
Abstract
Aims/hypothesis
Typical Western diets cause postprandial lipaemia for 18 h per day. We tested the hypothesis that postprandial lipaemia decreases insulin sensitivity.
Subjects, materials and methods
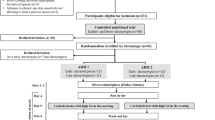
Employing a randomised crossover design, we administered two types of virtually isocaloric meals to ten healthy volunteers on two separate occasions. The meals (Meals 1 and 2) were both designed to produce a rise in triglycerides, but only Meal 1 generated a rise in NEFA, too. Insulin sensitivity, as quantified by an IVGTT with minimal model analysis, was calculated postabsorptively at 08.00 h and postprandially at 13.00 h, i.e. 3 h after meal ingestion.
Results
Triglycerides rose from 0.91±0.31 mmol/l postabsorptively to 2.08±0.70 mmol/l postprandially with Meal 1 (p=0.005) and from 0.92±0.41 to 1.71±0.79 mmol/l with Meal 2 (p=0.005). Neither the triglyceride levels at 13.00 h, nor the post-meal AUCs for triglycerides were statistically different between Meal 1 and Meal 2. NEFA rose from 0.44±0.17 mmol/l postabsorptively to 0.69±0.16 mmol/l postprandially with Meal 1 (p=0.005) and showed no significant change with Meal 2 (0.46±0.31 mmol/l postabsorptively vs 0.36±0.32 mmol/l postprandially, p=0.09). Both the NEFA level at 13.00 h and the post-meal AUC for NEFA were significantly higher after Meal 1 than Meal 2. Compared with the postabsorptive state, insulin sensitivity decreased postprandially after each of the two meals to a comparable degree (Meal 1: −53%, p=0.02; Meal 2: −45%, p=0.005).
Conclusions/interpretation
Our study reveals a drop in insulin sensitivity during postprandial lipaemia and strongly suggests that decreased insulin sensitivity is brought about by elevated plasma levels of triglyceride-rich lipoproteins independently of plasma NEFA levels.
Similar content being viewed by others


Introduction
The concept of atherosclerosis being a postprandial phenomenon was introduced some 20 years ago [1]. We and others have shown that pronounced postprandial lipaemia represents a risk factor for the development of coronary artery disease [2, 3] and elicits potential atherogenic effects including endothelial dysfunction [4] and increased blood coagulability [5].
Both plasma insulin levels [6, 7] and insulin sensitivity [8–11] have been suggested to be an important determinant of postprandial lipaemia, which is prolonged and exaggerated in patients suffering from insulin resistance and type 2 diabetes [12–14]. Whether postprandial lipaemia with its characteristic elevation of triglyceride-rich lipoproteins (TGRLs), in turn, affects insulin sensitivity is not known. As a major part of life is spent in the postprandial state, with the consumption of successive fat-rich meals being common in many societies, this open question remains very relevant. Some studies, however, indicate that high levels of postabsorptive and/or postprandial TGRLs can cause insulin resistance. For instance, in type 2 diabetic patients, lowering of plasma triglycerides by bilio-pancreatic diversion [15, 16] or by the use of fibrates [17] enhanced insulin sensitivity and glucose tolerance. Furthermore, in families with clustering of hypertriglyceridaemia, elevation of serum triglycerides constituted a risk marker for subsequent development of glucose intolerance and type 2 diabetes [18].
With regard to a potential plausible mechanism, TGRLs and their lipolytic products, i.e. NEFA and TGRL remnant particles, could indeed be capable of inducing a state of insulin resistance. In the postprandial phase, TGRL lipolysis occurs predominantly in the adipose tissue capillaries with NEFA uptake by adipose tissue, however, being incomplete; between 20 and 80% of NEFA escape adipose tissue entrapment [19] to be taken up by liver and muscle [20, 21], both major sites of insulin resistance. For these two tissues, numerous human and animal studies have shown that elevated plasma NEFA levels induce insulin resistance (for review see [22, 23]). The other two lipid components involved in the lipolytic process, i.e. TGRLs and TGRL remnants, have been shown by us to induce insulin resistance in cultured skeletal muscle cells independently of NEFA levels [24].
Therefore, the overall objective of this study was to investigate whether postprandial lipaemia induces a state of decreased insulin sensitivity. Our observations with cultured skeletal muscle cells [24] prompted us to employ an experimental design enabling plasma NEFA to be modulated as an independent variable.
Methods
Subjects
For the study, ten non-obese healthy male volunteers were recruited from the University of Innsbruck. The mean age of the study subjects was 29.0±5.2 years, and the mean BMI, calculated as the weight in kilograms divided by the square of the height in metres, was 22.4±0.8 kg/m2. Study subjects were in good health as assessed by medical history and physical examination. None of the individuals was receiving drug therapy, had markers of the insulin resistance syndrome or displayed any other metabolic abnormality. A routine laboratory test revealed normal limits. Mean fasting lipid parameters were as follows: total cholesterol 3.71±0.65 mmol/l, HDL cholesterol 1.35±0.26 mmol/l, LDL cholesterol 2.16±0.73 mmol/l, triglycerides 0.91±0.31 mmol/l and NEFA 0.44±0.17 mmol/l.
Study design
We performed a randomised study in crossover design to investigate the effect of postprandial lipaemia—as induced by ingestion of a standardised meal—on insulin sensitivity. Insulin sensitivity was determined twice on one day of the experiment; first, at around 08.00 h after an overnight fast of about 12 h and second, at around 13:00, 3 h after consumption of one of the two test meals. Two different experiments were performed on each subject on two occasions within 3 months; with the first experiment a high-fat meal (Meal 1) was used to induce postprandial elevations of both triglycerides and NEFA in plasma, whereas with the second experiment a virtually isocaloric mixed meal with a reduced fat abundance but increased carbohydrate content (Meal 2) was used to cause a postprandial rise in triglycerides, but no significant change in plasma NEFA levels. The composition of the second meal was chosen on the basis of the notion that adding carbohydrates to an oral fat load reduces postprandial NEFA levels, presumably by enhanced peripheral NEFA trapping [25].
Study subjects were blinded with regard to the meal type ingested and randomised as to the chronological order of ingesting first Meal 1 followed by Meal 2 or vice versa. Meals were ingested within 5–10 min at 10:00 h and tolerated without any adverse effects. Intravenous catheters were placed in the antecubital veins of each forearm, one catheter for infusion and the other for blood sampling. Physical activity during the study was controlled, and no food intake in addition to the meal was allowed. The volume loss resulting from multiple blood samples was substituted for immediately after blood sampling by infusion of appropriate amounts of 0.9% NaCl.
The study protocol was approved by the Ethic Commission of the Medical University of Innsbruck, and written informed consent was obtained from all subjects.
Materials and analytical procedures
Meal 1 consisted of 350 ml whipped cream, 10 g cacao powder and 1 ml artificial sweetener, and contained 11 g protein, 12 g carbohydrates, 106 g triglycerides and 315 mg cholesterol. From the total of 1,080 kcal, 4.2% were derived from protein, 4.6% from carbohydrates and 91.2% from fat.
Meal 2 consisted of 192 ml whipped cream, 10 g cacao powder, 1 ml artificial sweetener, 100 g low-fat milk powder, 10 g maltodextrin and 160 ml water, and contained 43 g protein, 65 g carbohydrates, 60 g triglycerides and 173 mg cholesterol. From the total of 1,001 kcal, 17.7% were derived from protein, 26.7% from carbohydrates and 55.6% from fat. Calculations of the composition of the meals were performed with the software package Prodi 4.0 from Nutri Science.
Plasma glucose was measured by the hexokinase method using a Cobas MIRA analyser, and plasma insulin by a micro-particle enzyme immunoassay from Abbott (Wiesbaden, Germany). The cross-reactivity with proinsulin of this assay was <0.005%. Serum NEFA concentration was determined by an enzymatic colorimetric method (Wako Chemicals, Neuss, Germany). Total cholesterol and triglycerides were quantified by specific enzymatic assays, HDL cholesterol by a precipitation method (all these assays were from Roche Diagnostics, Mannheim, Germany). Plasma LDL cholesterol levels were calculated by the Friedewald method. Free cholesterol was analysed with an enzymatic colorimetric method from Wako Chemicals, and esterified cholesterol was calculated as total cholesterol minus free cholesterol. Total ghrelin levels were determined with an RIA from Linco Research (St Charles, MO, USA) and growth hormone levels with an IRMA from Biosource (Nivelles, Belgium).
The post-meal AUCs for NEFA and triglycerides were calculated for the 3 h following meal ingestion, i.e. from 10.00 until 13.00 h, using iteration of trapezoidal areas.
IVGTTs
An IVGTT was performed postabsorptively at 08.00 h and postprandially at 13.00 h using the protocol described by Bergman et al. [26] with minor modifications. Briefly, after blood samples taken at 07.50 and 07.59 h and at 12.50 and 12.59 h, respectively, an i.v. bolus of 33% glucose (0.3 g/kg) was injected over a time-period of 60 s. Blood samples were taken from the contralateral forearm at 2, 3, 4, 6, 8, 10, 12, 14, 16, 19, 22, 23, 24, 25, 27, 30, 40, 50, 60, 70, 80 and 100 min.
Evaluation of insulin sensitivity
To provide quantitative estimates of insulin sensitivity, the IVGTTs were analysed with a minimal model [26] using MLAB (rev. 2002-04-18; Civilized Software, Silver Spring, MD, USA).
We decided not to use the hyperinsulinaemic–euglycaemic clamp method, considered the gold standard for determination of insulin sensitivity, because we wished to select a procedure that would interfere as little as possible with the metabolism of lipids. With the clamp method, the concurrent infusion of insulin and glucose over several hours would dramatically interfere with the metabolism of postprandial lipids and thus blur a possible relationship of lipids with insulin sensitivity.
Calculation of the glucose disappearance constant and the acute insulin response to glucose
The glucose disappearance constant (K G) is the absolute value of the slope of the regression line relating the natural log of glucose levels to time from 10 to 22 min after glucose injection, multiplied by 100. The acute insulin response to glucose (AIR) was calculated as the average suprabasal insulin concentration during the initial 2 to 10 min of the IVGTTs.
Statistical analysis
Statistical analysis was carried out using SPSS software, version 12. Analysed parameters were tested for normal distribution using the Shapiro–Wilk test. Since most variables were not normally distributed, the Wilcoxon matched-pairs signed-rank test was employed for all comparisons to detect statistically significant differences. We considered p values less than 0.05 to be significant. All data are presented as means±SD.
Results
The mean plasma profiles of glucose and insulin throughout the days of the experiments are illustrated in Fig. 1a,b. Postprandial plasma glucose levels showed no significant changes with ingestion of Meal 1. With Meal 2, postprandial plasma glucose levels reached a peak value of 6.28±1.33 mmol/l 30 min after meal ingestion and returned to postabsorptive levels about 90 min after the meal (Fig. 1a). Postprandial glucose tolerance, as assessed by the K G, increased after either of the two meals compared with respective postabsorptive values (Meal 1: 2.20±0.94/min postabsorptively vs 3.39±1.09/min postprandially, p=0.02; Meal 2: 2.90±0.92/min postabsorptively vs 5.21±2.09/min postprandially, p=0.005). Postprandial plasma insulin levels showed a delayed return to postabsorptive levels after ingestion of Meal 2 compared with Meal 1 (Fig. 1b). AIR increased significantly after either of the two meal types when compared with the respective postabsorptive values (55.89±26.87 vs 102.1±72.4 mU/l with Meal 1, p=0.004; and 42.8±14.3 vs 107.0±63.1 mU/l with Meal 2, p=0.005).

Mean±SD plasma profiles of glucose (a), insulin (b), triglycerides (c) and NEFA (d) on the days of the experiments. Intravenous glucose boli and meals were provided at the times indicated. Continuous line, Meal 1; broken line, Meal 2
The rationale underlying the formulation of the two test meals was to investigate whether sharply contrasting postprandial NEFA levels affected the degree of postprandial insulin sensitivity. Therefore, we searched in a series of pilot experiments for appropriate compositions of the test meals, such that both meals would give rise to marked postprandial triglyceride elevations, but only one, i.e. Meal 1, would produce elevated NEFA levels compared with respective postabsorptive values (Fig. 1c,d).
With Meal 1, NEFA levels rose from 0.44±0.17 mmol/l postabsorptively at 08.00 h to a maximum value of 0.69±0.16 mmol/l postprandially at 13.00 h (p=0.005). With Meal 2, NEFA levels showed no significant change between the postabsorptive level of 0.46±0.31 mmol/l and the postprandial one of 0.36±0.32 mmol/l (p=0.09) (Fig. 1d). The postprandial NEFA level at 13.00 h was significantly higher after Meal 1 compared with Meal 2 (p=0.04). Accordingly, the post-meal 0- to 3-h AUC for NEFA was higher after Meal 1 than after Meal 2 (72.1±21.1 vs 36.3±21.1 mmol·min·l−1, p=0.005).
Triglyceride levels rose from 0.91±0.31 mmol/l postabsorptively at 08.00 h to 2.08±0.70 mmol/l postprandially at 13.00 h with Meal 1 (p=0.005) and from 0.92±0.41 mmol/l postabsorptively to 1.71±0.79 mmol/l postprandially with Meal 2 (p=0.005) (Fig. 1c). Both the postprandial triglyceride levels analysed at 13.00 h (2.08±0.70 mmol/l with Meal 1 vs 1.71±0.79 mmol/l with Meal 2, p=0.20) and the post-meal 0- to 3-h AUCs for triglycerides (246±104 mmol·min·l−1 with Meal 1 vs 213±96 mmol·min·l−1 with Meal 2, p=0.09) were statistically not different between the two meal types.
We also analysed plasma total cholesterol levels, which showed no significant difference between the postabsorptive and the postprandial states of both Meal 1 (3.71±0.65 vs 3.84±0.50 mmol/l, p=0.16) and Meal 2 (3.64±0.52 vs 3.74±0.66 mmol/l, p=0.13). Free cholesterol levels, however, increased from 1.16±0.46 mmol/l postabsorptively to 1.62±0.28 mmol/l postprandially with ingestion of Meal 1 (p=0.007), whereas with Meal 2 there was no statistical difference between postabsorptive and postprandial levels (1.35±0.21 vs 1.39±0.35 mmol/l, p=0.29). In accord with free cholesterol, esterified cholesterol decreased from 2.60±0.62 mmol/l postabsorptively to 2.21±0.63 mmol/l postprandially with Meal 1 (p=0.02) and remained unchanged with ingestion of Meal 2 (2.29±0.55 mmol/l postabsorptively vs 2.35±0.34 mmol/l postprandially, p=0.72).
Next, we compared insulin sensitivity in the postabsorptive and postprandial states (Fig. 2). With Meal 1, insulin sensitivity decreased from 0.00124±0.000749 postabsorptively to 0.000565±0.000344 postprandially (p=0.02); with Meal 2, insulin sensitivity decreased from 0.00120±0.000629 postabsorptively to 0.000638±0.000297 postprandially (p=0.005).

Comparison of insulin sensitivity values between postabsorptive and postprandial states. Insulin sensitivity values were calculated from an IVGTT with minimal model analysis. p=0.02 (Meal 1); p=0.005 (Meal 2)
We also determined postabsorptive and postprandial plasma levels of growth hormone and ghrelin, both of which have been shown to be affected by food intake [27–29] and which may, in turn, affect insulin sensitivity [30, 31]. Growth hormone levels did not differ significantly between postabsorptive and postprandial states of both Meal 1 (0.11±0.044 vs 0.064±0.035 ng/ml, p=0.06) and Meal 2 (0.18±0.11 vs 0.091±0.036 ng/ml, p=0.09). Ghrelin levels, however, decreased from 1,264±373 pg/ml postabsorptively to 1,116±446 pg/ml with ingestion of Meal 2 (p=0.02), whereas with ingestion of Meal 1 there was no statistical difference between the postabsorptive and postprandial values (1,302±357 vs 1,076±404 pg/ml, p=0.07).
Discussion
Despite considerable progress in recent years, the mechanisms in the pathogenesis of insulin resistance, a major predictor of type 2 diabetes, are only poorly understood. Abnormalities in lipid metabolism, particularly overabundance of circulating NEFA, have been shown to play an important role in this process (for review, see [22, 23]).
In the present study, we describe a mechanism of lipid-induced deterioration of insulin sensitivity not appreciated to date, i.e. diminished insulin sensitivity induced by postprandial lipaemia. The reduction in insulin sensitivity during transition from the postabsorptive to the postprandial phase in our study was documented by using minimal model analysis of an IVGTT, which enables calculation of insulin sensitivity even at disequilibrium of glucose and insulin plasma levels. For the two meals employed, we observed a comparable decrease in postprandial insulin sensitivity. Since one meal led to a rise in plasma NEFA levels shown by others to elicit insulin resistance [32] and the second meal failed to raise NEFA above their postabsorptive levels, our data indicate that plasma NEFA were not involved in the impairment of postprandial insulin sensitivity in our study. Similarly to NEFA, free and esterified cholesterol showed significant changes only after one of the two meals. These findings point to TGRLs, whose postprandial levels at 13.00 h and post-meal AUCs were increased to a comparable degree following ingestion of either of the two test meals. TGRLs as a cause of insulin resistance would be in keeping with our recently published in vitro study which demonstrated that highly purified postprandial TGRLs/remnants—independently of NEFA levels—are capable of causing muscle insulin resistance as evidenced by impaired insulin signalling and glucose metabolism [24]. Numerous studies have shown that a large proportion of TGRLs and remnant particles are taken up not only by the liver but also by peripheral tissues including muscle [33, 34]. Whole lipoprotein uptake has been suggested to be of importance for TGRL removal by skeletal muscle [33, 35] where TGRLs and their remnants could cause intramyocellular accumulation of lipids [24] including lipid intermediates such as long-chain acyl-CoAs and ceramide, suggested as a cause of insulin resistance [36].
As far as lipids are concerned as a cause of insulin resistance, a considerable part of the experimental basis rests on data from i.v. infusion of lipid emulsions with subsequently rising plasma NEFA levels. With this experimental approach, however, one should keep in mind the presence of parent triglyceride-containing particles from the emulsion, which have been shown to function similarly to native TGRLs [34].
In addition to TGRLs, diurnal changes in insulin sensitivity ought to be taken into consideration as a cause of the decline in insulin sensitivity in the postprandial state. Based on the analyses of both AIR and glucose tolerance, we strongly believe, however, that the decrease in postprandial insulin sensitivity is not due to diurnal variations in insulin sensitivity. In most studies, it has been observed that glucose tolerance is decreased in the afternoon or evening compared with the morning [37–39]; decreased glucose tolerance, in these studies, was shown to result from decreased insulin secretion [38–40], decreased insulin sensitivity [41, 42] or both [37, 43]. Although we observed decreased insulin sensitivity at 13.00 h, there is a noticeable difference to the above-mentioned reports on diurnal variation in glucose tolerance/insulin sensitivity; in our study, decreased insulin sensitivity is compensated by enhanced insulin secretion resulting in an even improved postprandial glucose tolerance when compared with respective postabsorptive values analysed in the morning. Based on these differences, it seems unlikely that diurnal variations in insulin sensitivity account for the decrease in postprandial insulin sensitivity in our study. Moreover, the vast majority of published reports compared glucose tolerance/insulin sensitivity between morning and afternoon, and not between morning and noon, as we did. In this regard, a decline in insulin sensitivity from noon or afternoon to late afternoon or night has been demonstrated [44].
Another important point to be addressed is the implication of more than one glucose load administered on a single day and the so-called ‘Staub–Traugott effect’, i.e. the facilitating effect of glucose on the disposal of a consecutive glucose load [45]. In this respect, it has been shown that the acute insulin responses during the first minutes of two successive IVGTTs are statistically not different between the first and the second IVGTT [46, 47]. Similar results were obtained when an IVGTT was performed after an oral glucose load containing a glucose amount in the range of our test meals; after oral glucose, the K G value increased significantly whereas the insulin response in the IVGTT was unchanged when compared with the control condition, i.e. IVGTT preceded by saline drip [45]. A study in healthy men analysed insulin sensitivity 2–3 h following an oral glucose challenge, the glucose content of which was also in the range of our meals. In that study, glucose ingestion induced a rapid enhancement of glucose disposal and whole-body insulin sensitivity [48]. Insulin sensitivity and glucose disposal were calculated when postprandial blood glucose and insulin levels returned to baseline, as in our study. In line with this in vivo study, an in vitro study using human adipocytes also revealed increased insulin sensitivity following a glucose load [49]. Hence, the results of all these studies indicate that the facilitating effect of glucose on the disposal of a consecutive glucose load is based on improved insulin sensitivity without a significant change in the acute insulin response. Thus, the observations on repeated glucose loads are, in fact, very different from what we noted in our study; we found enhanced insulin responses and, most importantly, decreased postprandial insulin sensitivity after either of the two test meals. Similarly to diurnal changes in insulin sensitivity/glucose tolerance, these striking differences strongly suggest that, in our study, neither the glucose challenge of the first IVGTT nor that of the meal could explain the decrease of postprandial insulin sensitivity in the second IVGTT.
Of the potential hormones affecting insulin sensitivity, we quantified the plasma levels of growth hormone and ghrelin. Growth hormone levels showed a tendency to decrease postprandially with either of the two meals without reaching statistical significance. The decrease was more pronounced with the ingestion of Meal 1, which is in accord with the observation that NEFA exert an inhibitory effect on growth hormone secretion [27, 28]. Also ghrelin levels decreased after ingestion of either of the two meals, reaching statistical significance after Meal 2. These findings argue against changes in growth hormone and ghrelin as an explanation for the acute decrease in insulin sensitivity described in the present report.
Insulin resistance induced by postprandial lipaemia represents a phenomenon which, to the best of our knowledge, has not been appreciated to date. The physiological significance of the postprandial decline in insulin sensitivity seen in the present investigation, however, remains to be established and, of course, its quantitative contribution, if any, to insulin resistance in the metabolic syndrome or type 2 diabetes cannot be decided from the work presented here. Moreover, our data by no means preclude other causes of insulin resistance, including elevated plasma NEFA levels.
In summary, the present study reveals decreased insulin sensitivity during postprandial lipaemia, supporting the hypothesis that the postprandial state might be of importance in the development of insulin resistance. Our results strongly indicate that the postprandial decline in insulin sensitivity is induced by elevated plasma levels of TGRLs/remnants independently of plasma NEFA levels, and could be interpreted to suggest that not only atherosclerosis but also one of its major risk factors, i.e. insulin resistance, is a postprandial phenomenon.
Abbreviations
- AIR:
-
acute insulin response to glucose
- K G :
-
glucose disappearance constant
- TGRL:
-
triglyceride-rich lipoprotein
References
Zilversmit DB (1979) Atherogenesis: a postprandial phenomenon. Circulation 60:473–485
Groot PH, van Stiphout WA, Krauss XH et al (1991) Postprandial lipoprotein metabolism in normolipidemic men with and without coronary artery disease. Arterioscler Thromb 11:653–662
Patsch JR, Miesenbock G, Hopferwieser T et al (1992) Relation of triglyceride metabolism and coronary artery disease. Studies in the postprandial state. Arterioscler Thromb 12:1336–1345
Gaenzer H, Sturm W, Neumayr G et al (2001) Pronounced postprandial lipaemia impairs endothelium-dependent dilation of the brachial artery in men. Cardiovasc Res 52:509–516
Silveira A, Karpe F, Blomback M, Steiner G, Walldius G, Hamsten A (1994) Activation of coagulation factor VII during alimentary lipaemia. Arterioscler Thromb 14:60–69
Harbis A, Defoort C, Narbonne H et al (2001) Acute hyperinsulinism modulates plasma apolipoprotein B-48 triglyceride-rich lipoproteins in healthy subjects during the postprandial period. Diabetes 50:462–469
Harbis A, Perdreau S, Vincent-Baudry S et al (2004) Glycemic and insulinemic meal responses modulate postprandial hepatic and intestinal lipoprotein accumulation in obese, insulin-resistant subjects. Am J Clin Nutr 80:896–902
Boquist S, Ruotolo G, Tang R et al (1999) Alimentary lipaemia, postprandial triglyceride-rich lipoproteins, and common carotid intima-media thickness in healthy, middle-aged men. Circulation 100:723–728
Jeppesen J, Hollenbeck CB, Zhou MY et al (1995) Relation between insulin resistance, hyperinsulinemia, postheparin plasma lipoprotein lipase activity, and postprandial lipaemia. Arterioscler Thromb Vasc Biol 15:320–324
Moro E, Gallina P, Pais M, Cazzolato G, Alessandrini P, Bittolo-Bon G (2003) Hypertriglyceridaemia is associated with increased insulin resistance in subjects with normal glucose tolerance: evaluation in a large cohort of subjects assessed with the 1999 World Health Organization criteria for the classification of diabetes. Metabolism 52:616–619
van Oostrom AJ, Castro Cabezas M, Ribalta J et al (2000) Diurnal triglyceride profiles in healthy normolipidemic male subjects are associated to insulin sensitivity, body composition and diet. Eur J Clin Invest 30:964–971
Chen YD, Swami S, Skowronski R, Coulston A, Reaven GM (1993) Differences in postprandial lipaemia between patients with normal glucose tolerance and noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab 76:172–177
Lewis GF, O’Meara NM, Soltys PA et al (1991) Fasting hypertriglyceridaemia in noninsulin-dependent diabetes mellitus is an important predictor of postprandial lipid and lipoprotein abnormalities. J Clin Endocrinol Metab 72:934–944
Syvanne M, Hilden H, Taskinen MR (1994) Abnormal metabolism of postprandial lipoproteins in patients with non-insulin-dependent diabetes mellitus is not related to coronary artery disease. J Lipid Res 35:15–26
Mingrone G, DeGaetano A, Greco AV et al (1997) Reversibility of insulin resistance in obese diabetic patients: role of plasma lipids. Diabetologia 40:599–605
Mingrone G, Henriksen FL, Greco AV et al (1999) Triglyceride-induced diabetes associated with familial lipoprotein lipase deficiency. Diabetes 48:1258–1263
Damci T, Tatliagac S, Osar Z, Ilkova H (2003) Fenofibrate treatment is associated with better glycemic control and lower serum leptin and insulin levels in type 2 diabetic patients with hypertriglyceridaemia. Eur J Intern Med 14:357–360
Sane T, Taskinen MR (1993) Does familial hypertriglyceridaemia predispose to NIDDM? Diabetes Care 16:1494–1501
Evans K, Burdge GC, Wootton SA, Clark ML, Frayn KN (2002) Regulation of dietary fatty acid entrapment in subcutaneous adipose tissue and skeletal muscle. Diabetes 51:2684–2690
Roust LR, Jensen MD (1993) Postprandial free fatty acid kinetics are abnormal in upper body obesity. Diabetes 42:1567–1573
Frayn KN, Shadid S, Hamlani R et al (1994) Regulation of fatty acid movement in human adipose tissue in the post absorptive-to-postprandial transition. Am J Physiol 266:E308–E317
Boden G, Laakso M (2004) Lipids and glucose in type 2 diabetes: what is the cause and effect? Diabetes Care 27:2253–2259
Lewis GF, Carpentier A, Adeli K, Giacca A (2002) Disordered fat storage and mobilisation in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev 23:201–229
Pedrini MT, Kranebitter M, Niederwanger A et al (2005) Human triglyceride-rich lipoproteins impair glucose metabolism and insulin signalling in L6 skeletal muscle cells independently of non-esterified fatty acid levels. Diabetologia 48:756–766
van Oostrom AJ, van Dijk H, Verseyden C et al (2004) Addition of glucose to an oral fat load reduces postprandial free fatty acids and prevents the postprandial increase in complement component 3. Am J Clin Nutr 79:510–515
Bergman RN, Ider YZ, Bowden CR, Cobelli C (1979) Quantitative estimation of insulin sensitivity. Am J Physiol 236:E667–E677
Casanueva FF, Villanueva L, Dieguez C et al (1987) Free fatty acids block growth hormone (GH) releasing hormone-stimulated GH secretion in man directly at the pituitary. J Clin Endocrinol Metab 65:634–642
Imaki T, Shibasaki T, Shizume K et al (1985) The effect of free fatty acids on growth hormone (GH)-releasing hormone-mediated GH secretion in man. J Clin Endocrinol Metab 60:290–293
Cummings DE, Purnell JQ, Frayo RS, Schmidova K, Wisse BE, Weigle DS (2001) A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes 50:1714–1719
Ho KK, O’Sullivan AJ, Hoffman DM (1996) Metabolic actions of growth hormone in man. Endocr J 43(Suppl):S57–S63
Gauna C, Meyler FM, Janssen JA et al (2004) Administration of acylated ghrelin reduces insulin sensitivity, whereas the combination of acylated plus unacylated ghrelin strongly improves insulin sensitivity. J Clin Endocrinol Metab 89:5035–5042
Boden G, Chen X, Ruiz J, White JV, Rossetti L (1994) Mechanisms of fatty acid-induced inhibition of glucose uptake. J Clin Invest 93:2438–2446
Karpe F, Humphreys SM, Samra JS, Summers LK, Frayn KN (1997) Clearance of lipoprotein remnant particles in adipose tissue and muscle in humans. J Lipid Res 38:2335–2343
Hultin M, Carneheim C, Rosenqvist K, Olivecrona T (1995) Intravenous lipid emulsions: removal mechanisms as compared to chylomicrons. J Lipid Res 36:2174–2184
Merkel M, Heeren J, Dudeck W et al (2002) Inactive lipoprotein lipase (LPL) alone increases selective cholesterol ester uptake in vivo, whereas in the presence of active LPL it also increases triglyceride hydrolysis and whole particle lipoprotein uptake. J Biol Chem 277:7405–7411
Hegarty BD, Furler SM, Ye J, Cooney GJ, Kraegen EW (2003) The role of intramuscular lipid in insulin resistance. Acta Physiol Scand 178:373–383
Whichelow MJ, Sturge RA, Keen H, Jarrett RJ, Stimmler L, Grainger S (1974) Diurnal variation in response to intravenous glucose. Br Med J 1:488–491
Zimmet PZ, Wall JR, Rome R, Stimmler L, Jarrett RJ (1974) Diurnal variation in glucose tolerance: associated changes in plasma insulin, growth hormone, and non-esterified fatty acids. Br Med J 1:485–488
Carroll KF, Nestel PJ (1973) Diurnal variation in glucose tolerance and in insulin secretion in man. Diabetes 22:333–348
Boden G, Ruiz J, Urbain JL, Chen X (1996) Evidence for a circadian rhythm of insulin secretion. Am J Physiol 271:E246–E252
Verrillo A, De Teresa A, Martino C et al (1989) Differential roles of splanchnic and peripheral tissues in determining diurnal fluctuation of glucose tolerance. Am J Physiol 257:E459–E465
Gibson T, Stimmler L, Jarrett RJ, Rutland P, Shiu M (1975) Diurnal variation in the effects of insulin on blood glucose, plasma non-esterified fatty acids and growth hormone. Diabetologia 11:83–88
Lee A, Ader M, Bray GA, Bergman RN (1992) Diurnal variation in glucose tolerance. Cyclic suppression of insulin action and insulin secretion in normal-weight, but not obese, subjects. Diabetes 41:742–749
Schulz B, Ratzmann KP, Albrecht G, Bibergeil H (1983) Diurnal rhythm of insulin sensitivity in subjects with normal and impaired glucose tolerance. Exp Clin Endocrinol 81:263–272
Wajngot A, Grill V, Efendic S, Cerasi E (1982) The Staub–Traugott effect. Evidence for multifactorial regulation of a physiological function. Scand J Clin Lab Invest 42:307–313
Diltoer M, Camu F (1988) Glucose homeostasis and insulin secretion during isoflurane anesthesia in humans. Anesthesiology 68:880–886
Metz R, Friedenberg R (1970) Effects of repetitive glucose loads on plasma concentrations of glucose, insulin and free fatty acids: paradoxical insulin responses in subjects with mild glucose intolerance. J Clin Endocrinol Metab 30:602–608
Kingston WJ, Livingston JN, Moxley RT 3rd (1986) Enhancement of insulin action after oral glucose ingestion. J Clin Invest 77:1153–1162
Arner P, Bolinder J, Ostman J (1983) Marked increase in insulin sensitivity of human fat cells 1 hour after glucose ingestion. J Clin Invest 71:709–714
Acknowledgements
We are indebted to K. Salzmann for expert technical assistance and to the volunteers for participating in this study. This study was supported by a grant from the Austrian Science Fund (FWF): P15951-B07 to M. T. Pedrini.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pedrini, M.T., Niederwanger, A., Kranebitter, M. et al. Postprandial lipaemia induces an acute decrease of insulin sensitivity in healthy men independently of plasma NEFA levels. Diabetologia 49, 1612–1618 (2006). https://doi.org/10.1007/s00125-006-0262-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-006-0262-z