
Abstract
Aims/hypothesis
The precise mechanisms underlying the increased risk of cardiovascular disease (CVD) in type 2 diabetes are unclear. Fibrin clot structure has been related to CVD risk in the general population. We therefore assessed this in type 2 diabetic patients as a potential mechanism whereby diabetes influences CVD risk.
Methods
Fibrin clots were formed from fibrinogen purified from 150 subjects with type 2 diabetes and varying degrees of glycaemic control (assessed by HbA1c), and from 50 matched control subjects. Clot structure was assessed by turbidity, permeation and confocal microscopy. The specific effect of glucose itself was assessed by analysing the structure of clots formed from purified fibrinogen in the presence of increasing concentrations of the sugar.
Results
Clots formed by fibrinogen purified from type 2 diabetic subjects had a denser, less porous structure than those from control subjects. The structural changes found were related to the individual’s glycaemic control; HbA1c correlated negatively with permeation coefficient (K s) values (indicates clot pore size) (r=−0.57, p<0.0001) and positively with maximum absorbance (indicator of fibre size) (r=0.33, p<0.0001), branch point number (r=0.78, p<0.0001) and fibre density (r=0.63, p<0.0001). The ambient glucose level influenced clot structure; hypo- (<5 mmol) and hyperglycaemia (≥10 mmol/l) were both associated with a reduction in K s values and maximum absorbance, and with increased fibre density and branch point number within clots.
Conclusions/interpretation
The structural differences found to occur in type 2 diabetes and in association with hypo- and hyperglycaemia may confer increased resistance to fibrinolysis, and in consequence contribute to the increase in CVD risk in diabetic patients.
Similar content being viewed by others


Introduction
Cardiovascular disease (CVD) is the major cause of morbidity and mortality amongst individuals with type 2 diabetes [1]. Whilst this is in part related to the clustering of atheromatous risk factors in patients, diabetes has been shown to exert a significant effect on CVD risk independently of co-existent cardiovascular risk factors such as hypertension and dyslipidaemia [2]. This increased risk may be related to other metabolic abnormalities associated with the condition. The majority of individuals with type 2 diabetes and CVD are insulin-resistant [3], yet the precise cellular/molecular changes that result from insulin resistance/hyperinsulinaemia and link to the process of atherogenesis remain under investigation.
Despite these uncertainties, the pathophysiological processes underlying the development of atherosclerosis have become more clearly defined over recent years [4, 5]. The formation of an atherosclerotic plaque is a complex process that occurs over many decades and may remain clinically silent. The pivotal events leading to clinical presentation are the development of an unstable plaque and the formation of an occlusive platelet-rich thrombus on plaque rupture, which cause the acute complications of CVD (unstable angina, myocardial infarction and stroke) [6, 7]. The release of procoagulant debris on plaque rupture precipitates a series of events that result in the activation and adherence of platelets to the damaged vessel wall followed by local activation of coagulation and the formation of a cross-linked fibrin clot, which provides the key structural support for the thrombus. Fibrin has also been shown to be a consistent component of human atherosclerotic plaques [8, 9], and may play a role in promoting their development [10–12].
Prospective studies have consistently reported an association between elevated fibrinogen levels and cardiovascular risk [2, 13]. Assessment of the structure of the fibrin clot itself in relation to atherothrombotic disease development has revealed its structure to be heterogeneous, being influenced by genetic and environmental factors [14]. Clinical studies have reported an association between clot structure and the development of premature coronary heart disease [15]; patients suffering a myocardial infarction before the age of 45 years formed clots with a denser, less permeable structure than healthy control subjects [15], a structural change that may contribute to atherothrombotic risk by increasing resistance to fibrinolysis [16].
Analysis of the structure of the fibrin clot in relation to diabetes has revealed that fibrin clots formed from the plasma of subjects with type 1 diabetes have a denser structure, with smaller intrinsic pores than those formed by healthy control subjects [17, 18], a feature that has been shown to enhance a clot’s resistance to fibrinolysis [16, 19]. However, the mechanism(s) by which fibrin clot structure is altered in type 1 diabetes are unclear. The changes found were inversely correlated to glycaemic control (HbA1c) [17], leading the authors to suggest that this effect may result from glycation of the fibrinogen molecule. Indeed, fibrinogen has been shown to be glycated in vivo [20], and in vitro glycation of fibrinogen has been shown to influence its interaction with other coagulation/fibrinolytic proteins [21–23].
The aim of this study was therefore to investigate—using a purified system—whether the structure/function of the fibrin clot is altered in subjects with type 2 diabetes. Fibrin clots were formed from fibrinogen purified from subjects with type 2 diabetes and from healthy age- and sex-matched control subjects. The direct effect of glucose molecules on the formation and cross-linkage of the fibrin clot was also assessed in this purified system. In addition, we sought to determine possible mechanisms underlying the observed changes in clot structure by assessing the activity of thrombin and factor XIII (FXIII).
Subjects, materials and methods
Subjects
Type 2 diabetic subjects (n=150) were recruited through the Diabetes Centre at Leeds General Infirmary. Age- and sex-matched control subjects (n=50) were recruited from the Leeds Health Authority Family Health Service Register (Table 1). All subjects were white, north European, and gave informed consent according to a protocol approved by the Leeds Teaching Hospitals Research Ethics Committee. Patients with microalbuminuria, diabetic retinopathy, peripheral neuropathy and CVD were excluded from the study; none of the subjects was taking oral anticoagulants or aspirin.
Blood sampling
Venous blood was taken between 08.00 and 10.00 hours, after an overnight fast, into siliconised tubes with 15 IU per ml lithium heparin for purification of fibrinogen, and into EDTA for HbA1c measurement using a Glycomat autoanalyser (Ciba Corning, Halstead, UK; reference range 4.5–6.5%). Within 1 h, samples for fibrinogen purification were centrifuged (2,540 g for 20 min) at room temperature to obtain platelet-poor plasma, which was separated, frozen in liquid nitrogen, and stored at −40°C.
Fibrinogen purification
Fibrinogen was purified from the plasma of each subject using IF-1 affinity chromatography as described previously [24], and was standardised at a concentration of 1 mg/ml for the analysis of clot structure.
Factor XIII purification
Human FXIII was purified from plasma by ammonium sulphate precipitation and gel filtration, as previously described [25].
Clot permeation
Liquid permeation studies were performed on fibrinogen purified from all 200 subjects as described previously [26]. This technique measures the rate at which a buffer flows through a fibrin clot, and enables calculation of the permeation coefficient (K s), which represents the surface of the gel allowing flow through the network, giving an indication of the pore size within the fibrin clot. Clots were formed from purified fibrinogen (1 mg/ml) on addition of 22 μg/ml FXIII, 1 U/ml human α-thrombin (American Diagnostica, Stamford, CT, USA) and 5 mmol/l calcium. Two replicate clots were analysed for each subject.
Turbidity measurements
Turbidity experiments were performed as described previously [26], using fibrinogen (1 mg/ml) purified from all 200 subjects, and purified FXIII (22 μg/ml), 1 U/ml human α-thrombin and 5 mmol/l calcium. Two replicate measurements were performed for each sample. The parameters recorded were lag phase (s) (taken as the time taken for the absorbency to increase by 0.01 above baseline [i.e. time zero reading]), and maximum absorbency at full polymerisation (calculated as the absorbency value at 30 min minus the baseline [time zero] absorbency reading). The lag phase represents the length of time required for fibrin protofibrils to grow to sufficient length to allow lateral aggregation to occur and is sensitive to a variety of factors, including fibrinogen concentration and rate of fibrinopeptide A (FPA) cleavage [27]. Clot turbidity is directly proportional to the average cross-sectional area of the fibres comprising it, and the maximum absorbency reflects the average size of the fibrin fibres within the clot [27].
Confocal microscopy
Fibrin clots were formed from fibrinogen (1 mg/ml) purified from 30 randomly selected diabetic subjects and ten control subjects matched for all clinical parameters, and incubated with FXIII (22 μg/ml), 1 U/ml human α-thrombin, and 5 mmol/l CaCl2 for assessment by confocal microscopy as described previously [16]. All samples were prepared in duplicate. Specimens were scanned using a Leica TCS SP-2 laser scanning confocal microscope (Leica Microsystems, Heidelberg, Germany), and digital photographs were obtained from six regions in each clot (measuring 30×30×30 μm). Thirty optical sections were collected at intervals of 1 μm depth, and the images obtained were analysed to determine the number of branch points and fibres per standardised volume of clot (27,000 μm3). The average fibre diameter (n=500) within each clot was also determined.
Alpha- and gamma-chain cross-linking
Rates of α- and γ-chain cross-link formation within fibrin clots by FXIII were determined in all subjects. Purified fibrinogen (1 mg/ml) was incubated with 22 μg/ml FXIII, 0.5 IU/ml human α-thrombin, and 5 mmol/l calcium in TRIS-buffer saline (TBS) at room temperature for varying time periods of up to 120 min. The rate of cross-linkage was determined as described previously [28]. Cross-linking by FXIII caused the formation of multiple α-chain polymers. The diffuse nature of the bands representing α-polymers on SDS-PAGE made the measurement of band density inaccurate; therefore, the reduction in α-monomer chain band density over time (corresponding to increasing degrees of α-chain cross-linkage and α-polymer formation) was used as a surrogate marker of α-chain cross-linkage, and the results were expressed as a percentage of the α-monomer present at time zero (which was taken to be 100%).
Fibrinopeptide-A and B release
Thrombin-induced fibrinopeptide release from fibrinogen purified from 30 randomly selected diabetic subjects and 20 matched controls was measured by reverse-phase HPLC as previously described [25], using fibrinogen (1 mg/ml) incubated with 0.5 IU/ml human α-thrombin and 2.2 mmol/l calcium. Molar quantities of the fibrinopeptides were calculated from the chromatograms by integration of the respective peak areas at each time point, and comparison with the amount of fibrinopeptides obtained at maximum release (45 min). Catalytic efficiencies for FPA and fibrinopeptide B (FPB) release reactions by thrombin were calculated as described [25, 29].
Effects of ambient glucose levels on fibrin clot structure/function
The effect of glucose on fibrin clot structure was assessed in view of the significant correlation found between fasting blood glucose levels and fibrin clot structure found in diabetic and control subjects (Table 2). Human plasminogen-free fibrinogen (Calbiochem, San Diego, CA, USA) at a concentration of 1 mg/ml was used to determine the effect of ambient glucose on clot structure. Clots were formed in the presence of 0–20 mmol/l glucose (Sigma) in TBS; polymerisation was initiated immediately following addition of glucose. Fibrin clot structure was assessed using previously described techniques; clot permeation (15 clots per glucose concentration), turbidity analysis (96 clots per glucose concentration) and confocal microscopy (five replicate clots per glucose concentration). The effects of glucose on the cross-linkage of fibrin clots by FXIII and thrombin-induced cleavage of fibrinopeptides were also assessed using the methods described above.
Statistical analysis
Data were tested for conformity to the normal distribution using the Kolmogorov–Smirnov test. For the glycation study, data were not normally distributed and non-parametric tests were used. Spearman correlation coefficients were used to identify associations between fibrin clot structure/function and HbA1c. Differences in parameters of fibrin clot structure, FXIII cross-linkage and fibrinopeptide release between the diabetic and control subjects were compared using the Mann–Whitney U-test. For the glucose study, data showed a sufficient fit to the normal distribution (p>0.2) to allow parametric analysis. ANOVA with post hoc Bonferroni analysis was used to compare fibrin structures at different glucose concentrations. Statistical significance was taken as p values <0.05. All analyses were performed with the Statistics Package for Social Scientists for Windows version 11.5.
Results
Diabetic and control subjects were matched for age, sex and smoking habits (Table 1). Whilst significant differences in other clinical parameters occurred between the two groups (BMI, systolic BP, triglyceride and HDL cholesterol levels), only fasting glucose levels and HbA1c were found to correlate with the parameters of clot structure assessed (Table 2). A variety of different treatments were used to control blood glucose levels within the diabetic subjects (Table 3).
Assessment of fibrin clot structure in type II diabetes
Permeation and turbidity
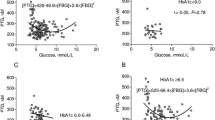
Significant differences in clot structure were identified between clots formed from fibrinogen purified from diabetic and from control subjects. Diabetic clots had a shorter lag phase in polymerisation, achieved a greater maximum absorbance at full polymerisation, and had a significantly lower mean permeation coefficient than those formed by controls (Table 4). The changes in clot structure found correlated with subjects’ glycaemic control (HbA1c), which correlated significantly with clot permeability: K s (r=−0.57, p 0.0001) (Fig. 1), maximum absorbance (r=+0.33, p 0.0001) and lag phase (r=−0.36, p 0.0001) in clot polymerisation.

Relationship between fibrin clot structure and chronic glycaemic control (HbA1c) analysed by permeation studies and confocal microscopy. a Permeation studies. Significant correlation between HbA1c and pore size within clots. b–d Confocal microscopy. Positive correlation between HbA1c and fibre number per unit volume (b), fibre diameter (c) and d branch-point number per unit volume of clot (27,000 μm3)
Confocal microscopy
Analysis of clot structure using confocal microscopy identified significant differences in all the parameters assessed between the two groups except fibre diameter (Table 4). Significant correlations were, however, found to exist between HbA1c and all parameters assessed (Fig. 1); as glycaemic control worsened the fibrin clots formed had a denser, less permeable structure.
Assessment of the influence of ambient glucose levels on fibrin clot structure
Permeation and turbidity
Fibrin structure was influenced by ambient glucose levels (Fig. 2). Pore size within clots and maximum absorbance of clots were significantly reduced at glucose concentrations above and below 5 mmol/l (p<0.0001) compared with those formed at 5 mmol/l. There was a significant increase in the lag phase recorded for clots formed at concentrations below 5 mmol/l (p<0.01), and exceeding 5 mmol/l (p<0.0001) over that of clots formed at 5 mmol/l.

Analysis of the structure of the fibrin clot formed at different glucose concentrations. a Permeation coefficient (K s). Significant reduction (p<0.0001) in pore size at glucose levels above and below 5 mmol. b,c Turbidity analysis. Significant increase in lag time (p<0.0001) (b) and reduction in maximum absorbance (p<0.0001) (c) at glucose concentrations above and below 5 mmol. d,e Confocal microscopy. Mean branch point (d) and fibre number (e) per unit volume of clot (27,000 μm3); significant differences between all groups (p<0.0001) except between 15 and 20 mmol glucose. Error bars indicate standard error of the mean
Confocal microscopy
As no significant difference was found (p=1.0) between the structures of the fibrin clots formed in the presence of 0 and 2.5 mmol/l glucose in permeation and turbidity experiments, only 0 mmol/l glucose was used in the analysis of clot structure by confocal microscopy. No significant difference in mean fibre diameter was found (data not shown); however, there were significant differences in fibre number and the extent of fibre branching within the clots formed at different glucose concentrations (Fig. 2). As with permeation and turbidity analysis, data for each parameter had a J-shaped distribution, with a significant increase in fibre density and branch points at 0 mmol glucose and at concentrations above 5 mmol/l. The extremes of glycaemic control (hyperglycaemia/hypoglycaemia) induce changes that result in the formation of clots with a denser, less porous structure.
Factor XIII induced cross-linkage of alpha- and gamma-chains within the fibrin clot
Whilst there was no difference in the rate of γ-chain cross-link formation between the two groups, and no relationship between HbA1c and the rate of γ-chain cross-linkage (data no shown), there were significant differences in α-chain cross-link formation. The rate of reduction in α-monomers (and hence of α-chain cross-linkage by FXIII to form α-polymers) over time was significantly faster in fibrinogen purified from diabetic (mean reduction rate 0.67% min−1) than from control subjects (mean reduction rate 0.5% min−1) (p<0.02), and a significant correlation was observed between α-chain cross-linkage rate and HbA1c (r=+0.35, p<0.0001).
Thrombin-induced cleavage of fibrinopeptides A and B
Although there was no difference in the kinetics of FPA release, there was a significant increase in the catalytic efficiency (K cat/K m) with which thrombin cleaved FPB from fibrinogen purified from diabetic subjects compared with that from control subjects, which was due to an increase in both the catalytic constant of the reaction and the affinity of thrombin for the fibrinogen β-chain, as shown by a decrease in K m (Table 5).
Acute glycaemic control
The activities of factor XIII to form covalent cross-links within the fibrin clot and thrombin to cleave fibrinopeptides from fibrinogen to initiate polymerisation were not influenced by the concentration of ambient glucose present during fibrin clot formation (data not shown).
Discussion
The present study was undertaken to determine whether the structure/function of the fibrin clot is influenced by type 2 diabetes. Although previous groups have demonstrated changes in clot structure to occur in association with type 1 diabetes [17, 18], these studies were performed on clots formed from plasma, so it cannot be ruled out that other constitutive plasma proteins (such as albumin and fibronectin [30, 31]) and other plasma constituents (the levels or structures of which may become altered in association with the metabolic changes that occur in diabetes) played a role in causing the changes in clot structure found in those studies. By using fibrinogen purified from diabetic and control subjects, we were able to assess the effect of posttranslational modifications of fibrinogen that occur in vivo in association with type 2 diabetes, while eliminating the potential impact of other proteins/plasma constituents, which would confound the results had a plasma system been used.
Our data indicate that the structure/function of the fibrin clot is altered in type 2 diabetes. Fibrin clots formed by diabetic subjects have a denser, less porous structure than those formed by controls, and the structural changes found appear to relate to the subjects’ glycaemic control (HbA1c). As chronic glycaemic control worsens, the clots are formed more rapidly (shorter lag phase) and have a structure that is less dense (increased fibre and branch point number per unit volume) and less porous (lower K s values). The lack of a significant difference in fibre diameters between the two groups, despite a correlation existing between fibre diameter and glycaemic control, may reflect the resolution limitations of confocal microscopy. Clot structure could be further assessed using scanning electron microscopy; however, clots are observed in a dehydrated state in this technique and the results obtained are therefore not directly comparable with those obtained with other techniques used to assess clot structure, and the relation of such results to the in vivo state would be harder to define. A number of different treatment modalities were used to control blood glucose levels in the diabetic subjects studied. Although previous in vitro studies undertaken by our group and others have found the addition of oral hypoglycaemic agents to purified fibrinogen/plasma to influence the structure of the fibrin clot that is formed [18, 28], our experiments were performed in a purified system, and so the various drugs would only be able to influence clot structure if they copurified with fibrinogen (by binding with high affinity) or if they directly influenced the fibrinogen molecule itself in vivo (posttranslational modification). The consistency of the fibrin structural changes amongst the diabetic subjects (irrespective of their treatment modality) suggests that the influence of medication on the changes found is likely to be minor; however, the influence of oral hypoglycaemic agents on the regulation of the coagulation cascade in vivo is an area worthy of further study.
In separate experiments, the structure of the fibrin clot was also found to be influenced by the ambient glucose concentration present during their formation. Glucose levels either above or below the equivalent of normoglycaemic values in vivo are associated with an increase in lag phase and the formation of a clot with a denser structure (increased fibre density and branching, and smaller intrinsic pores).
Fibrinogen circulates in the plasma as a dimer of three polypeptide chains, Aα, Bβ and γ, linked by disulphide bonds [32]. Fibrin clot formation is initiated by the cleavage of FPA from the Aα chains by thrombin, to yield fibrin monomers which spontaneously assemble into protofibrils, which in turn aggregate together to form fibres. The cleavage of FPB from the Bβ chains by thrombin occurs at a slower rate, and appears to be associated with the lateral aggregation of protofibrils [33]. FXIII catalyses the formation of covalent bonds between adjacent α- and γ-chains within fibres, enhancing the clot’s mechanical strength. Factors which influence the intermolecular interactions between the components involved in clot formation (e.g. fibrinogen, thrombin and FXIII) could in consequence influence the structure of the formed clot.
In type 2 diabetic subjects the faster, more extensive FXIII-induced α-chain cross-linkage that was found to occur within the fibrin clots formed (changes that correlated with glycaemic control) may result from an increase in FXIII activity. This enhanced activity may result from an enhancement of the cofactor activity of fibrin [34] for thrombin-induced activation of FXIII resulting from the modifications to fibrin(ogen) occurring in subjects with type 2 diabetes, although further assessment is needed to clarify the potential mechanisms involved. The lack of influence over γ-chain cross-link formation may reflect the sites on the fibrinogen molecule which are altered as a consequence of poor metabolic control in diabetes. These may be more extensive on the α-chain [35], or more likely to influence α-chain than γ-chain cross-linkage, as six cross-linkage sites exist on the α-chain compared with one on the γ-chain [36].
Associations between FPB cleavage rates, the degree of protofibril lateral aggregation and fibrin clot structure have been reported [37], although the causal relationships underlying these events are not entirely understood [37, 38]. Weisel and Nagaswami [27] have used computer modelling to predict how variation in the different parameters involved in fibrin polymerisation might influence clot structure. In their model, an increased FPB cleavage rate shortens the lag phase and increases the maximum rate of fibre assembly, resulting in the formation of shorter, thicker fibres, consistent with our results. An increased FPB cleavage rate resulting from an enhancement in thrombin’s binding affinity and/or catalytic efficiency for the fibrinogen β-chain could therefore contribute to the changes in fibrin clot structure found, by promoting protofibril aggregation.
The structure of the fibrin clot has been shown to influence its susceptibility to lysis [16]. Whilst individual thin fibrin fibres are cleaved at a faster rate, fibrin networks composed of thin densely packed fibres are lysed more slowly than those with loosely packed thicker fibres [16]. This implies that the tightly packed clots consisting of thicker fibres with a denser, more cross-linked structure that we have found to be formed by diabetic subjects are more resistant to fibrinolysis. An alteration in clot susceptibility to fibrinolysis, together with the profound suppression of fibrinolysis, due to high levels of the fibrinolytic inhibitor PAI-1, which is associated with type 2 diabetes [39], may be of considerable importance in determining the success of both physiological and pharmacological clot lysis, and may contribute to the increased atherothrombotic risk in these patients; indeed the results of clinical studies have shown type 2 diabetes to be a significant predictor of failure of thrombolytic therapy after myocardial infarction [40].
The mechanism(s) by which the structure of the fibrin clot is influenced by the ambient glucose concentration are as yet undetermined. Glycation of the fibrinogen molecule is unlikely to account for these changes, owing to the short incubation time used. Other possible explanations include alterations in fibrin(ogen) conformation and function induced by changes in ‘osmotic stress’ [41]. However, further investigation into potential mechanisms is warranted. The development of hyperglycaemia in patients with acute coronary syndromes is associated with an adverse outcome [42], implying that glucose is an important modifiable risk factor for morbidity and mortality after myocardial infarction. Our results suggest that part of this influence may be exerted via an effect on fibrin clot structure. Prospective epidemiological studies have also found glucose levels to predict the risk of cardiovascular disease mortality. A J-shaped relationship between fasting glucose levels and cardiovascular mortality has been reported, hypo- and hyperglycaemic values being associated with significant increases in cardiovascular risk [43]. A similar J-shaped relationship appears to exist between glucose levels and fibrin clot structure parameters. The extremes (hyper- and hypoglycaemia) induce structural changes associated with hypofibrinolysis [16], and suggest that the relationship between glucose levels and cardiovascular disease risk may result in part from changes in fibrin clot structure.
A potential limitation of our study is that although we worked with purified fibrinogen to exclude the potentially confounding effects of other metabolic changes and of other plasma proteins (which could be also influenced by hyperglycaemia) on fibrin clot structure [44], we did not define the exact changes that occur within the fibrinogen molecule following exposure to hyperglycaemia. Whilst persistent hyperglycaemia is associated with glycation of proteins (including fibrinogen [35]) the absolute level of fibrinogen glycation and the sites of glycation within the fibrinogen molecule have not been determined. Although the in vitro glycation of fibrinogen has been shown to influence its interaction with other coagulation/fibrinolytic proteins [21–23], there are potential limitations to these studies and their conclusions, which arise from the fact that this is a non-physiological process and may therefore result in glycation occurring at more sites per molecule, or at a different profile of sites than would occur in vivo. Poor glycaemic control is associated with other complex metabolic derangements in addition to hyperglycaemia, such as oxidative and carbonyl stress [45], which may alter the structure of the fibrinogen molecule [46], and hence fibrin structure/function. Relating fibrin structural changes to fibrinogen glycation alone, without taking the other associated metabolic influences into account, would not necessarily reflect the in vivo situation. Provisional results of mass spectroscopic analysis of individual fibrinogen chains purified from ten diabetic and ten control subjects (data not shown) appear to confirm this. Whilst the mean mass of each chain (α, β, γ) was significantly greater in diabetic that in control subjects, the actual increase in the mass of individual chains could often not be accounted for by individual glucose molecules, but rather by their subsequent modification by oxidation/reduction and dehydration. The combination of these events may be crucial in determining the final fibrin phenotype.
Our results indicate that the metabolic milieu that develops in association with type 2 diabetes interacts with factors regulating the conversion of fibrinogen to fibrin, and results in the formation of clots whose structure has been previously identified as being more resistant to fibrinolysis. These findings may contribute to understanding of the enhanced cardiovascular risk associated with type 2 diabetes. Further research is warranted to determine the mechanisms underlying these changes and their influence on susceptibility to clot lysis, as this may identify potential therapeutic targets to reduce macrovascular disease.
Abbreviations
- CVD:
-
cardiovascular disease
- FPA:
-
fibrinopeptide A
- FPB:
-
fibrinopeptide B
- FXIII:
-
factor XIII
- IU:
-
international units
- Ks :
-
permeation coefficient
- TBS:
-
TRIS-buffer saline
References
Kannel W, McGee D (2002) Diabetes and cardiovascular disease: the Framingham study. JAMA 241:2035–2038
Kannel W, D’Agostino R, Wilson P, Belanger A, Gagnon D (1990) Diabetes, fibrinogen, and risk of cardiovascular disease: the Framingham experience. Am Heart J 120:672–676
Reaven G (1988) Role of insulin resistance in human disease. Diabetes 37:1595–1607
Ross R (1999) Atherosclerosis—an inflammatory disease. N Engl J Med 340:115–126
Lusis A (2000) Atherosclerosis. Nature 407:233–241
DeWood M, Spores J, Notske R et al (1980) Prevalence of total coronary occlusion during the early hours of transmural myocardial infarction. N Engl J Med 303:897–902
Zack P, Ischinger T, Aker U, Dincer B, Kennedy H (1984) The occurrence of angiographically detected intracoronary thrombus in patients with unstable angina pectoris. Am Heart J 108:1408–1412
Smith E, Alexander K, Massie I (1976) Insoluble ‘fibrin’ in human aortic intima. Quantitative studies on the relationship between insoluble ‘fibrin’, soluble fibrinogen and low density lipoprotein. Atherosclerosis 23:19–39
Bini A, Fenoglio J, Mesa-Tejada R, Kudryk B, Kaplan K (1989) Identification and distribution of fibrinogen, fibrin, and fibrin(ogen) degradation products in atherosclerosis. Arteriosclerosis 9:109–121
Kadish J (1979) Fibrin and atherogenesis—a hypothesis. Atherosclerosis 33:409–413
Bini A, Fenoglio J, Sobel J, Owen J, Fejgl M, Kaplan K (1987) Immunohistochemical characterization of fibrinogen, fibrin I, and fibrin II in human thrombi and atherosclerotic lesions. Blood 69:1038–1045
Thompson W, Smith E (1989) Atherosclerosis and the coagulation system. J Pathol 159:97–106
Ernst E, Resch K (1993) Fibrinogen as a cardiovascular risk factor: a meta-analysis and review of the literature. Ann Intern Med 118:956–963
Dunn E, Ariens R, de Lange M et al (2004) Genetics of fibrin clot structure: a twin study. Blood 103:1735–1740
Fatah K, Silveira A, Tornvall P, Karpe F, Blomback M, Hamsten A (1996) Proneness to formation of tight and rigid fibrin gel structures in men with myocardial infarction at a young age. Thromb Haemost 76:535–540
Collet J, Park D, Lesty C et al (2000) Influence of fibrin network conformation and fibrin fibre diameter on fibrinolysis speed: dynamic and structural analysis approaches by confocal microscopy. Arterioscler Thromb Vasc Biol 20:1354–1361
Jorneskog G, Egberg N, Fagrell B et al (1996) Altered properties of the fibrin gel structure in patients with IDDM. Diabetologia 39:1519–1523
Nair C, Azhar A, Wilson J, Dhall D (1991) Studies of fibrin network structure in human plasma, part II—clinical application: diabetes and anti-diabetic drugs. Thromb Res 64:477–485
Gabriel D, Muga K, Boothroyd E (1992) The effect of fibrin structure on fibrinolysis. J Biol Chem 267:24259–24263
Lutjens A, te Velde A, v.d. Veen E, v.d. Meer J (1985) Glycosylation of human fibrinogen in vivo. Diabetologia 28:87–89
Brownlee M, Vlassara H, Cerami A (1983) Nonenzymatic glycosylation reduces the susceptibility of fibrin to degradation by plasmin. Diabetes 32:680–684
Ney K, Pasqua J, Colley K, Guthrow C, Pizzo S (1985) In vitro preparation of nonenzymatically glycosylated human transferrin, alpha2-macroglobulin, and fibrinogen with preservation of function. Diabetes 34:462–470
Bobbink I, Tekelenburg W, Sixma J, de Boer H, Banga J, de Groot P (1997) Glycated proteins modulate tissue-plasminogen activator-catalyzed plasminogen activation. Biochem Biophys Res Commun 240:595–601
Takebe M, Soe G, Kohno I, Sugo T, Matsuda M (1995) Calcium ion-dependent monoclonal antibody against human fibrinogen: preparation, characterization, and application to fibrinogen purification. Thromb Haemost 73:662–667
Ariens R, Philippou H, Nagaswami C, Weisel J, Lane D, Grant PJ (2000) The factor XIII V34L polymorphism accelerates thrombin activation of factor XIII and affects cross-linked fibrin structure. Blood 96:988–995
Mills J, Ariens R, Mansfield M, Grant PJ (2002) Altered fibrin clot structure in healthy relatives of patients with premature coronary artery disease. Circulation 106:1938–1942
Weisel J, Nagaswami C (1992) Computer modelling of fibrin polymerization kinetics correlated with electron microscope and turbidity observations: clot structure and assembly are kinetically controlled. Biophys J 63:111–128
Standeven K, Ariens R, Whitaker P, Ashcroft A, Weisel J, Grant PJ (2002) The effect of dimethylbiguanide on thrombin activity, FXIII activation, fibrin polymerization, and fibrin clot formation. Diabetes 51:189–197
Janus T, Lewis S, Lorand L, Shafer J (1983) Promotion of thrombin-catalyzed activation of factor XIII by fibrinogen. Biochemistry 22:6269–6272
Wilf J, Gladner J, Minton A (1985) Acceleration of fibrin gel formation by unrelated proteins. Thromb Res 37:681–688
Torbet J (1986) Fibrin assembly in human plasma and fibrinogen/albumin mixtures. Biochemistry 25:5309–5314
Doolittle R (1984) Fibrinogen and fibrin. Annu Rev Biochem 53:195–229
Weisel J (1986) Fibrin assembly: lateral aggregation and the role of the two pairs of fibrinopeptides. Biophys J 50:1079–1083
Muszbek L, Adany R, Mikkola H (1996) Novel aspects of blood coagulation factor XIII: I. Structure, distribution, activation, and function. Crit Rev Clin Lab Sci 33:357–421
McVerry B, Thorpe S, Joe F, Gaffney P, Huehns E (1981) Non-enzymatic glycation of fibrinogen. Haemostasis 10:261–270
Lorand L (2001) Factor XIII: structure, activation, and interactions with fibrinogen and fibrin. Ann NY Acad Sci 936:291–311
Weisel J, Veklich Y, Gorkun O (1993) The sequence of cleavage of fibrinopeptides from fibrinogen is important for protofibril formation and enhancement of lateral aggregation in fibrin clots. J Mol Biol 232:285–297
Weisel J, Medved L (2001) The structure and function of the alpha-C domains of fibrinogen. Ann NY Acad Sci 936:312–327
Juhan-Vague I, Roul C, Alessi M, Ardissone J, Heim M, Vague P (1989) Increased plasminogen activator inhibitor activity in non insulin dependent diabetic patients. Relationship with plasma insulin. Thromb Haemost 61:370–373
Zairis M, Lyras A, Makrygiannis S et al (2004) Type 2 diabetes and intravenous thrombolysis outcome in the setting of ST elevation myocardial infarction. Diabetes Care 27:967–971
Rand R (2004) Probing the role of water in protein conformation and function. Philos Trans R Soc Lond B Biol Sci 359:1277–1285
Capes S, Hunt D, Malmberg K, Gerstein H (2000) Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet 355:773–778
Wei M, Gibbons L, Mitchell T, Kampert J, Stern M, Blair S (2000) Low fasting plasma glucose level as a predictor of cardiovascular disease and all-cause mortality. Circulation 101:2047–2052
Galanakis D (1992) Anticoagulant albumin fragments that bind to fibrinogen/fibrin: possible implications. Semin Thromb Hemost 18:44–52
Baynes J, Thorpe S (1999) Role of oxidative stress in diabetic complications. A new perspective on an old paradigm. Diabetes 48:1–9
Shacter E, Williams J, Levine R (1995) Oxidative modification of fibrinogen inhibits thrombin catalyzed clot formation. Free Radic Biol Med 18:815–821
Acknowledgements
Dr E. Dunn is funded by Diabetes UK.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dunn, E.J., Ariëns, R.A.S. & Grant, P.J. The influence of type 2 diabetes on fibrin structure and function. Diabetologia 48, 1198–1206 (2005). https://doi.org/10.1007/s00125-005-1742-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-005-1742-2