
Abstract
Aims/hypothesis
The acute-phase proteins, serum amyloid As (SAA), are precursors of amyloid A, involved in the pathogenesis of AA amyloidosis. This work started with the characterisation of systemic AA amyloidosis concurrent with SAA overexpression in the subcutaneous white adipose tissue (sWAT) of an obese patient with a leptin receptor deficiency. In the present study a series of histopathological, cellular and gene expression studies was performed to assess the importance of SAA in common obesity and its possible production by mature adipocytes.
Materials and methods
Gene expression profiling was performed in the sWAT of two extremely obese patients with a leptin receptor deficiency. Levels of the mRNAs of the different SAA isoforms were quantified in sWAT cellular fractions from lean subjects and from obese subjects before and after a very-low-calorie diet. These values were subsequently compared with serum levels of SAA in these individuals. In addition, histopathological analyses of sWAT were performed in lean and obese subjects.
Results
In sWAT, the expression of SAA is more than 20-fold higher in mature adipocytes than in the cells of the stroma vascular fraction (p<0.01). Levels of SAA mRNA expression and circulating levels of the protein are sixfold (p<0.001) and 3.5-fold (p<0.01) higher in obese subjects than in lean subjects, respectively. In lean subjects, 5% of adipocytes are immunoreactive for SAA, whereas the corresponding value is greater than 20% in obese subjects. Caloric restriction results in decreases of 45–75% in levels of the transcripts for the SAA isoforms and in circulating levels of the protein.
Conclusions/interpretation
The results of the present study indicate that SAA is expressed by sWAT, and its production at this site is regulated by nutritional status. If amyloidosis is seen in the context of obesity, it is possible that production of SAA by adipocytes could be a contributory factor.
Similar content being viewed by others

Introduction
Obesity is associated with increased circulating levels of inflammatory markers such as acute-phase proteins and cytokines [1–5]. Increasing evidence suggests that cytokines released from adipose tissue contribute to the development of complications related to obesity, including metabolic [6, 7] and cardiovascular diseases [8, 9]. Indeed, adipose cells have been shown to secrete a wide number of proinflammatory cytokines (i.e. TNF-α, TGF-β, interferon γ and interleukins) and chemokines (macrophage inflammatory protein-1α, monocyte chemotactic protein-1) [10–13]. It was recently shown that macrophage infiltration is increased in the adipose tissue of obese rodents and represents an important source of inflammation in this tissue [14, 15]. Like atherosclerosis [16], obesity may be viewed as a metabolic as well as an inflammatory disease.
At present there is no clinical evidence of a direct relationship between obesity and inflammation-related diseases in humans. It is not known whether the production of inflammatory mediators by adipose tissue in extreme forms of adiposity may increase susceptibility to specific pathogenic processes [17]. Gene expression profiling of human genetic forms of obesity characterised by extreme adiposity may help to identify the consequences of early-onset fat mass accumulation on adipose tissue gene expression and the development of inflammation-related complications.
We have previously reported the clinical and biological features associated with extreme obesity due to a leptin receptor (LEPR) deficiency [18]. Since the identification of the mutation, the two sisters who participated in this study have been under regular clinical care, and adipose tissue studies were continued in order to gain further insight into this very severe clinical situation that was observed to progressively deteriorate.
More recently, we showed that the expression of key transcription factors involved in adipose tissue differentiation was normal in these two subjects [19]. In the present investigation we performed adipose tissue gene expression studies on a larger scale with no a priori hypothesis using DNA microarray technology in this human model of extreme obesity. This led to the unexpected finding of serum amyloid A (SAA) overexpression in subcutaneous white adipose tissue (sWAT) in both patients, concurrent with clinical and biological features of systemic amyloidosis. Based on these findings we performed a series of histopathological, cellular and gene expression studies to assess for the first time the importance of SAA in common forms of obesity and to determine whether the increased SAA expression was specific to extreme obesity due to LEPR deficiency.
Materials and methods
Clinical investigation protocols
The study included several groups of subjects. All subjects were premenopausal Caucasian women. None of the subjects had a familial or personal history of diabetes, and none were taking medication. All of the subjects had stable body weights, were at their maximal peak weight, and were not on a restrictive diet at entry to the different studies (Table 1). None were involved in an exercise programme. All clinical investigations were performed according to the Declaration of Helsinki and were approved by the ethical committees of the Hôtel-Dieu Hospital (Paris, France), Toulouse University Hospitals (Toulouse, France), the Third Faculty of Medicine (Charles University, Prague, Czech Republic) and Maastricht University (Maastricht, The Netherlands). Informed consent was obtained from all patients.
In the first set of experiments, the transcriptional profiles of sWAT samples from the two LEPR-mutated women (Patient 1 and Patient 2) were compared with those of sWAT samples from a group of healthy obese women (denoted as ‘reference pool’) who had circulating leptin levels consistent with their corpulence (range 44–98 ng/ml) (‘Gene profiling’ column in Table 1). This group of morbidly obese women were followed by the Department of Nutrition of the Hôtel-Dieu Hospital. The clinical characteristics of the LEPR-mutated subjects have been described previously [18, 19]. For Patient 1, sWAT abdominal biopsies were performed and metabolic parameters were determined in 1997 and 2000, when the patient was 19 and 22 years old, respectively. Samples of sWAT were collected from Patient 2 during an abdominal lipectomy performed in 2001, when the patient was 17 years old, justified by the mechanical consequences of extreme adiposity.
In the second set of experiments we investigated the regulation of SAA in weight-stable commonly obese patients and in non-obese subjects (‘Obese and control subjects’ column in Table 1), as well as during a nutritional challenge in obese women (‘Response to a very-low-calorie diet’ column in Table 1). The group of 34 healthy obese patients were prospectively recruited by the Department of Nutrition at the Hôtel-Dieu Hospital. Patients in this group were different to the obese women constituting the reference pool mentioned above. The 27 lean subjects (control subjects) were volunteers recruited by the Obesity Research Unit at Toulouse Hospital and the Department of Nutrition at the Hôtel-Dieu Hospital. Biopsy specimens of sWAT were obtained from a subset of nine control subjects (‘Control subjects, basal’ column in Table 1).
In order to assess the changes in the expression of the gene encoding SAA and circulating levels of the protein after caloric restriction, a group of 21 obese volunteers received a very-low-calorie diet (3.35 kJ/day) for 28 days at the Department of Sports Medicine and Obesity Unit at Charles University. Biopsy specimens of sWAT were obtained at Day 1 and Day 28 in a subset of ten patients (‘Response to a very-low-calorie diet’ column in Table 1).
The third set of experiments aimed to analyse the effects of a leptin injection on inflammatory markers and SAA expression. Six healthy male volunteers (mean age 25±5 years, mean BMI 24±1.7 kg/m2) recruited at the Department of Human Biology at Maastricht University (Maastricht, The Netherlands) received an injection of 60 mg of pegylated recombinant leptin (PEG-leptin) for 3 days. Blood and sWAT samples were obtained before and after leptin treatment (when plasma leptin concentrations were at their peak).
In the three sets of experiments, sWAT samples were obtained by needle biopsy from the peri-umbilical area under local anaesthesia (1% xylocaine) after an overnight fast. The same standard operating procedures were followed at the different participating centres for all groups studied. Biopsy specimens of sWAT were washed with sterile 0.9% sodium chloride solution, immediately frozen in liquid nitrogen and stored at −80°C until analysis.
Gene expression profiling
Total RNA was prepared using the RNeasy total RNA Mini Kit (Qiagen, Courtaboeuf, France). The RNA concentration and integrity of all individual samples were assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies, Massy, France). Aliquots (15 μg) of total RNA from each of the 14 obese women (see Table 1 for clinical data) without a LEPR mutation were pooled to provide a reference for microarray experiments. The MessageAmp RNA Kit (Ambion, Austin, TX, USA) was used to amplify mRNA [20, 21]. The CyScribe First-Strand cDNA Labelling Kit (Amersham Biosciences, Orsay, France) was used to label 15 μg of amplified RNA. The amplified RNA from the pool of obese women was labelled with Cy3, whereas the amplified RNA from the two patients with a LEPR mutation was labelled with Cy5. A duplicate of each sample of amplified RNA from the LEPR-mutated patients was hybridised against the amplified RNA from the pool (i.e. four hybridisations). The labelled cDNA mixtures were hybridised according to the protocol described at http://cmgm.stanford.edu/pbrown/protocols/index (accessed 8 March 2004). The cDNA microarrays consisted of PCR-amplified cDNAs printed on glass slides with 42,786 spots representing 29,308 UniGene clusters. The procedure has previously been described in detail [20, 21]. Briefly, after scanning the four arrays, the resulting images were analysed using Stanford Microarray Database Online resources (http://genome-www5.stanford.edu/MicroArray/SMD, accessed 8 March 2004). A uniform scale factor was applied to normalised signal intensities between Cy5 and Cy3, and then the data were filtered. Using an average intensity 2.5 times above the background, 21,471 cDNA ratios were recovered for all four experiments. After normalisation in log-space to set the mean to 0 and the standard deviation to 1, the data were analysed using the significance analysis of microarray (SAM) procedure (http://www-stat.stanford.edu/~tibs/SAM, accessed 8 March 2004). This method provides a list of significant genes and an estimate of the false discovery rate (FDR), which represents the percentage of genes that could have been identified by chance [22].
Quantitation of mRNA levels
Quantitative RT-PCR was performed using the GeneAmp 7000 Sequence Detection System (Applied Biosystems, Courtaboeuf, France) as previously described [20]. We used 18S rRNA as the control. Because of the very high degree of homology between SAA1 and SAA2, the primer pairs designed for SAA1 mRNA also detected SAA2 mRNA. Measurements of SAA2 and SAA4 mRNA levels were specific.
Studies on adipocytes and cell types of the stroma vascular fraction
Samples of sWAT were obtained from nine female subjects undergoing abdominal lipectomy in accordance with French laws on biomedical research. The specimens of subcutaneous fat were digested using collagenase (type 1; Biochrom, Berlin, Germany) as described previously [23]. Mature adipocytes and the stroma vascular fraction (SVF) were separated by centrifugation, lysed with denaturing buffer from the RNeasy total RNA Mini Kit (Qiagen), and then stored at −80°C prior to mRNA preparation.
Histological, immunohistochemical and ultrastructural analysis of adipose tissue
All samples were fixed in 4% paraformaldehyde, dehydrated, paraffin embedded and sectioned (thin sections of 5 μm). Biopsy specimens obtained from the kidney, antrum and duodenum of Patient 1 and sWAT samples from Patient 2 were processed for haematoxylin–eosin staining, for Congo-red staining (examined using light and polarised microscopy), and for immunohistochemical analysis of SAA protein as described below. Since morphological analyses cannot be performed on samples collected by needle biopsies, sWAT samples from seven morbidly obese patients (mean BMI 47.55±5 kg/m2), five obese patients (mean BMI 33.1±3.3 kg/m2) and two non-obese subjects (BMI 20 and 21 kg/m2) were obtained during plastic surgery performed at the Hôtel-Dieu Hospital. These subjects were different to the nine obese subjects described in the section ‘Studies on adipocytes and cell types of the stroma vascular fraction’. All samples of adipose tissue, skeletal muscle and gut (used as negative control), and a ganglia biopsy (positive control) from a deceased patient with amyloidosis as the cause of death, were stained with haematoxylin–eosin or processed for immunohistochemical detection of SAA proteins with an anti-SAA antibody (Dako, Trappes, France) according to a standard immunoperoxidase procedure. Adipose tissue explants (300 mg) from one morbidly subject (BMI 49 kg/m2) and two obese subjects (BMI 30.9 and 31 kg/m2) were incubated in 3 ml of DMEM containing 1% BSA, penicillin (100 U/ml), streptomycin (100 mg/ml) and Protease Inhibitor Cocktail (Roche Diagnostics, Germany) for 24. Following incubation, supernatants were collected and SAA secretion was measured using the ELISA Cytoscreen Immunoassay Kit (BioSource International, Camarilla, CA, USA).
Ultrastructural analysis (by transmission electron microscopy) of sWAT was also performed for Patient 2 and for 16 obese premenopausal female patients (mean BMI 43.5±7.5 kg/m2) from the Institute of Normal Human Morphology–Anatomy at the University of Ancona (Italy). Small fragments of sWAT were processed for transmission electron microscopy as described previously [24]. Electron microscopy was performed on several sections (at least four sections) of random samples of the tissue. Comparison of light microscopy sections with resin-embedded sections ensured that representative areas were studied.
Biochemical analysis
Concentrations of SAA were measured using the ELISA Cytoscreen Immunoassay Kit (BioSource International) [25]. Serum leptin concentrations were determined using a standard kit (Linco Research, St. Charles, MO, USA). Levels of IL-6 were evaluated using ELISA (Quantikine HS600B; R&D Systems Europe, Abingdon, UK). Plasma C-reactive protein was measured by an immunoturbidity method using an Olympus AU600 automated analyser (reference OS6147; Olympus Optical, Tokyo, Japan).
Statistical analysis
Data are expressed as means±SEM. Statistical analysis was performed using JMP statistical software (SAS Institute, Cary, NC, USA). Comparisons between mRNA levels before and after caloric restriction, before and after leptin administration, and between SAA plasma levels before and after caloric restriction were made using a non-parametric test (Wilcoxon signed-rank test). Plasma SAA concentrations in obese and non-obese groups were compared using the Mann–Whitney rank test. The significance of correlations between the levels of SAA circulating levels and clinical parameters was calculated using the non-parametric Spearman’s rank correlation test. A p value of less than 0.05 was considered statistically significant.
Results
Overexpression of SAA in LEPR-deficient women
The sWAT transcriptional profiles of the two patients with extreme obesity due to LEPR mutation were compared with those of 14 morbidly obese women (see Table 1 for clinical data). The SAM statistical procedure was used to control for multiple testing [22]. Of 21,471 cDNAs with signals recovered in all experiments and an estimated FDR of <1.2%, SAM isolated 645 cDNAs, among which 349 were overexpressed (mean fold change 1.8, range 1.5–3.5) and 296 were underexpressed (mean fold change 1.6, range 1.3–4.5) in the two LEPR-mutated patients. Fifty-four percent of the differentially expressed genes were expressed sequence tags (ESTs) or hypothetical proteins. Genes encoding immune and inflammatory response proteins were among the main categories represented; 10% of the differentially expressed genes were members of this category (see Table 1 of the electronic supplementary material). Four transcripts of plasma acute-phase proteins (SAA1, SAA2, SAA4 and haptoglobin) were overexpressed (fold change range 2.1–3.5), and SAAs were among the most highly overexpressed transcripts. The changes in concentrations of SAA1, SAA2 and SAA4 mRNAs were assessed using quantitative RT-PCR. The results confirmed the microarray data for SAA2 (fold change 1.3 and 1.7) and SAA4 (fold change 1.5 and 1.9) in Patient 1 and Patient 2, respectively. Combined detection of SAA1 and SAA2 mRNA showed that levels were increased (fold change 1.3) in Patient 2.
Overexpression of SAA in sWAT and systemic amyloidosis
Samples of sWAT were obtained from Patient 1 (LEPR-mutated subject) in 1997 and in 2000. Over this period of time the clinical condition of the patient deteriorated. In 1997, Patient 1 had proteinuria levels of <1 g/24 h; however, she rapidly developed a nephrotic syndrome in 2000, and developed renal insufficiency leading to dialysis 1 year later. An examination of the kidney biopsy under light (Fig. 1a), by polarised microscopy (data not shown), and by immunohistochemistry (Fig. 1b) using specific anti-SAA antibodies led unambiguously to the diagnosis of kidney AA amyloidosis. Amyloid A deposits were found in the duodenum and antrum (Fig. 1c, d). Exhaustive clinical, biological and radiological investigations did not detect any common cause of secondary amyloidosis (infection, neoplasia, inflammatory disease such as arthritis) at the time of diagnosis or during the 9 years of follow-up. The patient did not experience recurrent attacks of fever, pain or skin lesions, or any other symptoms indicative of hereditary periodic fever [26]. We found no evidence of mutations associated with hereditary periodic syndromes [27, 28].

Amyloid deposits in kidney and intestinal biopsies of Patient 1. a Congo-red-stained renal glomerulus examined by light microscopy (×20). b Deposits show immunoreactivity to anti-SAA antibodies (×10). c Congo-red-stained fragments of duodenum show large vascular SAA deposits (×20). d Congo-red-stained antrum biopsy shows vascular deposits (×20)
Since the mRNAs of all SAA isoforms were overexpressed in Patient 2, the younger patient, relative to the reference pool of morbidly obese women, we performed a morphological analysis of sWAT in order to determine whether any early features of amyloidosis and/or inflammation were present. Immunohistochemical investigations revealed the presence of SAA proteins within well-differentiated adipocyte cytoplasm (Fig. 2). Morphological features of inflammation were demonstrated on histological examination. Approximately 40% of the small arterioles had been infiltrated by lymphocytes, and most of them had thickened walls (data not shown). Electron microscopy revealed features unique to this patient compared with the 16 morbidly obese control subjects. Vascular walls and all intercellular spaces, including the narrow intercellular space between adipocytes, had been infiltrated by collagen fibrils (Fig. 3a), and activated fibroblasts were identified (Fig. 3b). A clear increase in collagen arranged in thick strands surrounding areas of variable size composed of unilocular small adipocytes was also observed (Fig. 3c–e). In the absence of any alteration in renal function in Patient 2, no renal biopsy has been performed for this subject as yet.

Amyloid protein in adipocytes of Patient 2. a The cytoplasm of mature adipocytes shows immunoreactivity to anti-SAA antibodies (×40). b Adipocytes are immunoreactive in the perinuclear area (×100)

Electron microscopy morphological analysis of sWAT of Patient 2. a Adipose tissue structure with three adipocytes indicated by an asterisk (×1,000). b Higher magnification of the black square shown in a (×4,900). The f indicates an activated fibroblast that produces collagen fibrils. c Numerous collagen fibrils are present and infiltrate the interstitial space between adipocytes (×2,570). d Higher magnification of the black square shown in c (×14,000). e Higher magnification of the black square shown in d (×35,000). The arrow indicates a collagen fibril
In order to document the low-grade inflammatory status of the two LEPR-mutated subjects, we measured plasma circulating levels of SAA on several occasions and evaluated other inflammation-related parameters. Plasma SAA concentrations were moderately increased in Patient 2 (range 12–30 μg/ml) and Patient 1 (range 10–60 μg/ml) compared with those in non-obese subjects (4.9±2.3 μg/ml, range 0.8–16.6 μg/ml), and were within the upper range of those in subjects with common obesity (Fig. 4a). Circulating levels of IL6 were also increased in the LEPR-mutated patients (5.7 and 6 pg/ml) compared with those in control subjects (2.1±1.5 pg/ml) and obese subjects (2.8±0.2 pg/ml) (‘Obese and control subjects’ in Table 1). Levels of C-reactive protein were also increased in Patient 2 (range 16–22 mg/l) relative to those in non-obese subjects (1.3±0.2 mg/l) and obese subjects (5±2.5 mg/l).

Circulating levels of SAA and SAA mRNA expression in sWAT of control subjects (open bars), obese subjects from the ‘Obese and control subjects’ group in Table 1 (grey bar), obese subjects from the ‘Response to a very-low-calorie diet’ group in Table 1 before the diet (black bars) and after the diet (striped bars). The data are means±SEM. a Concentration of SAA in 34 obese subjects, in 27 non-obese subjects, and in 21 obese patients before and after a very-low-calorie diet. *p<0·01; ***p<0.0002. b Expression of transcripts for the SAA isoforms (SAA1+2, SAA2, SAA4) in sWAT of nine control subjects and 10 obese women before and after a very-low-calorie diet. **p<0.001; NS=0.1
Expression of SAA in mature adipose cells of obese subjects
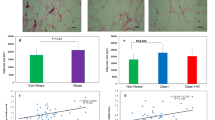
We wished to determine whether the SAA detected in adipose tissue was produced by adipocytes or by the cells of the SVF, which include preadipocytes, endothelial cells and monocytes/macrophages. We first investigated the presence and relative mRNA levels of all SAA isoforms. The expression of all SAA isoform mRNAs was higher in mature adipocytes than in SVF cells. The ratio of adipocyte/SVF concentrations was 49±24 (range 10–192) for SAA1+SAA2, 70±35 (range 10–275) for SAA2, and 22±7 (range 5–62) for SAA4 (p<0.01). The results of the gene expression analysis were confirmed by immunohistochemical detection of SAA in sections of sWAT. Strong positive staining was observed in the perinuclear area where the cytoplasm is richer in Golgi complexes and endoplasmic reticulum (Figs. 2, 5). In contrast, all the other cell types (fibroblasts, endothelial cells, smooth muscle cells of the vessel walls) tested negative, clearly indicating that the SAA protein detected in adipose tissue was derived from fully mature adipocytes.

Immunohistochemical detection of SAA protein in sWAT of obese subjects. Strong positive staining was observed in ganglia (a) (×20) and sWAT (b) (×40) of an amyloidosis-deceased patient, used as positive control. Gut (c) (×20) and skeletal muscle (d) (×20) from a massively obese patient (BMI 57 kg/m2) stained negative. In sWAT from a non-obese patient (BMI 20 kg/m2) rare, weakly positive adipocytes were observed (e) (×100), while a stronger positivity and a high number of positive adipocytes were found in obese (BMI 33 kg/m2) (f) (×100) and morbidly obese patients (BMI 57 kg/m2) (g) (×20) and (h) (×100). Capillaries are indicated by an asterisk, nuclei by a small n, and the arrow indicates cytoplasmic perinuclear positivity
Expression of SAA in common forms of obesity and variation with nutritional status
We further investigated whether adipose cell SAA overproduction was a feature specific to morbid obesity caused by LEPR mutation or was more generally related to the excess fat mass seen in common obesity. Expression of SAA in nine non-obese individuals was compared with that in ten weight-stable obese subjects before caloric restriction (‘Response to a very-low-calorie diet’ subjects in Table 1). As shown in Fig. 4b, the average levels of the transcripts for the SAA isoforms were statistically different between the obese and non-obese subjects. In all subjects, significant correlations were found between the mRNA levels of the three different SAA isoforms (r=0.9, p<0.001 for SAA1 vs SAA2; r=0.84, p<0.001 for SAA2 vs SAA4; and r=0.89, p<0.001 for SAA1 vs SAA4). Immunohistochemical analysis of sWAT samples obtained from obese and non-obese subjects showed that, in non-obese subjects, <5% of the adipose cells were immunoreactive for SAA, whereas the corresponding frequency in obese subjects was >20% (Table 2). Interestingly, the level of SAA immunoreactivity observed in morbidly obese subjects was comparable to that observed in Patient 2 (Table 2). We also detected the SAA protein in the media in which adipose tissue explants from three obese subjects undergoing plastic surgery were incubated. Over a 24-h period, 31.4 ng of SAA were released per gram of adipose tissue from explants from a morbidly obese subject (BMI 49 kg/m2). The corresponding figures for two obese subjects, one with a BMI of 30.9 kg/m2, the other with a BMI of 31 kg/m2, were 3.6 and 7.6 ng/g of adipose tissue, respectively.
Over the course of a 4-week very-low-calorie diet that induced a substantial decrease in body weight (‘Response to a very-low-calorie diet’ column in Table 1) the expression of the transcripts for all of the SAA isoforms decreased by 45–75% (Fig. 4b). The data on SAA expression in sWAT were consistent with the plasma levels of SAA (Fig. 4a). Circulating levels of SAA were significantly higher in the 34 obese patients (16.4±1.7 μg/ml, range 0.9–67 μg/ml) than in the 27 non-obese subjects (4.9±2.3 μg/ml, range 0.8–16.6 μg/ml) (Fig. 4a; ‘Obese and control subjects’ in Table 1). There was a significant correlation between circulating SAA levels and BMI (r=0.73, p<10−4). No relationship was observed between the age of the subject and the level of SAA expression. There were no differences between non-obese healthy men and women in terms of the concentration of circulating SAA levels (p=0.7).
Caloric restriction significantly reduced SAA plasma levels (Fig. 4a) in the 21 obese subjects who followed the caloric restriction protocol (‘Response to a very-low-calorie diet’ subjects in Table 1). In addition, we observed a significant correlation between the amount of weight loss and the change in circulating levels of SAA in this group (r=0.55, p=0.01).
SAA expression after leptin administration
We next studied the influence of the injection of leptin on SAA expression in sWAT. To avoid the confounding effect of obesity-induced leptin resistance, leptin was administered to six healthy subjects at a concentration that would produce a supraphysiological blood level of leptin. After 3 days of treatment, the mean serum leptin concentration increased from 3±1 μmol/l to 2112±186 μmol/l (p<0.016); however, the expression of SAA1+2, SAA2 and SAA4 did not change significantly (ratio of after/before leptin 0.83±0.1, 0.93±0.3 and 0.93±0.15, respectively, p>0.05). The measurements of circulating levels of SAA also showed no significant change (p=0.13).
Discussion
The presence of AA amyloidosis together with SAA overexpression in the sWAT of a subject with extreme adiposity due to a LEPR mutation prompted a series of investigations, the results of which indicate that SAA is expressed by adipocytes and is regulated by nutritional changes. This suggests that SAA could contribute to the systemic complications of obesity in humans. The SAA proteins are a family of apolipoproteins that were, until now, thought to be mainly expressed in hepatocytes. They may be involved in cholesterol transport and in the early response to injury. The SAAs are precursors of amyloid A, a constituent of the amyloid fibrils that are involved in AA amyloidosis [29, 30]. This form of amyloidosis is a complication that is associated with many inflammatory conditions (including rheumatoid arthritis and neoplasia). Amyloid deposits have also been found in some chronic diseases, such as Alzheimer’s disease, dialysis-related amyloidosis and atherosclerosis [30]. The causes and effects of amyloid deposition are uncertain in these pathologies, which are all characterised by a low-grade inflammation.
Our data from gene expression and immunohistochemical experiments demonstrate that SAA mRNA and protein are expressed in human adipocytes. Other cell types found in adipose tissue do not seem to significantly contribute to SAA expression. These findings are in agreement with in vitro and in vivo studies performed in rodents. Mouse adipose tissue expresses transcripts of the genes encoding SAA4 and SAA3, which correspond to pseudogenes in humans [31, 32]. The expression of SAA3 in murine 3T3-L1 adipocytes has been shown to be strongly induced by TNF-α and lipopolysaccharides in vitro [31].
In the present study we have demonstrated that the overproduction of SAA in adipose tissue is not exclusive to LEPR-mutated subjects as it is also observed in obese subjects with a functional leptin receptor, particularly in morbidly obese subjects. In addition, weight loss produced by calorie restriction in obese subjects is associated with reductions in levels of SAA mRNA and protein, confirming that SAA concentrations are highly regulated by the nutritional status of the individual. Thus, the overexpression of SAA in LEPR-mutated subjects is due to extreme early onset adiposity as opposed to the absence of leptin signalling. This was confirmed by the lack of SAA regulation observed in normal subjects following the administration of a supraphysiological dose of leptin. In accordance with these results, gene expression profiling in the adipose tissue of ob/ob mice showed that SAA overexpression was only partially corrected by leptin [32].
The relative contributions made by adipose tissue and the liver to the increase in circulating levels of SAA are not known. Using immunohistochemistry we showed that, in mature adipocytes, SAA protein is localised in cytoplasmic regions that are very rich in rough endoplasmic reticulum/Golgi apparatus (the cytoplasmic organelles involved in the synthesis of secreted proteins). Electron microscopy did not reveal any amyloid deposits in the intercellular spaces in sWAT samples from the LEPR-mutated patients. This suggests that, even if SAA is synthesised in adipose tissue, it could be delivered to the systemic circulation together with SAA from other tissues, particularly the liver. Finally, our preliminary observations in three obese subjects revealed that SAA protein is released into the media in which adipose tissue explants are incubated. Together, these observations provide further evidence that adipose tissue contributes to the increased circulating SAA levels observed in morbidly obese subjects.
The biological function of SAA, particularly its role in glucose and lipid metabolism, has yet to be fully established. When associated with HDL particles, SAA protein may impair reverse cholesterol transport, and it has been implicated in the decreased HDL levels observed during inflammation [33]. Transgenic mice overexpressing SAA have been shown to have similar lipoprotein profiles to wild-type mice [34]. In humans, circulating SAA levels were found to be correlated with atherogenic remnant-like lipoprotein particles in type 2 diabetic patients with coronary heart disease [35], and with HbA1c but not with HDL cholesterol in diabetic subjects [36]. The Tanis protein has recently been described as a putative receptor for SAA, and has been demonstrated to be deregulated in diabetic obese rats [37]. It has been shown that the expression of skeletal muscle and adipose tissue Tanis/SelS mRNA is positively correlated with circulating levels of SAA in humans, and that it is induced by insulin infusion in adipose tissue from type 2 diabetic subjects. These data suggest that SAA protein and its putative receptor could be involved in insulin signalling processes [38]. Our microarray data did not provide any evidence for Tanis/SelS regulation in the two LEPR-mutated patients. However, preliminary analysis in subjects with common obesity indicates trends towards correlations between the expression of SAA and fasting insulin status and HDL cholesterol levels (data not shown). These results need to be confirmed and extended to a larger cohort.
The link between the increase of SAA in the context of morbid obesity and the presence of AA amyloidosis in LEPR-mutated subjects is questionable. Although an early study on ob/ob mice reported the occurrence of amyloidosis in a model of massive obesity [39], our observations in LEPR-mutated patients appear to contradict previous findings linking AA amyloidosis to chronically high circulating concentrations of SAA [40]. Repetitive measurements of circulating SAA showed that this protein never reached extremely high levels in Patient 1. This finding is contrast to the peak acute-phase response and the markedly elevated levels of SAA (>1000 mg/l) associated with chronic inflammatory disease. No symptoms indicative of hereditary periodic fever [26, 27] were observed during 9 years of regular exhaustive clinical and biological checkups. At the present time classic inflammation sources of AA amyloidosis can be ruled out. Whether extreme obesity could have contributed to the pathogenesis of AA amyloidosis by creating a low-grade inflammation through the exacerbated production of proinflammatory molecules by adipose and liver tissues remains to be determined.
While increased concentrations of circulating SAA are considered to be a prerequisite for AA amyloidosis, several other mechanisms could be important. Enlargement of fat mass around organs could contribute to a local effect of SAA, as could dysfunction of SAA proteolytic processes and remodelling of the extracellular matrix [30, 41, 42]. Histopathological analysis of sWAT samples from Patient 2 revealed signs of inflammation, including fibroblast activation and infiltration by monocyte/macrophage cells (data not shown), as well extracellular matrix components with an unusual morphology.
Increased SAA mRNA and protein expression in adipocytes and the liver are associated with increased circulating levels of SAA and the increased expression of associated proteins in subjects with common obesity. This suggests that amyloid deposition may be a potential complication of morbid obesity. Secondary amyloidosis leads to proteinuria and microalbuminuria [43], which are urinary features seen in morbid obesity [44, 45]. Histological studies are scarce, particularly in morbid obesity where kidney biopsy is technically difficult. Hence, the presence of amyloidosis has not been systematically investigated.
In conclusion, our work reveals that adipocytes are able to express SAA mRNA and protein. In the present paper we have described a previously unsuspected link between extreme obesity and systemic AA amyloidosis associated with kidney dysfunction. These data suggest that the overproduction of the SAA family of proteins in human adipose tissue may contribute to morbidity in obese individuals. A systematic search for amyloidosis should be carried out on sWAT biopsies when kidney dysfunction secondary to obesity is suspected.
Abbreviations
- FDR:
-
False discovery rate
- LEPR:
-
Leptin receptor
- SAA:
-
Serum amyloid A
- SAM:
-
Significance analysis of microarray
- SVF:
-
Stroma vascular fraction
- sWAT:
-
Subcutaneous white adipose tissue
- VLCD:
-
Very-low-calorie-diet
References
Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB (1999) Elevated C-reactive protein levels in overweight and obese adults. JAMA 282:2131–2135
Engeli S, Feldpausch M, Gorzelniak K et al (2003) Association between adiponectin and mediators of inflammation in obese women. Diabetes 52:942–947
van Dielen FM, van’t Veer C, Schols AM, Soeters PB, Buurman WA, Greve JW (2001) Increased leptin concentrations correlate with increased concentrations of inflammatory markers in morbidly obese individuals. Int J Obes Relat Metab Disord 25:1759–1766
Danesh J, Muir J, Wong YK, Ward M, Gallimore JR, Pepys MB (1999) Risk factors for coronary heart disease and acute-phase proteins. A population-based study. Eur Heart J 20:954–959
Bastard JP, Jardel C, Delattre J, Hainque B, Bruckert E, Oberlin F (1999) Evidence for a link between adipose tissue interleukin-6 content and serum C-reactive protein concentrations in obese subjects. Circulation 99:2221–2222
Marette A (2002) Mediators of cytokine-induced insulin resistance in obesity and other inflammatory settings. Curr Opin Clin Nutr Metab Care 5:377–383
Lyon CJ, Law RE, Hsueh WA (2003) Minireview: adiposity, inflammation, and atherogenesis. Endocrinology 144:2195–2200
Yudkin JS, Kumari M, Humphries SE, Mohamed-Ali V (2000) Inflammation, obesity, stress and coronary heart disease: is interleukin-6 the link? Atherosclerosis 148:209–214
Theuma P, Fonseca VA (2003) Inflammation and emerging risk factors in diabetes mellitus and atherosclerosis. Curr Diab Rep 3:248–254
Gomez-Ambrosi J, Fruhbeck G (2001) Do resistin and resistin-like molecules also link obesity to inflammatory diseases? Ann Intern Med 135:306–307
Gerhardt CC, Romero IA, Cancello R, Camoin L, Strosberg AD (2001) Chemokines control fat accumulation and leptin secretion by cultured human adipocytes. Mol Cell Endocrinol 175:81–92
Rajala MW, Scherer PE (2003) Minireview: the adipocyte—at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology 144:3765–3773
Coppack SW (2001) Pro-inflammatory cytokines and adipose tissue. Proc Nutr Soc 60:349–356
Xu H, Barnes GT, Yang Q et al (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112:1821–1830
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112:1796–1808
Ross R (1999) Atherosclerosis—an inflammatory disease. N Engl J Med 340:115–126
Arsenijevic D, Onuma H, Pecqueur C et al (2000) Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet 26:435–439
Clement K, Vaisse C, Lahlou N et al (1998) A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 392:398–401
Clement K, Vega N, Laville M et al (2002) Adipose tissue gene expression in patients with a loss of function mutation in the leptin receptor. Int J Obes Relat Metab Disord 26:1533–1538
Clement K, Viguerie N, Diehn M et al (2002) In vivo regulation of human skeletal muscle gene expression by thyroid hormone. Genome Res 12:281–291
Rome S, Clement K, Rabasa-Lhoret R et al (2003) Microarray profiling of human skeletal muscle reveals that insulin regulates approximately 800 genes during a hyperinsulinemic clamp. J Biol Chem 278:18063–18068
Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A 98:5116–5121
Viguerie N, Millet L, Avizou S, Vidal H, Larrouy D, Langin D (2002) Regulation of human adipocyte gene expression by thyroid hormone. J Clin Endocrinol Metab 87:630–634
Cinti S, Zingaretti MC, Cancello R, Ceresi E, Ferrara P (2001) Morphologic techniques for the study of brown adipose tissue and white adipose tissue. Methods Mol Biol 155:21–51
McDonald TL, Weber A, Smith JW (1991) A monoclonal antibody sandwich immunoassay for serum amyloid A (SAA) protein. J Immunol Methods 144:149–155
Drenth JP, van der Meer JW (2001) Hereditary periodic fever. N Engl J Med 345:1748–1757
Grateau G (2004) Clinical and genetic aspects of the hereditary periodic fever syndromes. Rheumatology (Oxford) 43:410–415
Hoffman HM, Wanderer AA, Broide DH (2001) Familial cold autoinflammatory syndrome: phenotype and genotype of an autosomal dominant periodic fever. J Allergy Clin Immunol 108:615–620
Urieli-Shoval S, Linke RP, Matzner Y (2000) Expression and function of serum amyloid A, a major acute-phase protein, in normal and disease states. Curr Opin Hematol 7:64–69
Merlini G, Bellotti V (2003) Molecular mechanisms of amyloidosis. N Engl J Med 349:583–596
Lin Y, Rajala MW, Berger JP, Moller DE, Barzilai N, Scherer PE (2001) Hyperglycemia-induced production of acute phase reactants in adipose tissue. J Biol Chem 276:42077–42083
Soukas A, Cohen P, Socci ND, Friedman JM (2000) Leptin-specific patterns of gene expression in white adipose tissue. Genes Dev 14:963–980
Coetzee GA, Strachan AF, van der Westhuyzen DR, Hoppe HC, Jeenah MS, de Beer FC (1986) Serum amyloid A-containing human high density lipoprotein 3. Density, size, and apolipoprotein composition. J Biol Chem 261:9644–9651
Kindy MS, de Beer MC, Yu J, de Beer FC (2000) Expression of mouse acute-phase (SAA1.1) and constitutive (SAA4) serum amyloid A isotypes: influence on lipoprotein profiles. Arterioscler Thromb Vasc Biol 20:1543–1550
Hamano M, Saito M, Eto M et al (2004) Serum amyloid A, C-reactive protein and remnant-like lipoprotein particle cholesterol in type 2 diabetic patients with coronary heart disease. Ann Clin Biochem 41:125–129
Leinonen E, Hurt-Camejo E, Wiklund O, Hulten LM, Hiukka A, Taskinen MR (2003) Insulin resistance and adiposity correlate with acute-phase reaction and soluble cell adhesion molecules in type 2 diabetes. Atherosclerosis 166:387–394
Walder K, Kantham L, McMillan JS et al (2002) Tanis: a link between type 2 diabetes and inflammation? Diabetes 51:1859–1866
Karlsson HK, Tsuchida H, Lake S, Koistinen HA, Krook A (2004) Relationship between serum amyloid A level and Tanis/SelS mRNA expression in skeletal muscle and adipose tissue from healthy and type 2 diabetic subjects. Diabetes 53:1424–1428
Naeser P, Westermark P (1977) Amyloidosis in ageing obese-hyperglycemic mice and their lean litter-mates. A morphological study. Acta Pathol Microbiol Scand A 85:761–767
Gillmore JD, Lovat LB, Persey MR, Pepys MB, Hawkins PN (2001) Amyloid load and clinical outcome in AA amyloidosis in relation to circulating concentration of serum amyloid A protein. Lancet 358:24–29
Buxbaum JN, Tagoe CE (2000) The genetics of the amyloidoses. Annu Rev Med 51:543–569
Migita K, Yamasaki S, Shibatomi K et al (2001) Impaired degradation of serum amyloid A (SAA) protein by cytokine-stimulated monocytes. Clin Exp Immunol 123:408–411
Falk RH, Comenzo RL, Skinner M (1997) The systemic amyloidoses. N Engl J Med 337:898–909
Adelman RD (2002) Obesity and renal disease. Curr Opin Nephrol Hypertens 11:331–335
de Jong PE, Verhave JC, Pinto-Sietsma SJ, Hillege HL (2002) Obesity and target organ damage: the kidney. Int J Obes Relat Metab Disord 26(Suppl 4):S21–S24
Acknowledgements
This work was supported by the Direction de la Recherche Clinique/Assistance Publique-Hopitaux de Paris, Alfediam (clinical research contract), the Programme Hospitalier de Recherche Clinique (AOM 02076), the Servier Research Institute, the Grant Agency of the Ministry of Health (Czech Republic; IGA 4674-3/1998 and IGA 6836-3), the research programme VZ-1 of the Third Faculty of Medicine (Charles University, Prague; MSMT 1112200001), the French Ministry for Foreign Affairs (Barrande programme), and INSERM ATC-nutrition (grant no. 4 NU10G). C. Poitou and K. Clément received a grant from Alfediam/Merck-Lipha and the French Association for Research on Obesity (AFERO), respectively. R. Cancello received support from INSERM, ADR Paris 6, St Antoine and ‘Conseil Regional Ile de France’.
We thank V. Pelloux, G. Pulou and S. Avizou for excellent technical help. The authors are not aware of any conflicts of interest in this study.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Rights and permissions
About this article
Cite this article
Poitou, C., Viguerie, N., Cancello, R. et al. Serum amyloid A: production by human white adipocyte and regulation by obesity and nutrition. Diabetologia 48, 519–528 (2005). https://doi.org/10.1007/s00125-004-1654-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-004-1654-6