
Abstract
Aims/hypothesis
To examine the role of additional immune, genetic and metabolic risk markers in determining risk of diabetes in islet cell antibody (ICA)-positive individuals with a family history of type 1 diabetes recruited into the European Nicotinamide Diabetes Intervention Trial.
Methods
Five hundred and forty-nine first-degree relatives with confirmed ICA levels ≥20 Juvenile Diabetes Foundation units (mean age 15.9 years; interquartile range 10.4–33.7 years) were recruited from 20 countries. OGTTs and IVGTTs were performed at baseline, antibodies to glutamate decarboxylase (GADA), protein tyrosine phosphatase (IA–2A) and insulin (IAA) were determined by RIA, and HLA class II genotyping was performed by PCR of sequence-specific oligonucleotides.
Results
One hundred and fifty-nine participants developed diabetes within 5 years. Univariate analysis showed that the cumulative risk of development of diabetes within 5 years varied according to age, relationship to the proband, positivity for IAA, IA–2A and GADA, number and combination of islet antibodies, HLA class II genotype, baseline glucose tolerance, and first-phase insulin secretion, but not gender or incidence of childhood type 1 diabetes in the background population. Children aged ≤10 years had a 59% risk of diabetes within 5 years, compared with 11% in those ≥25 years (p<0.0001). Using multivariate analysis, independent determinants were age, first-phase insulin response, baseline glucose tolerance and number of additional antibody markers, but not antibody type or genotype. Individuals <25 years with two or more additional antibodies at baseline had a 62% risk of diabetes within 5 years and these combined criteria identified 81% of the cases in the whole cohort.
Conclusions/interpretation
We suggest that screening and recruitment for future intervention trials should be limited to family members aged <25 years, and should be based on islet autoantibodies alone.
Similar content being viewed by others


Introduction
Islet cell antibodies (ICA) and antibodies to insulin (IAA), glutamate decarboxylase (GADA) and protein tyrosine phosphatase IA-2 (IA–2A) appear many years before the clinical onset of type 1 diabetes and can be used to identify individuals at increased risk of disease [1]. Further stratification can be achieved by consideration of insulin secretion and glucose tolerance [2], and risk may vary with HLA-determined genetic susceptibility [3]. Two large multicentre intervention trials - the European Nicotinamide Diabetes Intervention Trial (ENDIT) and the North American Diabetes Prevention Trial-Type 1 (DPT-1) - have been completed in relatives of patients with type 1 diabetes identified as being at high risk on the basis of ICA positivity, which was chosen as the best-validated marker at the time when the trials were designed [4–7]. Although neither intervention was effective in preventing or delaying the clinical onset of diabetes, large study populations such as these allow us to validate and extend models of prediction that have been developed on the basis of retrospective analysis of smaller prospective studies.
In DPT-1, the inclusion criteria for the parenteral insulin arm of the trial required a low first-phase insulin response (FPIR), and those for the oral insulin trial required the presence of IAA. Relatives carrying the protective HLA-DQ allele DQA1*0602 were excluded from both intervention arms. Many ICA-positive individuals were not therefore eligible for randomisation and follow-up within a rigorous trial protocol [6]. In contrast, recruitment for ENDIT was based solely on the detection of high levels of ICA (≥20 Juvenile Diabetes Foundation [JDF] units). Data were collected on other potential markers, but follow-up was independent of these results. The trial therefore provides a unique opportunity to evaluate the role of additional immune, genetic and metabolic risk markers in determining risk of diabetes in an otherwise unselected, large cohort of ICA-positive individuals with a family history of type 1 diabetes recruited in 18 European countries, Canada and the USA.
Patients and methods
The protocol, screening stages and main results of ENDIT have been published [4, 5].
Participants
First-degree relatives of patients who developed type 1 diabetes before age 20, and who were themselves aged between 3 and 40 years, were eligible for screening. The original upper age limit of 60 years was lowered to 40 in 1995, when a multicentre analysis showed that relatives with ICA above that age were at low risk of progression to diabetes [2]. We screened more than 30,000 eligible relatives for ICA, recruited through 354 local centres in 18 European countries, Canada and the USA between 1990 and 1998, and enrolled 552 eligible individuals between June 1994 and May 1998. The inclusion criteria were ICA ≥20 JDF units in at least one sample as measured in the central laboratory and ICA ≥5 JDF units in the other sample. Those found to have diabetes on OGTT were excluded. The protocol was approved by the research ethics committee or equivalent in each participating centre, and also by the appropriate national drug regulatory authorities. Written informed consent was obtained from all participants.
Protocol
Participants were randomised to receive either oral modified-release nicotinamide (Niatrim; Ferrosan AC, Copenhagen, Denmark) at a dose of 1.2 g m−2 day−1 up to a maximum of 3 g/day or placebo for 5 years in two divided doses.
We reviewed participants at baseline, 1 month and 6 months after study entry and 6 months thereafter. OGTT was mandatory at baseline and 6, 18, 30, 42, 54 and 60 months. We tried to maintain annual contact with all participants who had withdrawn from the study for reasons other than development of diabetes, in order to determine their diabetes status. Our primary outcome was the development of diabetes by WHO criteria [8].
Testing procedures and assays
OGTT
Oral glucose (1.75 g/kg body weight, up to a maximum of 75 g) was administered following an overnight fast. Venous plasma samples collected at 0 and 120 min were tested for glucose in the local study centre laboratory. Diabetes and IGT were defined using WHO criteria [8].
IVGTT
IVGTTs were performed according to the Islet Cell Antibody Register Users Study protocol [9]. A glucose dose of 0.5 g/kg, up to a maximum of 35 g, was infused over 3 min±15 s, and blood samples collected at −5, 0, 1, 3, 5 and 10 min. The FPIR was calculated as the sum of the insulin levels at +1 and +3 min.
Islet autoantibodies
Baseline samples from all participants were tested for ICA, GADA, IA–2A and IAA in our laboratories at the University of Bristol as previously described, and considered positive if ≥97.5th centile of a control population of 2,860 schoolchildren [10]. The ICA assay achieved 81% sensitivity with 86% specificity in the First Immunology of Diabetes Society Combined Antibody Workshop, and radiobinding assays for GADA, IA–2A and IAA achieved 91% sensitivity with 99% specificity, and 74% sensitivity with 99% specificity and 58% sensitivity with 99% specificity, respectively [11].
HLA genotyping
HLA genotyping was performed in the Institute of Transplantation Immunology, National Hospital, Oslo, Norway. Typing of HLA-DQA1 and -DQB1 was mainly performed using PCR of sequence-specific oligonucleotides [12]. A few samples were also typed for DQA1 and DQB1 with an allele-specific PCR kit (Olerup SSP Genovision, Oslo, Norway) and/or with a reverse dot-blot kit (Reli-SSOP; Dynal, Oslo, Norway).
Insulin assay
Plasma insulin was measured in a single laboratory at the Steno Diabetes Center, Gentofte, Denmark using an enzyme-linked two-site immunoassay [13]. To allow comparison of IVGTT results with age-specific centiles for FPIR obtained in the University of Washington, Seattle and used for DPT-1 [6], 105 samples spanning the range 0–500 pmol/l were assayed in both laboratories. The regression equation of insulin concentration measured in Seattle on the insulin concentration measured in the Steno laboratory was used to derive a correction factor to standardise the measurement to the Seattle assay [14].
Statistical analysis
Antibody prevalences were expressed in relation to percentiles derived from a population of 2,860 schoolchildren in the Oxford region, UK [10]. HLA-DQ genotypes were defined as follows: high risk: DQA1*0501-DQB1*0201/DQA1*0301-DQB1*0302 (DQ2/DQ8) or DQA1*0301-DQB1*0302/DQA1*0301-DQB1*0302 (DQ8/DQ8); intermediate risk: DQA1*0501-DQB1*0201/DQA1*0501-DQB1*0201 (DQ2/DQ2), DQA1*0301-DQB1*0302/X (DQ8/X) or DQA1*0501-DQB1*0201/X (DQ2/X), where X is any haplotype other than DQA1*0501-DQB1*0201 (DQ2), DQA1*0301-DQB1*0302 (DQ8) or DQA1*0102-DQB1*0602 (DQ6); low risk: X/X; protective: all genotypes including DQA1*0102-DQB1*0602 (DQ6).
The background incidence of childhood type 1 diabetes for each national centre was derived from comparable studies collated by the Eurodiab or Diamond study groups covering the period 1990–1994 [15, 16]. Data for Russia and Switzerland were taken from other publications covering the same period [17, 18]. No data were available on the incidence of childhood diabetes in Turkey, which was included in the low incidence group on the advice of local investigators. Centres were divided into four incidence categories to give groups of approximately equal size: high (>27.5 cases 100,000−1 year−1), high intermediate (18.5–25.4 cases 100,000−1 year−1), low intermediate (9.6–15.5 cases 100,000−1 year−1) and low incidence (<9.1 cases 100,000−1 year−1 and Turkey).
The plasma glucose value at 120 min in the OGTT was categorised according to the WHO definition into IGT (≥7.8 mmol/l) and NGT.
Time-to-event curves were constructed using the Kaplan–Meier method. The follow-up period for each individual was calculated from the date of the baseline visit, and the end of follow-up was defined as the date of last contact or date of diagnosis of diabetes. Five-year risks were calculated to 5 years±1 month as the acceptable range for the study visit defined by the protocol. Diabetes was defined according to the WHO criteria [8].
Cox proportional hazards models were used to analyse the time-to-event outcome for individual and combined predictive markers, controlling for treatment group. The following variables were included in the regression model: gender, age at study entry, relationship to the proband, background incidence of childhood type 1 diabetes in the study centre, presence or absence of DQ2, DQ8 and DQ6 haplotype, HLA-DQ genotype risk category, presence or absence of IGT, FPIR above or below age-adjusted 10th centile, GADA, IA–2A and IAA above the 97.5th centile and number of islet autoantibodies above the 97.5th centile. Age at entry and number of additional islet autoantibodies were modelled as categorical variables as no additional linear effect on comparison of model fit was found when the variables were fitted as continuous variables. The model was built in stages following the general strategy for model selection suggested by Collett [19]. Variables found to be statistically significant at the 5% level in univariate analysis were included in the multivariate model. The effect of removing each variable from this multivariate model was assessed. Variables not significant on univariate analysis were also considered for inclusion in the final multivariate model. Finally, to be reassured that the final multivariate model was robust, the model was fitted using forward selection of variables, and yielded the same final model as the backwards selection we used. To overcome problems of collinearity, three multivariate models were constructed each including only one of the following: (1) number of additional antibodies; (2) combination of additional antibodies; or (3) GADA, IA–2A and IAA considered separately. Similarly, alternative models were constructed including either (1) age at study entry or (2) relationship to the proband. Inclusion in the final model was based on comparison of the changes in maximised likelihood associated with removal of the respective variable from the model. First-order interactions between variables were also examined. The assumption of proportionality was evaluated by the inclusion of time-dependent covariates in each model.
Statistical significance was taken as p<0.05. Results are expressed as relative risk (95% CI). Results are reported as number of participants and events for each risk group, and hazard ratios (95% CI) and p values from the Cox model analyses. Statistical analysis was performed using SPSS 11.5 (SPSS Inc., Chicago, IL, USA).
Results
Baseline characteristics
Of 552 individuals randomised, two withdrew consent and one developed diabetes before starting the trial medication. No follow-up data were available for these individuals, who have not been included in the present analysis. The baseline characteristics of the remaining 549 participants are given in Table 1.
As shown in Table 2, the baseline prevalence of additional immune markers of risk varied with age. GADA, IA–2A and IAA were detected more frequently in younger participants (p<0.0001 for each antibody), and these individuals were also more likely to be positive for two or three antibodies in addition to ICA (p<0.0001). HLA class II genotype also varied with age within this cohort (p=0.007) (data not shown). Metabolic markers of increased risk did not, however, vary with age. FPIR was below the age-specific 10th centile in 39% of participants below the median age (15.9 years) and 34% of older participants (p=0.192), and the baseline OGTT showed IGT in 12% and 7%, respectively (p=0.08).
The presence of additional antibodies was also associated with HLA class II haplotype and genotype (Table 2). Individuals with at least one DQ8 haplotype had a higher prevalence of GADA, IA–2A and IAA (p<0.0001 for each antibody), and were more likely to have at least two antibodies in addition to ICA (p<0.0001) than those without DQ8. In contrast, individuals with at least one DQ6 haplotype had a lower prevalence of GADA (p=0.014) and IA–2A (p=0.008), and of IAA (p=0.005) than those without DQ6, and at least two antibodies in addition to ICA were detected in 26% of those with DQ6 compared with 52% of those without (p<0.0001). At least one additional antibody was detected in 75% of individuals whose genotype was classified as high risk, 67% intermediate, 52% low risk, and 48% protective (p<0.0001), and two or more additional markers were detected in 63% of individuals whose genotype was classified as high risk, 53% intermediate, 34% low risk and 26% protective (p<0.0001).
Progression to diabetes
The median duration of follow-up of the cohort was 4.95 years. The primary outcome variable of diabetes status after 5 years of follow-up was established in 477 of 549 participants (87%). Of 549 participants, 159 developed diabetes within 5 years. The baseline characteristics of this group are shown in Table 1.
The 5-year risk of progression to diabetes estimated from Kaplan–Meier survival analysis and the corresponding hazard ratios in univariate Cox regression analysis according to baseline characteristics are shown in Table 1 and Fig. 1. Univariate analysis showed that risk varied with demographic characteristics (age at randomisation and relationship to the diabetic proband), other autoantibodies (presence of GADA, IA–2A or IAA, number of markers in addition to ICA and antibody combination), HLA class II (presence of at least one DQ8 or DQ6 haplotype and HLA-DQ genotype) and baseline metabolic status (IGT and loss of FPIR). Risk did not vary with gender, background incidence of childhood type 1 diabetes, or DQ2 haplotype. Multivariate analysis (Table 3) showed that age at randomisation, number of additional antibodies, glucose tolerance and loss of FPIR were independent determinants of risk, but genetic risk category and relationship to the proband were not. When relationship to the proband was substituted for age in the model to overcome partial collinearity between these variables, the adjusted hazard ratios were 3.00 (1.37–6.55) for siblings, 3.50 (1.41–8.70) for offspring and 2.84 (1.02–7.88) in fathers when compared with mothers of a person with diabetes. None of the variables displayed a statistically significant time-dependent covariate when tested in univariate analysis except for FPIR and IGT, which were of borderline significance (p=0.03 and p=0.02, respectively). These became non-significant when tested in the multivariate analysis and, for simplicity, were not considered further. There were no significant interactions between the variables. The use of forward selection of variables resulted in the same final model.

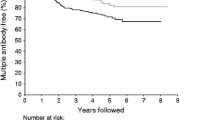
The cumulative risk of diabetes in ICA-positive relatives in relation to a age, b number of additional autoantibodies ≥97.5th centile, c OGTT and d first-phase insulin response on IVGTT. For all analyses shown, p<0.0001 on log-rank testing
To evaluate the contribution of individual autoantibody markers in determining risk, positivity for GADA, IA–2A and IAA were entered separately into the multivariate model in place of number of additional antibodies. The adjusted hazard ratio for positivity for IA–2A was 4.80 (3.11–7.38, p<0.0001), for GADA was 9.94 (4.80–20.56, p<0.0001) and for IAA was 4.63 (2.91–7.37, p<0.0001). Similarly, a model was constructed substituting the eight possible combinations of autoantibody positivity for number of antibodies. In comparison with ICA alone, the adjusted hazard ratio for positivity for a single additional antibody was 6.46 (1.65–25.24, p=0.007) for ICA/GADA and 6.28 (1.03–38.28, p=0.047) for ICA/IAA. None of eight individuals with ICA/IA–2A developed diabetes. For positivity for two additional antibodies the adjusted hazard ratio for ICA/GADA/IAA was 18.77 (5.39–65.34, p<0.0001), for ICA/GADA/IA–2A was 24.17 (6.99–83.55, p<0.0001) and for ICA/IA–2A/IAA was 18.66 (3.63–95.89, p<0.0001).
Sensitivity of risk assessment
Of the 159 participants who developed diabetes within 5 years, 140 (88%) were aged <25 years at study entry, 141 (89%) were siblings or offspring of probands, and 145 (91%) had two or three antibodies in addition to ICA; 129 cases (81%) were both aged <25 and had two or three additional antibodies. Of these 129 individuals, 74 (57%) had an FPIR below the 10th centile at entry, 27 (21%) had IGT and 17 (13%) were in both of these groups.
Discussion
Both ENDIT and DPT-1 emphasised that diabetes can be predicted effectively, and in ENDIT 29% of first-degree relatives, identified simply on the basis of high titres of ICA, progressed to diabetes within 5 years [5, 6]. This analysis has confirmed that risk assessment in ICA-positive relatives can be refined by additional testing for other islet autoantibodies, HLA class II genotype, stimulated insulin secretion and glucose tolerance, and that, irrespective of other markers, age or relationship to the proband are also determinants of risk. Family members with ICA alone had a low risk of progression to diabetes within 5 years, and, as expected, risk increased with the number of markers detected. Multivariate analysis showed that risk of developing diabetes was increased sixfold in those with ICA and one additional marker compared with those with ICA only, 21-fold in those with two additional antibody markers, and 33-fold in those with ICA and GADA, IA–2A and IAA. Of those who developed diabetes, 91% came from the subgroup with two or three additional markers. Low FPIR was confirmed as an important determinant of risk, although 36% of those who developed diabetes within 5 years had an age-adjusted FPIR above the 10th centile at baseline, and IGT was associated with very high risk in the short term, giving an estimated one-year risk of 35% compared with 4% in those with NGT.
This large dataset revealed a complex interaction of risk factors, in particular between age, genotype and islet autoantibodies. Possession of a DQ8 haplotype was associated with an increased prevalence of GADA, IA–2A and IAA, while DQ6 was associated with a decreased prevalence of these antibodies, and individuals with this haplotype were more likely to have ICA alone. Multivariate analysis showed that age and the number of additional antibodies detected were independent determinants of risk of progression, but that HLA class II genotype was not. For example, within the subgroup of participants with at least two antibodies in addition to ICA, the five-year risk of diabetes was similar in those with and without DQ8 (62 [53–72%] vs 58% [50–66%], p=0.2743) and the risk within 4 years was similar between the small group with DQ6 and those without this protective haplotype (50 [15–85%] vs 49% [42–56%], p=0.9832). These observations would be consistent with the speculation that the major role of HLA-associated genetic susceptibility may be in determining the pattern of islet autoimmunity rather than subsequent beta-cell destruction and progression to clinical diabetes-a situation analogous to the NOD mouse in which there are independent genetic determinants of insulitis and diabetes [20].
A major strength of this study is its size and high level of follow-up. Diabetes status after 5 years of follow-up was established in 477 of 549 participants (87%). In addition all procedures were performed to standardised protocols, and autoantibody assays, genotyping and insulin measurement were each performed in a single reference laboratory. A further strength is that the study was performed in European and North American populations with widely differing background risks of childhood diabetes, allowing us to examine the influence of this variable on risk of progression in antibody-positive relatives. A potential problem in applying the results of this study in future intervention trials is that participants were identified on the basis of ICA, which are no longer used as the primary screen. Other studies have, however, shown that individuals at the highest risk, i.e. those with multiple antibodies, could be identified using alternative strategies based on screening for GADA and IA–2A [21–24]. It will, however, be of interest to validate these findings in individuals at high risk identified by screening based on GADA, IA–2A and IAA. Nicotinamide was taken by half the participants, but the trial demonstrated that this had no effect upon progression to diabetes [5] and, since we have taken the additional precaution of controlling for treatment group in univariate and multivariate analyses, we think it is valid to analyse the prognostic significance of additional markers in the whole population.
Our findings confirm the importance of age, which was closely associated with positivity for additional islet autoantibodies and FPIR in enhancing risk assessment based on ICA [2]. Risk fell very abruptly in early adult life, and was essentially unchanged after age 25. Loss of FPIR was once again found to be predictive, but we would have excluded more than a third of the individuals who developed diabetes within 5 years if this had been used as an entry criterion. Risk stratification by number of islet autoantibodies was confirmed in this large dataset [25–28]. Those with ICA alone or one additional marker were at low risk, and risk was concentrated among family members with two or three additional markers. Since this group included almost everyone who developed diabetes, further metabolic testing of these individuals would have provided little additional information. This model predicted diabetes in populations with widely varying background incidence of childhood diabetes, and therefore appears to be robust. Number of islet autoantibodies was the major determinant of risk, and very few ICA-positive adults had two or more additional autoantibodies. If screening for ENDIT had been restricted to the 60% of family members aged <25 [29], we would have identified 84% of those with two or three antibodies in addition to ICA and 88% of those who developed diabetes within 5 years.
Some of our findings differed from those of smaller studies. It has been suggested that IA–2A are particularly associated with risk of imminent diabetes and confer additional risk in multiple-antibody-positive siblings, to the extent that those without IA–2A could be considered at low risk (<10% within 5 years) [30]. We did not see this in ICA-positive ENDIT participants, whether analysed as a single group or in those aged <20 years, using either the 97.5th or 99th centile to define IA–2A positivity (data not shown), perhaps because the cohort was preselected on the basis of high ICA levels. In contrast to some other studies [31] we found no suggestion that risk differed between those with ICA and GADA/IAA, GADA/IA–2A or IA–2A/IAA; the adjusted hazard ratios for these combinations compared with ICA alone were 18, 24 and 19, respectively.
We have focused upon antibody number and combination in the present analysis, but the ENDIT dataset and sample bank allows us to validate other antibody markers of risk. Prediction may potentially be refined by inclusion of other antibody characteristics such as antibody titre, epitope specificity and isotype [31–33]. Risk has also been related to antibody affinity, particularly with respect to IAA [34], and consideration of metabolic variables such as insulin resistance could also enhance prediction [35]. In addition, more detailed genotyping, to include, for example, HLA-DR4 subtypes and IDDM2 alleles, might improve upon the limited range of genetic markers included in this analysis.
In summary, this analysis of the ENDIT dataset has validated predictive models developed by retrospective analysis of smaller studies, has confirmed that age and islet antibody testing allow sensitive and specific identification of relatives at high risk of developing diabetes, and has shown that ICA-positive family members >25 years are at low risk. It has demonstrated that HLA class II markers add little to prediction based on age and autoantibodies in relatives with multiple islet autoantibodies, and has confirmed that loss of FPIR is a specific but relatively insensitive marker of risk of progression to diabetes over 5 years. We suggest that screening and recruitment for future intervention trials should be limited to family members aged <25 years, and based on islet autoantibodies alone.
Abbreviations
- DPT-1:
-
Diabetes Prevention Trial—Type 1
- ENDIT:
-
European Nicotinamide Diabetes Intervention Trial
- FPIR:
-
first-phase insulin response
- GADA:
-
glutamate decarboxylase antibodies
- IAA:
-
insulin autoantibodies
- IA–2A:
-
antibodies to protein tyrosine phosphatase IA-2
- ICA:
-
islet cell antibodies
- JDF:
-
Juvenile Diabetes Foundation
References
Atkinson MA, Eisenbarth GS (2001) Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet 358:221–229
Bingley PJ, for the ICARUS Group (1996) Interactions of age, islet cell antibodies, insulin autoantibodies, and first-phase insulin response in predicting risk of progression to IDDM in ICA+ relatives: the ICARUS data set. Diabetes 45:1720–1728
Kulmala P, Savola K, Reijonen H et al (2000) Genetic markers, humoral autoimmunity, and prediction of type 1 diabetes in siblings of affected children. Diabetes 49:48–58
The European Nicotinamide Diabetes Intervention Trial (ENDIT) group (2003) Intervening before the onset of type 1 diabetes: baseline data from the European Nicotinamide Diabetes Intervention Trial (ENDIT). Diabetologia 46:339–346
The European Nicotinamide Diabetes Intervention Trial (ENDIT) group (2004) European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet 363:925–931
Diabetes Prevention Trial—Type 1 Diabetes Study Group (2002) Effects of insulin in relatives of patients with type 1 diabetes. N Engl J Med 346:1658–1691
Skyler JS, Krischer JP, Wolfsdorf J et al (2005) Effects of oral insulin in relatives of patients with type 1 diabetes: The Diabetes Prevention Trial—Type 1. Diabetes Care 28:1068–1076
World Health Organization (1985) Diabetes mellitus: report of a WHO study group. World Health Organization, Geneva
Bingley PJ, Colman PG, Eisenbarth GS et al (1992) Standardization of IVGTT to predict IDDM. Diabetes Care 15:93–102
Bingley PJ, Bonifacio E, Williams AJK, Genovese S, Bottazzo GF, Gale EAM (1997) Prediction of IDDM in the general population. Strategies based on combinations of autoantibody markers. Diabetes 46:1701–1710
Verge CF, Stenger D, Bonifacio E et al (1998) Combined use of autoantibodies (IA-2 autoantibody, GAD autoantibody, insulin autoantibody, cytoplasmic islet cell antibodies) in type 1 diabetes: Combinatorial Islet Autoantibody Workshop. Diabetes 47:1857–1866
Undlien D, Friede T, Rammansee H-G et al (1997) HLA-encoded genetic predisposition in IDDM. DR4 subtypes may be associated with different degrees of protection. Diabetes 46:143–149
Andersen L, Dinesen B, Jørgensen PN, Poulsen F, Røder ME (1993) Enzyme immunoassay for intact human insulin in serum or plasma. Clin Chem 39:578–582
McCulloch DK, Bingley PJ, Colman PG, Jackson RA, Gale EAM, The ICARUS Group (1993) Comparison of bolus and infusion protocols for determining acute insulin response to intravenous glucose in normal humans. Diabetes Care 16:911–915
Green A, Patterson CC, EURODIAB TIGER Study Group (2001) Trends in the incidence of childhood-onset diabetes in Europe 1989–1993. Diabetologia 44(Suppl 3):B3–B8
Karvonen M, Viik-Kajander M, Moltchanova E, Libman I, Laporte R, Tuomilehto J, Diabetes Mondiale (DiaMond) Project Group (2000) Incidence of childhood type 1 diabetes worldwide. Diabetes Care 23:1516–1526
Kondrashova A, Reunanen A, Romanov A et al (2005) A six-fold gradient in the incidence of type 1 diabetes at the eastern border of Finland. Ann Med 37:67–72
Schoenle EJ, Lang-Muritano M, Gschwend S et al. (2001) Epidemiology of type 1 diabetes in Switzerland: steep rise in incidence in under 5 year old children in the past decade. Diabetologia 44:286–289
Collett D (1994) Modelling survival data in medical research. Chapman and Hall, London
Wicker LS, Todd JA, Peterson LB (1995) Genetic control of autoimmune diabetes in the NOD mouse. Annu Rev Immunol 13:179–200
Bingley PJ, Williams AJK, Gale EAM (1999) Optimized autoantibody-based risk assessment in family members. Diabetes Care 22:1796–1801
Seissler J, Morgenthaler NG, Achenbach P et al (1996) Combined screening for autoantibodies to IA-2 and antibodies to glutamic acid decarboxylase in first degree relatives of patients with IDDM. Diabetologia 39:1351–1356
Pastore MR, Bazzigaluppi E, Bonfanti R et al (1998) Two-step islet autoantibody screening for risk assessment of type 1 diabetes in relatives. Diabetes Care 21:1445–1450
Dittler J, Seidel D, Schenker M, Ziegler AG (1998) GADIA2-combi determination as first line screening for improved prediction of type 1 diabetes in relatives. Diabetes 47:592–597
Bingley PJ, Christie MR, Bonifacio E et al (1994) Combined analysis of autoantibodies improves prediction of IDDM in islet cell antibody-positive relatives. Diabetes 43:1304–1310
Verge CF, Gianani R, Kawasaki E et al (1996) Prediction of type 1 diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes 45:926–933
Kulmala P, Savola K, Petersen JS et al (1998) Prediction of insulin-dependent diabetes in siblings of children with diabetes. A population-based study. J Clin Invest 101:327–336
Christie MR, Roll U, Payton MA, Hatfield ECI, Ziegler AG (1997) Validity of screening for individuals at risk for type 1 diabetes by combined analysis of antibodies to recombinant proteins. Diabetes Care 20:965–970
Williams AJK, Bingley PJ, Moore WPT, Gale EAM, The ENDIT screening group (2002) Islet autoantibodies, nationality and gender: a multinational screening study in first-degree relatives of patients with type 1 diabetes. Diabetologia 45:217–223
Decochez K, De Leeuw IH, Keymeulen B et al (2002) IA-2 antibodies predict impending type 1 diabetes in siblings of patients. Diabetologia 45:1658–1666
Achenbach P, Warncke K, Reiter J et al (2004) Stratification of type 1 diabetes risk on the basis of islet autoantibody characteristics. Diabetes 53:384–392
Hoppu S, Harkonen T, Ronkainen MS, Akerblom HK, Knip M (2004) IA-2 antibody epitopes and isotypes during the prediabetic process in siblings of children with type 1 diabetes. J Autoimmun 23:361–370
Hoppu S, Ronkainen MS, Kulmala P, Akerblom HK, Knip M (2004) GAD65 antibody isotypes and epitope recognition during the prediabetic process in siblings of children with type 1 diabetes. Clin Exp Immunol 136:120–128
Achenbach P, Koczwara K, Knopff A, Naserke H, Ziegler AG, Bonifacio E (2004) Mature high-affinity immune responses to (pro)insulin anticipate the autoimmune cascade that leads to type 1 diabetes. J Clin Invest 114:589–597
Fourlanos S, Narendran P, Byrnes GB, Colman PG, Harrison LC (2004) Insulin resistance is a risk factor for progression to type 1 diabetes. Diabetologia 47:1661–1667
Acknowledgements
We thank the many technical, nursing and medical staff in the participating centres for all their help in running the trial, Clare Emmett for data preparation and Paul Ewings for statistical advice. The final phase of ENDIT was funded by the Juvenile Diabetes Research Foundation grant 4-2000-943; earlier phases were funded by European Union grants PL92 0957 and PL95 0771. We are grateful to Novo Nordisk for additional financial support throughout the period of the trial.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
See Appendix for ENDIT Group.
Appendix
Appendix
The ENDIT Group:
National Coordinators: Austria: E. Schober; Belgium: F. K. Gorus; Canada: J. Dupre, J. L. Mahon; Croatia: V. Profozic; Denmark: J. I. Reimers; T. Mandrup-Poulsen; Finland: M. Knip; France: C. Levy-Marchal; Germany: C. Jaeger; Greece: C. Bartsocas, A. Vazeou; Hungary: M. Györko, G. Soltesz, L. Madacsy; Italy: M. R. Pastore, P. Pozzilli; Norway: K. Dahl-Jørgensen, G. Joner; Poland: I. Kinalska, A. Mrozikiewicz; Russia: Y. Vaykhonsky; Spain: A. de Leiva, M. T. Martinez-Larrad, D. Mauricio, M. Serrano-Rios; Sweden: J. Ludvigsson; Switzerland: E. J. Schoenle; Turkey: M. T. Yilmaz; UK: P. J. Bingley (England), D. J. Carson, H. Tennet (Northern Ireland), K. Robertson (Scotland); USA: W. J. Riley (Texas).
Laboratory Support: University of Bristol: K. M. Gillespie, H. Gillmor, W. P. T. Moore, A. Norcross, A. J. K. Williams; Steno Diabetes Center, Gentofte: B. Dinesen, S. Kjellberg, T. Mandrup-Poulsen, H. Niebling; National Hospital, Oslo: H. E. Akselsen, E. Thorsby, D. E. Undlien.
Study Administrators: C. L. Emmett, H. Swankie, S. Fewell, P. Kearsey.
Data Safety and Monitoring Committee: A.L. Drash, O. Aagenes, G. Dahlquist, A. Laupacis, A.E.M. McLean, J. Nerup, B. Weber.
Rights and permissions
About this article
Cite this article
The European Nicotinamide Diabetes Intervention Trial (ENDIT) Group., Bingley, P.J. & Gale, E.A.M. Progression to type 1 diabetes in islet cell antibody-positive relatives in the European Nicotinamide Diabetes Intervention Trial: the role of additional immune, genetic and metabolic markers of risk. Diabetologia 49, 881–890 (2006). https://doi.org/10.1007/s00125-006-0160-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-006-0160-4