (circle) residues was determined as described in Materials and methods. All data represent the mean±SEM (n = 3)
Effects of glycation on the physical properties of apoA-I in discoidal (A-I)rHDL
Incubation with MG did not affect discoidal (A-I)rHDL stoichiometry (Table 3). However, the diameter of the particles decreased progressively from 9.7 to 9.0 nm (not shown), probably as a consequence of alterations in the conformation of apoA-I (see below). As judged by agarose gel electrophoresis, the rHDL surface also became more negatively charged as the apoA-I became progressively glycated (Table 3).
To ensure that the changes in rHDL surface charge and particle size were not due to glycoxidation, POPC and apoA-I oxidation was assessed as the formation of lipid hydroperoxides and protein-bound DOPA, o-o′-di-tyrosine and o-tyrosine, respectively. The results for the unmodified and glycated particles were comparable, indicating that the changes in (A-I)rHDL size and surface charge were due to glycation and not to glycoxidation (data not shown).
To determine whether incubation with MG produced cross-linked apoA-I, discoidal (A-I)rHDL were incubated in absence or presence of 3 mmol/l MG for 1, 2, 6, 12 or 24 h (Fig. 2). As judged by SDS-PAGE, incubation with MG progressively decreased the concentration of monomeric apoA-I in association with a concomitant increase in oligomer formation (Fig. 2). The increase in discoidal (A-I)rHDL autofluorescence (Table 3), indicated that the oligomers represent cross-linked, rather than aggregated apoA-I. Comparable results were obtained when lipid-free apoA-I was incubated with MG, indicating that the cross-linking could not be attributed to the lipid content of the particles (data not shown).

Methylglyoxal-mediated cross-linking of apoA-I in discoidal (A-I)rHDL. Discoidal (A-I)rHDL (final apoA-I concentration 1 mg/ml) were incubated at 37°C in the absence (Track 6) or presence of 3 mmol/l MG for 1, 2, 6, 12 or 24 h (tracks 1–5 respectively). A track containing low molecular mass standards is shown on the left (unnumbered track). The samples were delipidated, subjected to SDS-PAGE and stained with Coomassie Blue R. A scan of a representative stained gel is shown. MM, molecular mass
To determine whether AGE adducts were formed when apoA-I was incubated with MG, lipid-free apoA-I was incubated for 24 h in the presence or absence of 1.5 mmol/l MG, subjected to SDS-PAGE and immunoblotted with an mAb that recognises MG-AGE adducts (Fig. 3). Adducts were evident in the apoA-I that had been incubated in the presence, but not in that incubated in the absence of MG. As judged by gas chromatography/mass spectrometry (for carboxymethyllysine and carboxyethyllysine) and HPLC (for pentosidine), the apoA-I that had been incubated with MG contained 0.19 mmol carboxyethyllysine/mol lysine, 0.07 mmol carboxymethyllysine/mol lysine and 417 pmol pentosidine/mg apoA-I.

Immunoblot of methylglyoxal-modified lipid-free apoA-I. Lipid-free apoA-I (final concentration 1 mg/ml) was incubated for 24 h at 37°C in the presence or absence of MG (final concentration 1.5 mmol/l). When the incubations were complete the samples were subjected to SDS-PAGE and immunoblotted for MG-AGE adducts as described under Materials and methods
Effects of glycation on the conformation of apoA-I in the lipid-free form and in discoidal (A-I)rHDL
Surface plasmon resonance was used to assess the binding of a panel of seven well defined, epitope-specific apoA-I mAbs to discoidal (A-I)rHDL that had been incubated for 24 h in the absence or presence of 30 mmol/l MG (Fig. 4a). Lipid-free apoA-I was incubated for 24 h in the absence or presence of 15 mmol/l MG (Fig. 4b). The discoidal (A-I)rHDL were incubated with twice as much MG as the lipid-free apoA-I to ensure that the arginine, lysine and tryptophan residues in both of the samples were modified to the same extent (see Tables 1 and 2). Incubation with MG dramatically altered the affinity of all the mAbs for their epitopes in lipid-associated apoA-I. With the exception of mAb AI-11, this was also the case for lipid-free apoA-I. The affinity of all seven mAbs for their epitopes in lipid-free apoA-I differed from the finding for lipid-associated apoA-I, confirming that association with lipid alters the conformation of apoA-I.

Effect of glycation on the conformation of lipid-free and lipid-associated apoA-I. Discoidal (A-I)rHDL (a) and lipid-free apoA-I (b) were incubated at 37°C for 24 h in the absence (closed bars) or presence (open bars) of 30 and 15 mmol/l MG, respectively. Surface plasmon resonance was used to determine association rate constants (k a) of seven unique, apoA-I-specific mAbs for the lipid-free and lipid-associated apoA-I. *p < 0.05, **p < 0.01, ***p < 0.001 versus controls incubated in the absence of MG. aa, amino acid
Effect of glycation on the ability of apoA-I to activate LCAT: time and concentration dependence
Aliquots of discoidal (A-I)rHDL were incubated for up to 24 h with 3 mmol/l MG. Increasing amounts of the resulting (A-I)rHDLglyc were then incubated for 1 h with a constant amount of LCAT (Fig. 5). The (A-I)rHDLglyc that had been incubated for 1 h with MG exhibited a slight, non-statistically significant increase in the rate of cholesterol esterification compared with unmodified (A-I)rHDL (Fig. 5). When (A-I)rHDL that had been incubated with MG for 2, 3 and 6 h were subsequently incubated with LCAT, the rate of cholesterol esterification progressively decreased relative to the control rHDL (p < 0.0001) (Fig. 5).

LCAT-mediated cholesterol esterification in discoidal (A-I)rHDL and discoidal (A-I)rHDLglyc. Discoidal (A-I)rHDL were incubated at 37°C in the absence (open circles) or presence of 3 mmol/l MG for 1 (closed circles), 2 (closed squares), 3 (open squares) and 6 h (closed triangles). The rHDL (0.5–14 μmol/l UC) were then incubated at 37°C for 1 h with LCAT (5 μl of 1/50 dilution of a preparation that esterified 1,600 nmol CE ml LCAT−1 h−1) and the rate of cholesterol esterification was determined. Values represent the mean±SEM of triplicate determinations. *p < 0.0001 versus (A-I)rHDL incubated in the absence of MG
The V max for the (A-I)rHDLglyc that were incubated with MG for 3 or 6 h was significantly decreased compared with the control (A-I)rHDL and the (A-I)rHDLglyc that were incubated with MG for 1 h (Table 4). The affinity, K m(app), of LCAT for (A-I)rHDL was independent of the duration of the incubation with 3 mmol/l MG. Kinetic parameters could not be determined for rHDL incubated with MG for more than 12 h because the rate of cholesterol esterification was very low. The ability of the discoidal (A-I)rHDL that had been incubated with a more physiologically relevant MG concentration (0.5 mmol/l) to esterify cholesterol was also significantly impaired compared with control (A-I)rHDL (Table 5).
Competitive inhibition of cholesterol esterification in unmodified (A-I)rHDL by (A-I)rHDLglyc
The preceding kinetic studies suggested that discoidal (A-I)rHDLglyc may competitively inhibit LCAT-mediated cholesterol esterification in unmodified discoidal (A-I)rHDL. This possibility was investigated by incubating discoidal (A-I)rHDL with 3 mmol/l MG for 24 h. When the resulting (A-I)rHDLglyc (final UC concentration 1.5 or 3 μmol/l) were incubated with unmodified [3H]UC-labelled (A-I)rHDL (final UC concentration 0.5–5 μmol/l) and a constant amount of LCAT, the rate of cholesterol esterification in the [3H]UC-labelled (A-I)rHDL decreased as the concentration of unlabelled (A-I)rHDLglyc in the incubation mixture increased (Table 6).
The values obtained from Eqs. (4) and (5), which predicted the ability of (A-I)rHDLglyc to decrease the rate of cholesterol esterification in the unmodified [3H]UC-labelled (A-I)rHDL, agreed closely with the experimentally determined values (Table 6). This indicates that (A-I)rHDLglyc competitively inhibits LCAT-mediated cholesterol esterification in unmodified, [3H]UC-labelled (A-I)rHDL.
Discussion
Many plasma proteins and apolipoproteins, including apoA-I, are non-enzymatically glycated in both type 1 and type 2 diabetes [7]. Given that protein glycation is closely associated with adverse metabolic outcomes, as evidenced by the strong association of HbA1c concentrations with cardiovascular disease and mortality in the EPIC-Norfolk study [40], it is important to understand the underlying mechanisms of these modifications and to identify exactly which metabolic processes are adversely affected.
A key aim of the present study was to determine how glycation with MG influences the ability of apoA-I to activate LCAT, a plasma factor that plays a key role in HDL metabolism and reverse cholesterol transport [41]. Plasma MG levels as high as 0.4–0.5 mmol/l have been reported in diabetic subjects, irrespective of the level of glycaemic control [42, 43]. The present studies show that, although incubation of discoidal (A-I)rHDL with 0.5 mmol/l MG, does not appreciably modify the apoA-I arginine, lysine and tryptophan residues, it does markedly impair the ability of the discs to act as substrates for LCAT (Tables 2 and 5). These observations raise the possibility that minor modifications of apolipoproteins that are mediated by α-oxoaldehyde could have a major impact on HDL function in vivo.
In order to investigate LCAT activation, it was important to use apoA-I-containing discoidal HDL, the primary substrates for LCAT in human plasma [16]. As sufficient amounts of these particles cannot be isolated readily from normolipidaemic plasma, well characterised preparations of discoidal (A-I)rHDL that are structurally and functionally comparable to their plasma counterparts were used for the study [44]. As judged by agarose gel electrophoresis and surface plasmon resonance, incubation with MG altered both the surface charge, and the conformation of apoA-I in the discoidal rHDL. The change in surface charge was probably due to the modification of lysine and arginine residues reducing the overall positive charge of apoA-I. Similar changes in surface charge have been reported for MG-modified LDL [28, 45].
It is likely that changes in rHDL surface charge as well as the conformation of apoA-I both contributed to the decreased ability of discoidal (A-I)rHDLglyc to activate LCAT. Alexander et al. have shown that mutation of negatively charged residues in apoA-I significantly reduces LCAT activation [46]. The present results suggest that this may also be the case when positively charged residues are modified. Additional evidence that alterations in the conformation of apoA-I affect LCAT activation comes from studies of the apoA-I in human interstitial fluid, which activates LCAT poorly compared with plasma apoA-I [47]. That result was attributed to a change in the conformation of an epitope of apoA-I that is recognised by the mAb A1–11, one of the antibodies used in the present study.
Incubation with MG also dramatically altered the exposure of the epitope that is recognised by mAb AI-17, which spans residues 143–165 of apoA-I and is known to activate LCAT [48]. It is also noteworthy that other regions of apoA-I that changed in conformation during incubation with MG, including the C-terminal domain, where the epitopes for mAbs AI-178.1, AI-187.1 and AI-141.7 are located, regulate additional functional properties of HDL such as lipid binding, cholesterol efflux and apoA-I stability [49].
The observation that LCAT binds to discoidal rHDL in which the apoA-I is extensively glycated, but is unable to esterify cholesterol in these particles, suggests that rHDL which contain glycated apoA-I may competitively inhibit the LCAT reaction. The results in Table 6 confirm that this is the case. The possibility that this result reflects the transfer of radiolabelled UC from unmodified (A-I)rHDL to rHDL that contain glycated apoA-I was considered to be unlikely because the transfer of cholesterol from a donor to an acceptor is independent of the acceptor concentration [50]. In other words, if significant transfer of radiolabelled UC between the unmodified (A-I)rHDL and the (A-I)rHDLglyc had occurred, any reduction in the experimentally determined rate of cholesterol esterification would have been independent of how much glycated rHDL was present in the incubation mixture. The results in Table 6 show clearly that this is not the case. The close agreement between the calculated and experimentally determined rates of cholesterol esterification in the unmodified (A-I)rHDL also argues against significant transfer of radiolabelled UC between the two types of particles.
In conclusion, this study explains previous conflicting results where glycation of apoA-I either enhances, inhibits or has no effect on LCAT-mediated cholesterol esterification. As the LCAT-mediated esterification of cholesterol in HDL is one of the key events in the reverse cholesterol transport pathway, the present findings indicate that, depending on the extent to which it is glycated, apoA-I may either impair or enhance removal of cholesterol from peripheral tissues and macrophages. The impaired removal of cholesterol from tissues and macrophages in poorly controlled diabetic subjects, in whom the extent of apoA-I glycation is likely to be considerable, may contribute directly to the premature atherosclerosis that is frequently observed in these subjects. It remains to be seen if glycation of apoA-I also affects other processes involved in cholesterol transport and HDL metabolism.
Abbreviations
- (A-I)rHDL:
-
reconstituted HDL containing phosphatidylcholine and apoA-I
- (A-I)rHDLglyc :
-
reconstituted HDL containing non-enzymatically glycated apoA-I
- apoA-I:
-
apolipoprotein A-I
- CE:
-
cholesteryl ester
- DOPA:
-
3,4-dihydroxy phenylalanine
- k arg :
-
rate constant for MG-mediated modification of apoA-I arginine
- k lys :
-
rate constant for MG-mediated modification of apoA-I lysine
- k trp :
-
rate constant for MG-mediated modification of apoA-I tryptophan
- LCAT:
-
lecithin:cholesterol acyltransferase
- mAb:
-
monoclonal antibody
- MG:
-
methylglyoxal
- POPC:
-
1-palmitoyl-2-oleoyl phosphatidylcholine
- rHDL:
-
reconstituted HDL
- UC:
-
unesterified cholesterol
- [3H]UC:
-
1α,2α-3H-labelled unesterified cholesterol
References
Gu K, Cowie CC, Harris MI (1999) Diabetes and decline in heart disease mortality in US adults. JAMA 281:1291–1297
Tan KC, Chow WS, Ai VH, Metz C, Bucala R, Lam KS (2002) Advanced glycation end products and endothelial dysfunction in type 2 diabetes. Diabetes Care 25:1055–1059
Lin RY, Reis ED, Dore AT et al (2002) Lowering of dietary advanced glycation endproducts (AGE) reduces neointimal formation after arterial injury in genetically hypercholesterolemic mice. Atherosclerosis 163: 303–311
McLellan AC, Thornalley PJ, Benn J, Sonksen PH (1994) Glyoxalase system in clinical diabetes mellitus and correlation with diabetic complications. Clin Sci 87:21–29
Lo TW, Westwood ME, McLellan AC, Selwood T, Thornalley PJ (1994) Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with N alpha-acetylarginine, N alpha-acetylcysteine, and N alpha-acetyllysine, and bovine serum albumin. J Biol Chem 269:32299–32305
Zeng J, Davies MJ (2005) Evidence for the formation of adducts and S-(carboxymethyl)cysteine on reaction of alpha-dicarbonyl compounds with thiol groups on amino acids, peptides, and proteins. Chem Res Toxicol 18:1232–1241
Curtiss LK, Witztum JL (1985) Plasma apolipoproteins AI, AII, B, CI, and E are glucosylated in hyperglycemic diabetic subjects. Diabetes 34:452–461
Shishino K, Murase M, Makino H, Saheki S (2000) Glycated apolipoprotein A-I assay by combination of affinity chromatography and latex immunoagglutination. Ann Clin Biochem 37:498–506
Winlove CP, Parker KH, Avery NC, Bailey AJ (1996) Interactions of elastin and aorta with sugars in vitro and their effects on biochemical and physical properties. Diabetologia 39:1131–1139
Che W, Asahi M, Takahashi M et al (1997) Selective induction of heparin-binding epidermal growth factor-like growth factor by methylglyoxal and 3-deoxyglucosone in rat aortic smooth muscle cells. The involvement of reactive oxygen species formation and a possible implication for atherogenesis in diabetes. J Biol Chem 272:18453–18459
Duell PB, Oram JF, Bierman EL (1990) Nonenzymatic glycosylation of HDL resulting in inhibition of high-affinity binding to cultured human fibroblasts. Diabetes 39:1257–1263
Duell PB, Oram JF, Bierman EL (1991) Nonenzymatic glycosylation of HDL and impaired HDL-receptor-mediated cholesterol efflux. Diabetes 40:377–384
Witztum JL, Fisher M, Pietro T, Steinbrecher UP, Elam RL (1982) Nonenzymatic glucosylation of high-density lipoprotein accelerates its catabolism in guinea pigs. Diabetes 31:1029–1032
Passarelli M, Catanozi S, Nakandakare ER et al (1997) Plasma lipoproteins from patients with poorly controlled diabetes mellitus and “in vitro” glycation of lipoproteins enhance the transfer rate of cholesteryl ester from HDL to apo-B-containing lipoproteins. Diabetologia 40: 1085–1093
Quintao EC, Medina WL, Passarelli M (2000) Reverse cholesterol transport in diabetes mellitus. Diabetes Metab Res Rev 16:237–250
Fielding CJ, Shore VG, Fielding PE (1972) A protein cofactor of lecithin:cholesterol acyltransferase. Biochem Biophys Res Commun 46:1493–1498
Ohta T, Nishiyama S, Nakamura T, Saku K, Maung KK, Matsuda I (1998) Predominance of large low density lipoprotein particles and lower fractional esterification rate of cholesterol in high density lipoprotein in children with insulin-dependent diabetes mellitus. Eur J Pediatr 157:276–281
Schernthaner G, Kostner GM, Dieplinger H, Prager R, Muhlhauser I (1983) Apolipoproteins (A-I, A-II, B), Lp(a) lipoprotein and lecithin-cholesterol acyltransferase activity in diabetes-mellitus. Atherosclerosis 49:277–293
Riemens S, van Tol A, Sluiter W, Dullaart R (1998) Elevated plasma cholesteryl ester transfer in NIDDM: relationships with apolipoprotein B-containing lipoproteins and phospholipid transfer protein. Atherosclerosis 140:71–79
Calvo C, Ulloa N, Campos M, Verdugo C, Ayrault-Jarrier M (1993) The preferential site of nonenzymatic glycation of human apolipoprotein-A-I in-vivo. Clin Chim Acta 217:193–198
Gugliucci A, Stahl AJ (1991) In vitro glycation of human apolipoprotein AI reduces its efficiency in lecithin:cholesterol acyltransferase activation. Clin Chim Acta 204:37–42
Fournier N, Myara I, Atger V, Moatti N (1995) Reactivity of lecithin-cholesterol acyl transferase (LCAT) towards glycated high-density lipoproteins (HDL). Clin Chim Acta 234:47–61
Rye KA, Garrety KH, Barter PJ (1993) Preparation and characterization of spheroidal, reconstituted high-density lipoproteins with apolipoprotein A-I only or with apolipoprotein A-I and A-II. Biochim Biophys Acta 1167:316–325
Osborne JC Jr (1986) Delipidation of plasma lipoproteins. Methods Enzymol 128:213–222
Weisweiler P (1987) Isolation and quantitation of apolipoproteins A-I and A-II from human high-density lipoproteins by fast-protein liquid chromatography. Clin Chim Acta 169:249–254
Matz CE, Jonas A (1982) Micellar complexes of human apolipoprotein A-I with phosphatidylcholines and cholesterol prepared from cholate-lipid dispersions. J Biol Chem 257:4535–4540
Piran U, Morin RJ (1979) A rapid radioassay procedure for plasma lecithin-cholesterol acyltransferase. J Lipid Res 20:1040–1043
Knott HM, Brown BE, Davies MJ, Dean RT (2003) Glycation and glycoxidation of low-density lipoproteins by glucose and low-molecular mass aldehydes. Formation of modified and oxidized particles. Eur J Biochem 270:3572–3582
Sparks DL, Phillips MC (1992) Quantitative measurement of lipoprotein surface charge by agarose gel electrophoresis. J Lipid Res 33:123–130
Curtiss LK, Bonnet DJ, Rye KA (2000) The conformation of apolipoprotein A-I in high-density lipoproteins is influenced by core lipid composition and particle size: a surface plasmon resonance study. Biochemistry 39:5712–5721
Rawn JD (1989) Enzyme catalysis and enzyme kinetics. In: Biochemistry, Int Ed. Neil Patterson, Burlington, NC, pp 148–193
Takayama M, Itoh S, Nagasaki T, Tanimizu I (1977) A new enzymatic method for determination of serum choline-containing phospholipids. Clin Chim Acta 79:93–98
Stahler F, Gruber W, Stinshoff K, Roschlau P (1977) A practical enzymatic cholesterol determination. Med Lab 30:29–37
Smith PK, Krohn RI, Hermanson GT et al (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150:76–85
Rainwater DL, Andres DW, Ford AL, Lowe F, Blanche PJ, Krauss RM (1992) Production of polyacrylamide gradient gels for the electrophoretic resolution of lipoproteins. J Lipid Res 33:1876–1881
Dunn JA, McCance DR, Thorpe SR, Lyons TJ, Baynes JW (1991) Age-dependent accumulation of N epsilon-(carboxymethyl)lysine and N epsilon-(carboxymethyl)hydroxylysine in human skin collagen. Biochemistry 30:1205–1210
Ahmed MU, Brinkmann Frye E, Degenhardt TP, Thorpe SR, Baynes JW (1997) N-epsilon-(carboxyethyl)lysine, a product of the chemical modification of proteins by methylglyoxal, increases with age in human lens proteins. Biochem J 324:565–570
Dyer DG, Blackledge JA, Thorpe SR, Baynes JW (1991) Formation of pentosidine during nonenzymatic browning of proteins by glucose. Identification of glucose and other carbohydrates as possible precursors of pentosidine in vivo. J Biol Chem 266:11654–11660
Malmendier CL, Delcroix C, Ameryckx JP (1983) In vivo metabolism of human apoprotein A-I-phospholipid complexes. Comparison with human high density lipoprotein-apoprotein A-I metabolism. Clin Chim Acta 131:201–210
Khaw KT, Wareham N, Luben R et al (2001) Glycated haemoglobin, diabetes, and mortality in men in Norfolk cohort of European prospective investigation of cancer and nutrition (EPIC-Norfolk). BMJ 322:15–18
Santamarina-Fojo S, Lambert G, Hoeg JM, Brewer HB Jr (2000) Lecithin-cholesterol acyltransferase: role in lipoprotein metabolism, reverse cholesterol transport and atherosclerosis. Curr Opin Lipidol 11:267–275
Lapolla A, Flamini R, Dalla Vedova A et al (2003) Glyoxal and methylglyoxal levels in diabetic patients: quantitative determination by a new GC/MS method. Clin Chem Lab Med 41:1166–1173
McLellan AC, Phillips SA, Thornalley PJ (1992) The assay of methylglyoxal in biological systems by derivatization with 1,2-diamino-4,5-dimethoxybenzene. Anal Biochem 206:17–23
Matz CE, Jonas A (1982) Reaction of human lecithin cholesterol acyltransferase with synthetic micellar complexes of apolipoprotein A-I, phosphatidylcholine, and cholesterol. J Biol Chem 257:4541–4546
Brown BE, Dean RT, Davies MJ (2005) Glycation of low-density lipoproteins by methylglyoxal and glycolaldehyde gives rise to the in vitro formation of lipid-laden cells. Diabetologia 48:361–369
Alexander ET, Bhat S, Thomas MJ et al (2005) Apolipoprotein A-I helix 6 negatively charged residues attenuate lecithin-cholesterol acyltransferase (LCAT) reactivity. Biochemistry 44:5409–5419
Wong L, Curtiss LK, Huang J, Mann CJ, Maldonado B, Roheim PS (1992) Altered epitope expression of human interstitial fluid apolipoprotein A-I reduces its ability to activate lecithin cholesterol acyl transferase. J Clin Invest 90:2370–2375
Jonas A (1998) Regulation of lecithin cholesterol acyltransferase activity. Prog Lipid Res 37:209–234
Frank PG, Marcel YL (2000) Apolipoprotein A-I: structure-function relationships. J Lipid Res 41:853–872
McLean LR, Phillips MC (1981) Mechanism of cholesterol and phosphatidylcholine exchange or transfer between unilamellar vesicles. Biochemistry 20 2893–2900
Acknowledgements
This research was supported by the National Health and Medical Research Council of Australia (Grant 222722) to K.-A. Rye and P. J. Barter, the Australian Research Council (M. J. Davies), the Diabetes Australia Research Trust (to M. J. Davies) and the National Heart Foundation of Australia (to A. J. Jenkins).
Duality of interest
None of the authors have a conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nobecourt, E., Davies, M.J., Brown, B.E. et al. The impact of glycation on apolipoprotein A-I structure and its ability to activate lecithin:cholesterol acyltransferase. Diabetologia 50, 643–653 (2007). https://doi.org/10.1007/s00125-006-0574-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-006-0574-z