
Abstract
Aims/hypothesis
Metformin has been shown to increase fatty acid oxidation, an effect mediated by AMP activated protein kinase (AMPK). We hypothesised that metformin could prevent both caspase-3 activation and apoptosis when induced by palmitic acid.
Materials and methods
Cardiomyocytes were incubated with 1 mmol/l palmitic acid, in the absence or presence of metformin (1–5 mmol/l). Following 1 to 16 h, cell damage was evaluated by measuring lactate dehydrogenase released into the incubation medium, and Hoechst staining. To investigate the mechanism of metformin’s effect on cardiomyocytes, substrate utilisation and phosphorylation of AMPK and acetyl-CoA carboxylase were measured. Intracellular mediators of apoptosis were also evaluated.
Results
Incubation of myocytes with palmitic acid for 16 h increased apoptosis, an effect that was partly blunted by 1 and 2 mmol/l metformin. This beneficial effect of metformin was associated with increased AMPK phosphorylation, palmitic acid oxidation and suppression of high-fat-induced increases in (1) long chain base biosynthesis protein 1 levels, (2) ceramide levels, and (3) caspase-3 activity. Unexpectedly, 5 mmol/l metformin dramatically increased apoptosis in myocytes incubated with high fat. This effect was associated with a robust increase in glycolysis, lactate accumulation, and a significant drop of pH in the myocyte incubation medium.
Conclusions/interpretation
Our study demonstrates that metformin reduces high-fat-induced cardiac cell death, probably through inhibition of ceramide synthesis. However, at high concentrations, metformin causes proton and lactate accumulation, leading to cell damage that is independent of caspase-3.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heart disease is a leading cause of death in diabetic patients, with coronary vessel disease and atherosclerosis being primary reasons for the increased incidence of cardiovascular dysfunction [1, 2]. However, a number of diabetic patients also suffer from a specific impairment of heart muscle (termed diabetic cardiomyopathy), a condition also evident in rodent models of chronic diabetes [3, 4]. Several factors have been put forward to explain the development of diabetic cardiomyopathy including an increased stiffness of the left ventricular wall, and abnormalities of various proteins that regulate ion flux, specifically intracellular calcium [5, 6]. More recently, it has been suggested that diabetic cardiomyopathy could also occur as a consequence of apoptosis, a process of cell death that occurs subsequent to the activation of a genetically programmed, energy-dependent mechanism [7, 8].
The ability of the heart and other non-adipose tissues to handle excess fatty acid is inadequate. For this reason, an increase in intracardiac fatty acid concentration can overwhelm fatty acid oxidation. In this situation, fatty acids accumulate and can, either by themselves, or via their channelling towards the production of second messengers like ceramide, provoke apoptosis [9, 10]. Enlargement of the intracardiac fatty acid pool during diabetes may similarly overpower the tissue capacity for utilisation, resulting in an accumulation of cardiotoxic lipid, which may be implicated in cardiac myocyte apoptosis [8, 11].
Metformin, a drug widely used in the treatment of type 2 diabetes, decreases plasma glucose and lipids. Recently, it has been proposed that activation of AMP activated protein kinase (AMPK) mediates the effects of metformin [12, 13]. Once activated, AMPK stimulates glucose uptake and subsequent glycolysis through translocation of glucose transporter 4 (GLUT4) and activation of 6-phosphofructo-2-kinase (PFK2) [14–16]. Moreover, through its control of lipoprotein lipase, CD36, and acetyl-CoA carboxylase (ACC), AMPK facilitates fatty acid delivery and oxidation [17–19]. In addition to its effect on substrate metabolism, metformin is also known to regulate endothelial cell death [20]. Whether metformin regulates cardiomyocyte cell death is unknown. Given the role of metformin in the regulation of lipid metabolism, we hypothesised that it would prevent cardiomyocyte cell death induced by high fatty acid levels.
Materials and methods
Experimental animals
The investigation conforms to the guide for the care and use of laboratory animals published by the US National Institutes of Health and the University of British Columbia (animal care certificate A00-0291). Adult male Wistar rats (250–280 g) were obtained from the University of British Columbia Animal Care Unit and given free access to a standard laboratory diet (PMI Feeds, Richmond, VA, USA) and water.
Materials
Total AMPK-α and phospho-AMPK-α, and ACC antibodies were obtained from Cell Signaling (Beverly, MA, USA). Anti-inducible nitric oxide synthase antibody was obtained from Santa Cruz Biotech (Santa Cruz, CA, USA). Anti-long chain base biosynthesis protein 1 (LCB1) was obtained from BD Sciences (San Diego, CA, USA). Medium 199 was obtained from Sigma (St. Louis, MO, USA). DMEM was obtained from Invitrogen (Burlington, ON, Canada). 32P, 5-3H-glucose and 3H-palmitic acid were obtained from New England Nuclear (Woodbridge, ON, Canada). The ECL detection kit was obtained from Amersham (Piscataway, NJ, USA).
Cardiomyocyte isolation and culturing
Ventricular myocytes were prepared by a previously described procedure [21]. Briefly, myocytes were made calcium-tolerant by successive exposure to increasing concentrations of calcium. Our method of isolation yields a highly enriched population of calcium-tolerant myocardial cells that are rod-shaped (over 80%) in the presence of 1 mmol/l Ca2+, with clear cross striations. Intolerant cells are intact but hypercontract into vesiculated spheres. Yield of myocytes was determined microscopically using an improved Neubauer haemocytometer (Fisher Scientific Canada, Ottawa, ON, Canada). Myocyte viability was assessed as the percentage of elongated cells with clear cross striations that excluded 0.2% trypan blue.
Cardiomyocytes were plated on laminin-coated six-well culture plates to a density of 100,000 cells/well. Cells were maintained using Media-199, in the presence or absence of 1 mmol/l albumin-bound palmitic acid (1:2), and incubated at 37°C under an atmosphere of 95%O2/5%CO2 for 16 h. Where indicated, metformin (1–5 mmol/l) was added to the culture medium, and myocytes kept for either 1 or 16 h. In some experiments, myocytes were incubated with metformin for 16 h in the absence of glucose.
Lactate dehydrogenase release
Lactate dehydrogenase (LDH) is a cellular enzyme released upon membrane damage and a recognised marker of cell damage or death [22]. LDH released into the myocyte incubation medium was estimated using an assay kit (Sigma). In brief, LDH reduces nicotinamide adenine dinucleotide, which then converts a tetrazolium dye to a soluble, coloured formazan derivative; this was measured using a spectrophotometer (Biochrom, Cambridge, UK) (490 nm).
Hoechst 33342 staining
To study high-fat-induced apoptosis and the influence of metformin on this process, cells were examined for morphological evidence of apoptosis using the fluorescent DNA-binding dye, Hoechst 33342 (Sigma). Cells were stained with 5 μg/ml Hoechst 33342, and viewed on a Zeiss IM fluorescence microscope (×400) (Carl Zeiss Canada, Toronto, ON, Canada). Cells were scored as apoptotic if they exhibited unequivocal nuclear chromatin condensation and/or fragmentation. To quantify apoptosis, 500 nuclei from different isolations (three different isolations were prepared) were randomly picked and examined, and the results presented as apoptotic cells per 1,000 cells.
Estimation of reactive oxygen species
The redox-sensitive dye 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA) was used to assess reactive oxygen species (ROS) levels. CM-H2DCFDA is a cell-permeant indicator that is oxidised in the presence of ROS such as H2O2 thereby emitting green fluorescence. Following 16 h incubation, the medium was discarded and wells were loaded with CM-H2DCFDA (5 μmol for 30 min at 37°C). Subsequently, cells were washed and incubated for an additional 45 min, after which green fluorescence was measured in a fluorimeter (Perspective Biosystems, Framingham, MA, USA) at 485/530-nm wavelengths.
Western blot analysis
Western blot was carried out as described previously [19]. Briefly, 50 mg of heart tissue was ground under liquid nitrogen, and homogenised. After centrifugation at 5,000 g for 20 min, the protein content of the supernatant was quantified using a Bradford protein assay (Bio-Rad, Hercules, CA, USA). Samples were diluted, boiled with sample loading dye, and 50 μg used in SDS-polyacrylamide gel electrophoresis. After transfer, membranes were blocked in 5% skim milk in Tris-buffered saline containing 0.1% Tween-20. Membranes were incubated with rabbit AMPK-α, phospho-AMPK (Thr-172), phospho-ACC, and long LCB1 antibodies, and subsequently with secondary goat anti-rabbit or goat anti-mouse horseradish peroxidase-conjugated antibodies, and visualised using the ECL detection kit.
Ceramide assay
Intracellular ceramide was estimated using a DAG kinase assay as described before [23]. Briefly, following 16 h incubation, phospholipids were extracted using chloroform/methanol from the same number of cells in different groups (1:2, v/v). Samples were then incubated with reaction mixture containing 5 mmol/l imidazole/HCl, pH 6.6, 1 mmol/l diethylenetriaminepenta-acetic acid, pH 6.6, 50 mmol/l NaCl, 12.5 mmol/l MgCl2, 1 mmol/l EGTA, 10 mmol/l dithiothreitol, 1 mmol/l ATP, [γ-32P]ATP, DAG kinase (15.5 mU/assay), 2.5% octyl-β-d-glucoside, 1 mmol/l cardiolipin. Following incubation, lipids were extracted with 470 μl of chloroform:methanol:10 mmol/l HCl (15:30:2, v/v/v) containing 20 μg/ml of phosphatidate as a carrier. Samples were separated on plastic thin layer plates of silica gel 60 (EM Industries, Hawthorne, NY, USA) by developing with chloroform:methanol:NH4OH (65:35:7.5, v/v/v), drying and then developing in chloroform:methanol:acetic acid:acetone:water (10:2:3:4:1 by volume). 32P-labelled ceramide 1-phosphate was identified with authentic standards and a radio-imager (Bio-Rad).
Caspase-3 activity
Activity of cardiac caspase-3 was determined using a fluorescent kit (EnzChek Caspase-3 Assay Kit; Molecular Probes, Eugene, OR, USA). Briefly, myocytes were lysed and protein extracted by centrifugation at 5,000 g for 5 min. Then 50 μl of protein were added to an equal volume of reaction buffer that contained 50 μmol/l of the respective substrate, and incubated at room temperature for 30 min. The enzyme-catalysed release of aminomethylcoumarin was quantified in a fluorimeter (Perspective Biosystems, Framingham, MA, USA) at 380/450-nm wavelengths. Protein was determined using a Bradford assay (Bio-Rad, Hercules, CA, USA). Caspase-3 activity is presented as activity per milligram of protein for each sample.
Rates of glycolysis and palmitic acid oxidation
To measure rates of glycolysis and fatty acid oxidation, 5-3H-glucose or 3H-palmitic acid were added separately into the incubation medium. After 16 h incubation, medium samples were collected. The 3H2O generated from glycolysis or fatty acid oxidation was separated from 5-3H-glucose or 3H-palmitic acid by previously described methods [24]. Radioactivity was measured by liquid scintillation counting. Protein was quantified using a Bradford protein assay (Bio-Rad, Hercules, CA, USA). Results were expressed as picomole per minute per milligram per protein.
Lactate assay
Glycolysis results in the formation of protons and pyruvate. Build up of these metabolites augments lactic acid generation, a process catalysed by LDH [25]. Therefore, lactate accumulation is recognised as a marker of acidosis. Lactate released into the myocyte incubation medium was determined using a kit from Sigma. Briefly, 100 μl of culture medium was incubated with 1 ml reaction medium at 30°C for 10 min, and lactate concentration measured using a spectrophotometer (Biochrom, Cambridge, UK).
Statistical analysis
One-way ANOVA followed by the Tukey tests was used to determine differences between group mean values. The level of statistical significance was set at p<0.05.
Results
High-fat-induced LDH release and consequence of metformin
Myocytes incubated with palmitic acid for 16 h augmented LDH release, an effect that was partly blunted by a low concentration of metformin (Fig. 1a). Unexpectedly, 5 mmol/l metformin dramatically increased LDH release in myocytes incubated with high fat (Fig. 1a). In the absence of high fat, metformin alone had no effect on LDH release (data not shown).

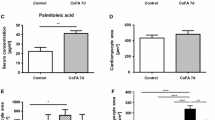
LDH release and cell apoptosis following incubation with high fat and metformin. Cardiomyocytes were incubated with 1 mmol/l palmitic acid (PA), in the presence or absence of metformin (MET) (1–5 mmol/l). Following 16 h, LDH released into the incubation medium was measured (a). Cells were also examined for morphological evidence of apoptosis (b, c) using the fluorescent DNA-binding dye Hoechst 33342. Data are means±SE; n=3–8 myocyte preparations from different animals. One-way ANOVA followed by the Tukey test was used to determine differences between means. * p<0.05 for difference from control; # p<0.05 for difference from the high-fat-treated group; @ p<0.05 for difference from all other groups. Open bars (a, c), control; closed bars, palmitic acid
High-fat-induced apoptosis and consequence of metformin
Figure 1b,c describes evidence of apoptosis and the effect of metformin. In the control group, less than 50 of 1,000 cells were scored as apoptotic. High fat significantly increased apoptosis (241/1,000 cells). Introducing low concentrations of metformin into the medium significantly lowered the number of apoptotic cells (1 mmol/l, 170/1,000; 2 mmol/l, 185/1,000). At a higher concentration of metformin (5 mmol/l), a larger number of cells (635/1,000) underwent apoptosis.
ROS generation following provision of high fat and metformin
ROS generated in the mitochondria is known to induce oxidative stress and apoptosis. In the present study, incubation of cardiomyocytes with 1 mmol/l palmitic acid for 16 h decreased ROS levels compared to control (without palmitic acid) (Fig. 2). Introducing a low concentration of metformin (1 or 2 mmol/l) into the medium increased ROS to the level similar to control (Fig. 2). In contrast, the lowest level of ROS was observed when a high concentration (5 mmol/l) of metformin was used (Fig. 2).

ROS levels following incubation with high fat and metformin. Cardiomyocytes were incubated with 1 mmol/l palmitic acid, in the presence or absence of metformin (MET) (1–5 mmol/l). Following 16 h, ROS levels were measured. Data are means±SE; n=4 myocyte preparations from different animals. One-way ANOVA followed by the Tukey test was used to determine differences between means. *Significantly different from control, p<0.05. * p<0.05 for difference from control; # p<0.05 for difference from the high-fat-treated group; @ p<0.05 for difference from all other groups. Open bars, control; closed bars, palmitic acid; DCFDA, 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester
Effects of metformin on AMPK phosphorylation in cardiomyocytes
Previous studies have reported that metformin activates AMPK in skeletal muscle, liver and cardiomyocytes [12, 13, 26]. In the present study, following 1 h incubation with metformin, an approximately two-fold increase in AMPK phosphorylation was observed (Fig. 3a). No difference in AMPK phosphorylation was observed between groups treated with 1, 2 and 5 mmol/l metformin. Increasing the incubation time with metformin to 16 h further increased AMPK phosphorylation (approx. 4–6-fold) (Fig. 3b), with 5 mmol/l metformin achieving the highest level of AMPK activation.

Time-dependent phosphorylation of AMPK by metformin. Cardiomyocytes were treated with metformin (MET) (1–5 mmol/l) for either 1 (a) or 16 (b) h. Protein was extracted and total (T) and phosphorylated (P) AMPK-α was measured using western blotting. Data are means±SE; n=4 myocyte preparations from different animals. One-way ANOVA followed by the Tukey test was used to determine differences between means. * p<0.05 for difference from control; @ p<0.05 for difference from all other groups. CON, control
ACC phosphorylation and fatty acid oxidation in cardiomyocytes incubated with metformin
AMPK phosphorylates and inhibits ACC, the key enzyme that controls generation of malonyl-CoA from acetyl-CoA. As malonyl-CoA decreases fatty acid oxidation through inhibition of carnitine palmitoyl transferase-1 (CPT-1), phosphorylation of ACC relieves the inhibition of CPT-1, favouring fatty acid oxidation. In the current study, metformin increased ACC phosphorylation in cardiomyocytes within 1 h (Fig. 4a). Prolonging the incubation time to 16 h further increased ACC phosphorylation (Fig. 4b). Coupled to this phosphorylation of ACC, metformin increased the oxidation rate of palmitic acid, with 5 mmol/l metformin having the highest effect (Fig. 4c).

Time-dependent phosphorylation of ACC by metformin and palmitic acid oxidation. Cardiomyocytes were treated with metformin (MET) (1–5 mmol/l) for either 1 (a) or 16 (b) h. Protein was extracted and phosphorylated (P) ACC was measured using western blotting (a, b). Palmitic acid (PA) oxidation (c) was measured using tritium-labelled PA. Data are means±SE; n=4 myocyte preparations from different animals. One-way ANOVA followed by the Tukey test was used to determine differences between means. * p<0.05 for difference from control; @ p<0.05 for difference from all other groups. Open bars, control
Effects of metformin on LCB1 expression and intracellular ceramide level
High fat increases ceramide, a pro-apoptotic mediator, through activation of serine palmitoyltransferase (SPT) [27]. Incubation of cardiomyocytes with palmitic acid (1 mmol/l) for 16 h increased LCB1 protein, a subunit of SPT (Fig. 5a), and intracellular ceramide (Fig. 5b). Introduction of metformin into the culture media reduced high-fat-induced LCB1 protein expression (Fig. 5a) and ceramide levels (Fig. 5b).

Apoptotic mediators in isolated cardiomyocytes. Cardiomyocytes were incubated with 1 mmol/l palmitic acid (PA), in the presence or absence of metformin (MET) (1–5 mmol/l). Following 16 h, cardiac LCB1 expression was examined by western blot (a). Ceramide levels (b) were measured using a DAG kinase assay. Caspase-3 activity (c) was determined using an assay kit. Data are means±SE; n=3–4 myocyte preparations from different animals. One-way ANOVA followed by the Tukey test was used to determine differences between means. * p<0.05 for difference from control; # p<0.05 for difference from the high-fat-treated group. Open bars, control; filled bars, PA
Effects of metformin on high-fat-induced caspase-3 activity
Accumulation of ceramide induces apoptosis in a manner that is dependent on caspase-3 [28, 29]. Incubation of cardiomyocytes with palmitic acid (1 mmol/l) for 16 h increased caspase activity (Fig. 5c). Consistent with the lowering of ceramide, introduction of metformin into the cardiomyocyte incubation medium also reduced the increases in caspase-3 activity that were mediated by high fat (Fig. 5c).
Lactate production and pH changes after metformin
AMPK activation enhances glucose uptake and glycolysis through stimulation of GLUT4 and PFK2, respectively [14–16]. Uncoupling of glycolysis from glucose oxidation leads to lactate production and proton accumulation [30]. In the current study, high fat did not change lactate or pH (control: 7.40±0.01; high fat: 7.38±0.02) in the medium following 16 h incubation. Although low concentrations of metformin increased lactate (Fig. 6) and decreased pH (1–7.26±0.02 mmol/l; 2–7.25±0.03 mmol/l, p<0.05), myocytes incubated with 5 mmol/l metformin showed the highest lactate concentration (Fig. 6) and lowest pH (7.16±0.03, p<0.05) in the medium.

Lactate release in the cardiomyocyte incubation medium. Cardiomyocytes were incubated with 1 mmol/l palmitic acid (filled bars), in the presence or absence of metformin (MET) (1–5 mmol/l). Following 16 h, lactate released in the culture medium was measured using an assay kit. Data are means±SE; n=8 myocyte preparations from different animals. One-way ANOVA followed by the Tukey test was used to determine differences between means. * p<0.05 for difference from control; @ p<0.05 for difference from all other groups. Open bar, control
Effects of metformin on glycolysis
AMPK activation is known to stimulate glycolysis through phosphorylation and activation of PFK2. Provision of 1 mmol/l palmitic acid significantly reduced the rate of glycolysis in cardiomyocytes (Fig. 7). Metformin dramatically increased glycolysis in a dose-dependent manner (Fig. 7).

Rate of cardiomyocyte glycolysis following metformin incubation. Cardiomyocytes were incubated with 1 mmol/l palmitic acid (filled bars), in the presence or absence of metformin (MET) (1–5 mmol/l). Following 16 h of incubation, glycolysis was measured using 3H-glucose. Data are means ± SE; n=4 myocyte preparations from different animals. One-way ANOVA followed by the Tukey test was used to determine differences between means. * p<0.05 for difference from control; # p<0.05 for difference from the high-fat-treated group; @ p<0.05 for difference from all other groups. Open bar, control
Effects of glucose removal on LDH release
We used glucose-free medium and repeated our incubations with palmitic acid, in the absence or presence of metformin. Absence of glucose normalised pH (data not shown) and prevented the increase induced by metformin (5 mmol/l) in LDH release (Fig. 8). In fact, in this glucose-free medium, the release of LDH with metformin (5 mmol/l) was similar to the control.

LDH release in the absence of glucose following high fat and metformin. Cardiomyocytes were incubated in a glucose-free medium containing 1 mmol/l palmitic acid (filled bars) in the presence or absence of metformin (MET) (1–5 mmol/l). Following 16 h, LDH was measured. Data are means±SE; n=4 myocyte preparations from different animals. One-way ANOVA followed by the Tukey test was used to determine differences between means. * p<0.05 for difference from control; # p<0.05 for difference from the high-fat-treated group. Open bar, control
Discussion
Metformin, a drug widely used in the treatment of type 2 diabetes, lowers plasma glucose without causing hypoglycaemia or weight gain. Recently, activation of AMPK has been implicated in mediating this effect on glucose, either through a reduction in hepatic glucose production, or an increase in glucose uptake and utilisation [12, 13]. Through activation of AMPK, metformin also enhances fatty acid uptake and oxidation [31]. Whether metformin, as a consequence of its modulated metabolism, influences cardiomyocyte cell death remained unknown. In the present study, metformin itself had no effect on cell death. At low concentrations, it reduced the number of high-fat-induced apoptotic cells and LDH release, whereas a high concentration of metformin dramatically increased the apoptotic cell number and LDH secretion. Our data suggest that the dual effects of metformin on high-fat-induced cell death are dose dependent.
ROS plays a key role in increasing mitochondrial cytochrome C release and inducing apoptosis. In endothelial cells, metformin has been shown to reduce hyperglycaemia-induced ROS generation and oxidative stress [32, 33]. Therefore, we examined whether the same mechanisms are also involved in protecting cardiomyocytes against high-fat-induced cell death. Surprisingly, incubation of cardiomyocytes with 1 mmol/l palmitic acid lowered, rather than increased ROS levels. This observation is consistent with previous studies, which suggest that a high concentration of palmitic acid decreases mitochondrial membrane potential, and the ability of the mitochondria to produce ROS [34, 35]. Provision of a low concentration of metformin (1 or 2 mmol/l) restored ROS to control levels, probably due to the effect of metformin in promoting fatty acid oxidation and electron flow through the mitochondrial electron transport chain [35]. In contrast, in the presence of palmitic acid, cardiomyocytes treated with 5 mmol/l metformin exhibited the lowest ROS level. One explanation is that following 16 h treatment, 5 mmol/l metformin induces apoptosis in over 60% of cardiomyocytes, probably leading to impaired oxidative metabolism and ROS generation. Overall, our data suggest that metformin prevents high-fat-induced apoptosis in cardiomyocytes through a mechanism independent of ROS.
In the heart, when supply of fatty acid exceeds tissue oxidative capacity, the accumulation of lipids decreases cardiolipin synthesis [36] and induces myofibrillar degeneration [10], leading to lipotoxicity and apoptosis. Given that exogenous C2-ceramide mimics the deleterious effects of fatty acid, and that inhibition of ceramide formation blocks these toxic effects caused by high fat, several studies have suggested that de novo ceramide formation plays an important contributory role in apoptosis [8, 37, 38]. Direct interaction with cytochrome C, followed by its release from the mitochondria and activation of caspase, is one mechanism proposed to explain this effect of ceramide in propagating apoptosis [39]. In the present study, we demonstrate that metformin prevented the high-fat-induced activation of caspase-3, probably through its inhibition of ceramide synthesis. Ceramide levels can increase as a result of SPT-mediated de novo synthesis, in the presence of excessive fatty acyl CoA. Given the role of AMPK in decreasing the expression of SPT [40], activation of AMPK by metformin would be expected to decrease ceramide formation. Moreover, through activation of AMPK and inhibition of ACC, metformin also promotes palmitic acid oxidation. It is possible that this increased fatty acid oxidation diverts fatty acid from ceramide generation to energy consumption, thus contributing to reduced ceramide levels. Interestingly, several studies have shown that activation of AMPK protects endothelial cells or astrocytes against cell death induced by hyperglycaemia or high fat, probably through lowering ceramide generation or caspase-3 activity [40, 41]. In the current study, myocytes incubated with metformin demonstrated higher AMPK phosphorylation and inhibition both of high-fat-induced LCB-1 (a subunit of SPT) expression and intracellular ceramide levels. Our data suggest that the beneficial effects of low doses of metformin in preventing cell death probably occur through modulation by metformin of ceramide formation and caspase-3 activation. It should be noted that although AMPK mediated the protective effects of metformin against high-fat-induced cell death, further studies are required to substantiate this hypothesis. It remains to be determined whether metformin can also prevent cell death by mechanisms other than ceramide formation.
It should be noted that despite the drop in ceramide and caspase-3 activity, high concentrations of metformin elicited substantial apoptosis. These data suggest that mechanisms other than lipotoxicity could play a central role in mediating these injurious effects of metformin. Metformin has been associated with lactic acidosis [42, 43]. In the present study, 5 mmol/l metformin caused the highest change in lactate and pH measured in the cardiomyocyte incubation medium, suggesting that lactic acidosis could be the major explanation of this toxic effect of metformin.
Metformin, associated with its activation of AMPK, stimulates glucose uptake by inducing GLUT4 recruitment to the plasma membrane, and subsequent glycolysis through activation of PFK2 [14–16, 44]. Metformin has also been implicated in fatty acid oxidation through activation of AMPK and inhibition of ACC [45]. Indeed, measurement of glycolysis and fatty acid oxidation demonstrated a dose-dependent increase in utilisation of these substrates in myocytes treated with metformin. It should be noted that fatty acid oxidation is known to inhibit glucose oxidation; acetyl-coA generated from fatty acid can inhibit pyruvate dehydrogenase complex, a rate-limiting enzyme in pyruvate oxidation [46, 47]. Thus, even though metformin promotes glycolysis, glucose oxidation is probably compromised, leading to a dissociation of glycolysis from glucose oxidation. In this situation, the protons and lactate generated from glycolysis accumulate, decrease intracellular pH, leading to intracellular calcium overload and cell death. To confirm that protons generated from glycolysis dramatically increased apoptosis, glucose was removed from the culture medium. Absence of glucose prevented the increase induced by metformin (5 mmol/l) in LDH release. In fact, in this glucose-free medium, the release of LDH with metformin was similar to control. Overall, even though high doses of metformin significantly reduced ceramide and caspase-3 activity, its protective effects against high-fat-induced cell death are probably masked by its ability to augment proton production.
A limitation of the present study was the use of quiescent non-beating myocytes. These cells have a lower overall oxidative capacity than the beating heart, probably leading to excessive lactate and proton accumulation. Additionally, in vivo, the lactate and protons generated are diluted and removed by body fluids, a situation that is unlikely in our in vitro cell culture. Whether the above limitations explain the LDH release even at low concentrations of metformin is yet to be determined.
In summary, our study demonstrates that low doses of metformin reduce high-fat-induced cardiac cell death, probably by decreasing ceramide formation and caspase-3 activity (Fig. 9). However, through its role in increasing proton accumulation and lactic acidosis, metformin can also induce cardiomyocyte cell damage in a manner that is independent of caspase-3 (Fig. 9).

Proposed mechanism of how metformin influences cardiomyocyte cell death. Metformin (MET), probably through activation of AMPK, decreases ceramide formation and caspase-3 activity, thereby preventing high-fat-induced LDH release. However, as metformin also promotes fatty acid (FA) oxidation, the high glycolysis is not matched by glucose oxidation (high fat oxidation inhibits glucose oxidation). This causes dissociation of glycolysis from glucose oxidation, leading to lactic acidosis, and cell death that is independent of caspase-3
Abbreviations
- ACC:
-
acetyl-CoA carboxylase
- AMPK:
-
AMP activated protein kinase
- CM-H2DCFDA:
-
5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester
- CPT-1:
-
carnitine palmitoyl transferase-1
- GLUT4:
-
glucose transporter 4
- LCB1:
-
long chain base biosynthesis protein 1
- LDH:
-
lactate dehydrogenase
- PFK2:
-
6-phosphofructo-2-kinase
- ROS:
-
reactive oxygen species
- SPT:
-
serine palmitoyltransferase
References
Lteif AA, Mather KJ, Clark CM (2003) Diabetes and heart disease an evidence-driven guide to risk factors management in diabetes. Cardiol Rev 11:262–274
Sowers JR, Epstein M, Frohlich ED (2001) Diabetes, hypertension, and cardiovascular disease: an update. Hypertension 37:1053–1059
Jackson CV, McGrath GM, Tahiliani AG, Vadlamudi RV, McNeill JH (1985) A functional and ultrastructural analysis of experimental diabetic rat myocardium. Manifestation of a cardiomyopathy. Diabetes 34:876–883
Vadlamudi RV, McNeill JH (1983) Effect of experimental diabetes on rat cardiac cAMP, phosphorylase, and inotropy. Am J Physiol 244:H844–H851
Hardin NJ (1996) The myocardial and vascular pathology of diabetic cardiomyopathy. Coron Artery Dis 7:99–108
Cai L, Kang YJ (2003) Cell death and diabetic cardiomyopathy. Cardiovasc Toxicol 3:219–228
Cai L, Li W, Wang G, Guo L, Jiang Y, Kang YJ (2002) Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes 51:1938–1948
Zhou YT, Grayburn P, Karim A et al (2000) Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci USA 97:1784–1789
Hickson-Bick DL, Buja ML, McMillin JB (2000) Palmitate-mediated alterations in the fatty acid metabolism of rat neonatal cardiac myocytes. J Mol Cell Cardiol 32:511–519
Dyntar D, Eppenberger-Eberhardt M, Maedler K et al (2001) Glucose and palmitic acid induce degeneration of myofibrils and modulate apoptosis in rat adult cardiomyocytes. Diabetes 50:2105–2113
Unger RH, Zhou YT (2001) Lipotoxicity of beta-cells in obesity and in other causes of fatty acid spillover. Diabetes 50(Suppl 1):S118–S121
Zhou G, Myers R, Li Y et al (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108:1167–1174
Musi N, Hirshman MF, Nygren J et al (2002) Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes 51:2074–2081
Hayashi T, Hirshman MF, Fujii N, Habinowski SA, Witters LA, Goodyear LJ (2000) Metabolic stress and altered glucose transport: activation of AMP-activated protein kinase as a unifying coupling mechanism. Diabetes 49:527–531
Kurth-Kraczek EJ, Hirshman MF, Goodyear LJ, Winder WW (1999) 5′ AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes 48:1667–1671
Marsin AS, Bertrand L, Rider MH et al (2000) Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol 10:1247–1255
Kudo N, Barr AJ, Barr RL, Desai S, Lopaschuk GD (1995) High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5′-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J Biol Chem 270:17513–17520
Luiken JJ, Coort SL, Willems J et al (2003) Contraction-induced fatty acid translocase/CD36 translocation in rat cardiac myocytes is mediated through AMP-activated protein kinase signaling. Diabetes 52:1627–1634
An D, Pulinilkunnil T, Qi D, Ghosh S, Abrahani A, Rodrigues B (2005) The metabolic “switch” AMPK regulates cardiac heparin-releasable lipoprotein lipase. Am J Physiol Endocrinol Metab 288:E246–E253
Detaille D, Guigas B, Chauvin C et al (2005) Metformin prevents high-glucose-induced endothelial cell death through a mitochondrial permeability transition-dependent process. Diabetes 54:2179–2187
Rodrigues B, Cam MC, Jian K, Lim F, Sambandam N, Shepherd G (1997) Differential effects of streptozotocin-induced diabetes on cardiac lipoprotein lipase activity. Diabetes 46:1346–1353
Das A, Xi L, Kukreja RC (2005) Phosphodiesterase-5 inhibitor sildenafil preconditions adult cardiac myocytes against necrosis and apoptosis. Essential role of nitric oxide signaling. J Biol Chem 280:12944–12955
Martin A, Duffy PA, Liossis C et al (1997) Increased concentrations of phosphatidate, diacylglycerol and ceramide in ras- and tyrosine kinase (fps)-transformed fibroblasts. Oncogene 14:1571–1580
Lopaschuk GD, Barr RL (1997) Measurements of fatty acid and carbohydrate metabolism in the isolated working rat heart. Mol Cell Biochem 172:137–147
Robergs RA, Ghiasvand F, Parker D (2004) Biochemistry of exercise-induced metabolic acidosis. Am J Physiol Regul Integr Comp Physiol 287:R502–R516
Chan AY, Soltys CL, Young ME, Proud CG, Dyck JR (2004) Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J Biol Chem 279:32771–32779
Shimabukuro M, Higa M, Zhou YT, Wang MY, Newgard CB, Unger RH (1998) Lipoapoptosis in beta-cells of obese prediabetic fa/fa rats. Role of serine palmitoyltransferase overexpression. J Biol Chem 273:32487–32490
Takeda Y, Tashima M, Takahashi A, Uchiyama T, Okazaki T (1999) Ceramide generation in nitric oxide-induced apoptosis. Activation of magnesium-dependent neutral sphingomyelinase via caspase-3. J Biol Chem 274:10654–10660
Unger RH, Orci L (2002) Lipoapoptosis: its mechanism and its diseases. Biochim Biophys Acta 1585:202–212
Hopkins TA, Dyck JR, Lopaschuk GD (2003) AMP-activated protein kinase regulation of fatty acid oxidation in the ischaemic heart. Biochem Soc Trans 31:207–212
Ruderman NB, Saha AK, Kraegen EW (2003) Minireview: malonyl CoA, AMP-activated protein kinase, and adiposity. Endocrinology 144:5166–5171
Gallo A, Ceolotto G, Pinton P et al (2005) Metformin prevents glucose-induced protein kinase C-beta2 activation in human umbilical vein endothelial cells through an antioxidant mechanism. Diabetes 54:1123–1131
Kukidome D, Nishikawa T, Sonoda K et al (2006) Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes 55:120–127
Hickson-Bick DL, Sparagna GC, Buja LM, McMillin JB (2002) Palmitate-induced apoptosis in neonatal cardiomyocytes is not dependent on the generation of ROS. Am J Physiol Heart Circ Physiol 282:H656–H664
Sparagna GC, Jones CE, Hickson-Bick DL (2004) Attenuation of fatty acid-induced apoptosis by low-dose alcohol in neonatal rat cardiomyocytes. Am J Physiol Heart Circ Physiol 287:H2209–H2215
Ostrander DB, Sparagna GC, Amoscato AA, McMillin JB, Dowhan W (2001) Decreased cardiolipin synthesis corresponds with cytochrome c release in palmitate-induced cardiomyocyte apoptosis. J Biol Chem 276:38061–38067
Shimabukuro M, Zhou YT, Levi M, Unger RH (1998) Fatty acid-induced beta cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci USA 95:2498–2502
Obeid LM, Hannun YA (1995) Ceramide: a stress signal and mediator of growth suppression and apoptosis. J Cell Biochem 58:191–198
Ghafourifar P, Klein SD, Schucht O et al (1999) Ceramide induces cytochrome c release from isolated mitochondria. Importance of mitochondrial redox state. J Biol Chem 274:6080–6084
Blazquez C, Geelen MJ, Velasco G, Guzman M (2001) The AMP-activated protein kinase prevents ceramide synthesis de novo and apoptosis in astrocytes. FEBS Lett 489:149–153
Ido Y, Carling D, Ruderman N (2002) Hyperglycemia-induced apoptosis in human umbilical vein endothelial cells: inhibition by the AMP-activated protein kinase activation. Diabetes 51:159–167
Bailey CJ, Turner RC (1996) Metformin. N Engl J Med 334:574–579
Brown JB, Pedula K, Barzilay J, Herson MK, Latare P (1998) Lactic acidosis rates in type 2 diabetes. Diabetes Care 21:1659–1663
Yang J, Holman GD (2006) Long-term metformin treatment stimulates cardiomyocyte glucose transport through an AMPK-dependent reduction in GLUT4 endocytosis. Endocrinology 147:2728–2736
Zang M, Zuccollo A, Hou X et al (2004) AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J Biol Chem 279:47898–47905
Randle PJ, Garland PB, Hales CN, Newsholme EA (1963) The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1:785–789
Hansford RG, Cohen L (1978) Relative importance of pyruvate dehydrogenase interconversion and feed-back inhibition in the effect of fatty acids on pyruvate oxidation by rat heart mitochondria. Arch Biochem Biophys 191:65–81
Acknowledgements
The studies described in this paper were supported by an operating grant from Heart and Stroke Foundation of British Columbia and Yukon. The financial support of the Health Research Foundation/Canadian Institutes of Health Research and the Heart and Stroke Foundation of Canada for Graduate Research Scholarships to T. Pulinilkunnil, S. Ghosh, and D. Qi is gratefully acknowledged. We thank D. Brindley, M. Allard and R. Wambolt for their invaluable technical support.
Duality of interest
The authors declare that they have no duality of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
An, D., Kewalramani, G., Chan, J.K.Y. et al. Metformin influences cardiomyocyte cell death by pathways that are dependent and independent of caspase-3. Diabetologia 49, 2174–2184 (2006). https://doi.org/10.1007/s00125-006-0338-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-006-0338-9