
Abstract
Aims/hypothesis
We investigated the effects of rosiglitazone on NEFA and triglyceride metabolism in type 2 diabetes.
Methods
In a double-blind, placebo-controlled, cross-over study of rosiglitazone in diet-treated type 2 diabetic subjects, we measured arteriovenous differences and tissue blood flow in forearm muscle and subcutaneous abdominal adipose tissue, used stable isotope techniques, and analysed gene expression. Responses to a mixed meal containing [1,1,1-13C]tripalmitin were assessed.
Results
Rosiglitazone induced insulin sensitisation without altering fasting NEFA concentrations (−6.6%, p=0.16). Postprandial NEFA concentrations were lowered by rosiglitazone compared with placebo (−21%, p=0.04). Adipose tissue NEFA release was not decreased in the fasting state by rosiglitazone treatment (+24%, p=0.17) and was associated with an increased fasting hormone-sensitive lipase rate of action (+118%, p=0.01). Postprandial triglyceride concentrations were decreased by rosiglitazone treatment (−26%, p<0.01) despite unchanged fasting concentrations. Rosiglitazone did not change concentrations of triglyceride-rich lipoprotein remnants. Adipose tissue blood flow increased with rosiglitazone (+32%, p=0.03). Postprandial triglyceride [13C]palmitic acid concentrations were unchanged, whilst NEFA [13C]palmitic acid concentrations were decreased (p=0.04). In muscle, hexokinase II mRNA expression was increased by rosiglitazone (+166%, p=0.001) whilst the expression of genes involved in insulin signalling was unchanged. Adipose tissue expression of FABP4, LPL and FAT/CD36 was increased.
Conclusions/interpretation
Rosiglitazone decreases postprandial NEFA and triglyceride concentrations. This may represent decreased spillover of NEFAs from adipose tissue depots. Decreased delivery of NEFAs to the liver may lead to lowered postprandial triglyceride concentrations. Upregulation of hexokinase II expression in muscle may contribute to insulin sensitisation by rosiglitazone.
Similar content being viewed by others


Introduction
The thiazolidinedione class of drugs is used to induce insulin sensitisation in insulin resistance. Despite their widespread use in type 2 diabetes, there is still debate over the physiological mechanisms by which thiazolidinediones exert their effects.
Some of the steps of thiazolidinedione action are known. Thiazolidinediones bind to and activate the peroxisome proliferator-activated receptor (PPARγ) nuclear receptor [1]. As a transcription factor, PPARγ regulates the expression of a variety of genes involved in metabolism [2]. A co-ordinated change in gene expression might be expected to increase the ‘efficiency’ of glucose handling and to increase insulin sensitivity.
The mechanisms of action of thiazolidinediones are less clear in vivo in humans: how do thiazolidinediones act on genes highly expressed in adipose tissue, yet induce insulin sensitisation in skeletal muscle? A commonly proposed answer is based on the interaction between adipose tissue and skeletal muscle: the glucose-fatty acid (Randle) cycle [3]. Glucose and fatty acids compete for use as a substrate by skeletal muscle. Thus, if circulating NEFA concentrations are increased, muscle glucose uptake would be less efficient. In this way, an increased output of NEFAs from adipose tissue in obesity would cause insulin resistance [4]. It is thought that thiazolidinediones decrease NEFA output from adipose tissue, thus modulating a signal of insulin resistance between adipose tissue and muscle [5, 6]. A recently proposed mechanism for this is the induction of glycerol kinase in adipose tissue by thiazolidinediones, thus establishing a futile metabolic cycle in adipocytes [7]. However, we have shown that this mechanism is not detectable in vivo in humans or in vitro in human adipocytes [8].
The concept that thiazolidinediones cause insulin sensitisation by affecting NEFA metabolism is supported by a large number of rodent in vivo studies. For example, in rodents, rosiglitazone and troglitazone decrease fasting NEFA concentrations by 40 to 90% [9, 10]. In contrast, in human studies, fasting NEFA concentrations increase, or fall by up to 30% [11–14], and these changes are often not statistically significant.
By reversing insulin resistance, do thiazolidinediones reverse the hypertriglyceridaemia of type 2 diabetes? Reports of the effects of different thiazolidinediones on triglyceride metabolism are inconsistent. Rosiglitazone has no effect on fasting triglyceride concentrations [15–19]. One study has looked at postprandial triglyceride concentrations and found no change with rosiglitazone [17]. In contrast, pioglitazone lowers fasting and postprandial triglyceride concentrations by approximately 15 to 25% [15, 20–22], which is presumably partly a result of its PPARα activity [23]. Indeed, PPARα activation by other drugs, such as the fibrate class of drugs [24], lowers fasting and postprandial triglyceride concentrations [25]. Troglitazone reduces fasting (−2 to −30% [11, 15, 26–30]) and postprandial triglyceride concentrations by 17 to 70% [11, 31]. However, it is unknown whether, like pioglitazone [23], troglitazone has any PPARα activity in addition to its PPARγ action. Rosiglitazone, a potent and specific agonist for PPARγ, which dissociates the effects of triglyceride lowering and insulin sensitisation, seems to be a good tool to dissect the connections between insulin resistance and triglyceride metabolism.
In vitro studies examining the mechanisms underlying changes in postprandial lipid metabolism with thiazolidinediones have produced conflicting results in adipocytes and pre-adipocytes. Lipoprotein lipase (LPL) mRNA expression and mass increased [32, 33] or were unchanged [33, 34], depending on the dose of the thiazolidinedione and model used. The activity of LPL generally decreased with treatment [33, 35], although this reduction was not always statistically significant [35].
We therefore set out to investigate the effects of rosiglitazone on fasting and postprandial NEFA and triglyceride metabolism. We used arteriovenous difference and stable isotope techniques in a double-blind, placebo-controlled, cross-over study design. To identify potential molecular mechanisms underlying the physiological responses, we augmented the metabolic studies with analyses of gene expression related to insulin signalling, and glucose and fat metabolism in skeletal muscle and adipose tissue biopsies.
Data on glycerol kinase from this study have been previously published and are not included here [8].
Subjects and methods
Patients
Twenty-four patients with type 2 diabetes, treated with diet alone participated in the study. Entry criteria included an age of 30–70 years, a fasting plasma glucose of 7–12 mmol/l and a BMI greater than 24 kg/m2. Patients previously treated with oral hypoglycaemic agents were excluded, as were those taking any medication known to affect glucose metabolism. Patients were in good health without cardiac, hepatic, renal or other chronic diseases, and without microvascular complications of diabetes as determined by history, clinical examination and routine blood investigations. Of the 24 subjects, one subject smoked (a pipe). This subject did not smoke for 12 h prior to the metabolic investigation days and did not change his smoking habits during the study period. All subjects had a residual insulin secretory capacity as defined by a fasting C-peptide concentration of 0.5 nmol/l or more. Measurements of weight, HbA1c, and fasting insulin and glucose concentrations were taken at baseline and at each visit.
Study design
This was a double-blind, placebo-controlled, cross-over study. Subjects were randomised into one of two groups: rosiglitazone 4 mg twice daily for 12 weeks followed by placebo for 12 weeks, or vice versa. Twelve of the 24 subjects had rosiglitazone as their first treatment and 12 had placebo as their first treatment. Compliance was assessed at regular intervals by tablet counts. At the end of each 12-week treatment period, patients attended the clinical research unit after a 10-h overnight fast for a full-day metabolic investigation. Vigorous exercise and alcohol were avoided for 24 h before each study.
The study was approved by the Oxfordshire Clinical Research Ethics Committee and all subjects gave their written informed consent.
Metabolic investigation protocol
Tissue-specific arteriovenous techniques To assess the in vivo metabolism of adipose tissue and skeletal muscle in humans, we measured arteriovenous differences across these tissues. Differences between the composition of venous blood draining a particular tissue and arterialised blood reflect the metabolic activity of the specific tissue. Inclusion of tissue blood flow in the calculations allows quantification of net metabolic activity. On the day of the study, serial blood samples were taken in the fasting state and for 6 h after metabolic provocation with a standardised mixed meal.
Blood sampling Arterialised blood was obtained from a vein draining a heated hand. Venous blood from muscle was taken retrogradely from a vein draining the deep structures of the forearm. Venous blood from adipose tissue was obtained from the superficial epigastric vein, as described previously [36]. This vein drains subcutaneous abdominal adipose tissue with negligible contribution from other tissues [37]. Oxygen saturation and ultrasonography were used to confirm adequate siting of the cannulae. Blood samples were taken simultaneously from all three sites. To prevent contamination of the blood from the forearm vein with blood from the hand, a wrist cuff was inflated to above the systolic blood pressure (either to 30 mmHg above the systolic blood pressure or to 200 mmHg, whichever value was higher) for 3 min before samples were taken. Two sets of baseline blood samples were taken 30 min apart. Patients then consumed a meal of a fat emulsion (40 g olive oil, 600 mg [1,1,1-13C]tripalmitin [CK Gas Products, Wokingham, UK], 400 mg emulsifier, flavourings), skimmed milk (200 ml) and Rice Krispies (Kellogg Company, Manchester, UK) containing 40 g fat, 40 g carbohydrate and 600 mg [1,1,1-13C]tripalmitin. All patients had been on a diet low in 13C prior to the study day. Further blood samples were taken for 6 h after the meal.
Tissue blood flow Subcutaneous abdominal adipose tissue blood flow was measured by 133Xe washout [38]. Forearm muscle blood flow was assessed by strain-gauge plethysmography [39].
Tissue biopsies Skeletal muscle and adipose tissue biopsies were taken under local anaesthesia (1% lignocaine) at a standardised time (+300 min) in the postprandial period. Subcutaneous abdominal adipose tissue was taken with a 12-gauge needle, and muscle biopsies were taken from the vastus lateralis muscle using a percutaneous needle technique. Samples were immediately frozen in liquid nitrogen and stored at −70°C for later mRNA quantification.
Biochemical and gene expression analysis
Plasma triglyceride, NEFA, glucose and insulin, and blood glycerol concentrations were measured as described previously [40].
The postprandial increase in 13C in the circulation was derived from the [1,1,1-13C]tripalmitin in the meal. In a representative subset of subjects (n=6), NEFAs and triglycerides were extracted from fasting and postprandial arterialised plasma samples for gas chromatography and gas chromatography-combustion-isotope ratio mass spectrometry (GC-C-IRMS). Enrichments of [13C]palmitate in plasma NEFA and triglyceride fractions were measured. The results were multiplied by the concentrations of palmitate-NEFA and palmitate-triglyceride respectively, to give the concentrations of the tracer in these fractions, as previously described [40].
Remnant lipoprotein cholesterol (RLP-C) concentrations were measured by an immunoaffinity-based method as previously described [41].
Specific mRNAs were quantified by RT-cPCR and real-time PCR techniques, correcting for the housekeeping gene, cyclophilin, as previously described [42]. The mRNA of the PPARγ-responsive gene, fatty acid binding protein 4 (FABP4) (aP2), was quantified in adipose tissue as a positive control for PPARγ action in adipose tissue, along with the mRNA of genes involved in fatty acid metabolism in adipose tissue, such as LPL, hormone-sensitive lipase (HSL), fatty acid translocase (FAT)/CD36), fatty acid synthase (FAS) and PPARγ2. In muscle, expression of mRNA in genes involved in the key steps of glucose metabolism (GLUT4, hexokinase II, pyruvate dehydrogenase kinase 4 [PDK4] and glycogen synthase) and insulin signalling (phosphatidyl-inositol-3 kinase [PI3 kinase] p85α, p110α, p110β subunits, insulin receptor [IR], IRS-1 and IRS-2) were measured.
Statistical analysis, power and calculations
Adipose tissue blood flow was calculated as described previously [38]. Arteriovenous (A-V) and venoarterial (V-A) differences in metabolite concentrations were calculated. Absolute flux was calculated as the product of the A-V or V-A difference and adipose tissue blood flow [36]. LPL rate of action in vivo was calculated from triglyceride removal across each tissue [36]. The rate of action of HSL in adipose tissue in vivo (i.e. the rate of adipose tissue lipolysis) was calculated from the total adipose tissue glycerol release after subtraction of LPL rate of action [36]. The net inward flow of fatty acids from capillary to tissue (transcapillary flux) was calculated from the net rate of triglyceride removal and the release or removal of NEFAs across the tissue [43], and is calculated as: 3×(A-V)triglyceride−(V-A)NEFA. This represents the net movement of fatty acids across the capillary wall, expected to be outward (negative) in the fasting state and inward (positive), representing fat storage, in the postprandial state. This net exchange includes fatty acids derived from triglycerides as well as plasma NEFAs. Insulin sensitivity in the fasting state was calculated by homeostasis model assessment (HOMA %S) [44].
The study was powered to detect a 30% difference in fasting NEFA output from adipose tissue with a power of 90% at a statistical significance level of 0.05.
Data were analysed using SPSS for Windows v10 (SPSS UK, Chertsey, UK) and statistical significance was set at p<0.05 for all tests. Variables are presented as means±SEM unless otherwise stated. Repeated measures ANOVA with time and drug (rosiglitazone or placebo) as within-subject factors was used to identify time effects, drug effects, and time and drug interactions. Potential carry-over effects were analysed using the Mann–Whitney U-test by comparing placebo data with data at randomisation, taking treatment order into account.
Results
Subjects
Of the 24 subjects recruited (17 male), complete physiological data sets were available for 18 subjects (13 male), complete adipose tissue gene expression data for 23 subjects (16 male), and muscle gene expression data for 18 subjects (15 male). The age and sex distributions of the subgroups did not differ significantly from those of the whole group.
Baseline characteristics and general responses to treatment
The mean age of the subjects at entry was 52.3±10.1 years (mean±SD). After enrolment, all subjects had a range of baseline measurements recorded. These variables were not different from results after placebo treatment. There was no suggestion of a carry-over effect. As in other studies, rosiglitazone treatment was associated with an increase in BMI (Table 1) and a fall in haematocrit (Table 1). There was no difference in response to rosiglitazone treatment between male and female, or between older and younger subjects. As in other studies in humans, rosiglitazone significantly increased total cholesterol by 8% (p=0.05) and LDL cholesterol by 10% (p<0.05), whereas HDL cholesterol was unchanged (p=0.12).
Glucose and insulin responses
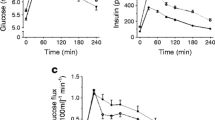
As expected, rosiglitazone lowered HbA1c (Table 1) and induced insulin sensitisation, as demonstrated by lowered fasting and postprandial plasma glucose and insulin concentrations (Fig. 1a, b) and an increase in the calculated median HOMA %S from 37.5 (interquartile range 23.8–50.3) with placebo treatment to 53.2 (43.5–63.9) (p=0.01) with rosiglitazone treatment.

Plasma glucose and insulin concentrations in response to a mixed meal (given at time 0) after placebo treatment (open circles) and rosiglitazone treatment (solid circles). a Plasma glucose concentrations (placebo vs rosiglitazone, p<0.005). b Plasma insulin concentrations (placebo vs rosiglitazone, p<0.005)
Insulin sensitisation was also seen in muscle: the rate of forearm glucose uptake was maintained despite lower plasma glucose and insulin concentrations. There was a 30% increase in muscle glucose clearance per pmol/l of insulin (from 0.066±0.017 to 0.086±0.023 ml/hour per 1000 ml tissue per pmol/l of insulin, p=0.05), quantified as a time-averaged area under the curve for the fasting and postprandial periods. There was no significant difference in glucose clearance by adipose tissue, assessed as a time-averaged area under the curve for the fasting and postprandial periods (p=0.38).
Changes in plasma NEFAs, triglycerides and RLP-C concentrations
Fasting plasma NEFA concentrations were not significantly changed by thiazolidinedione treatment (633±45–591±38 μmol/l, P=0.16) (Fig. 2a). Postprandial plasma NEFA concentrations were reduced by 21% (time-averaged AUC from 483±28 to 383±20 μmol/l, P=0.04), primarily in the late postprandial period (Fig. 2a).

Plasma triglyceride and NEFA concentrations in response to a mixed meal (given at time 0) after placebo treatment (open circles) and rosiglitazone treatment (solid circles). a Plasma NEFA concentrations (placebo fasting vs rosiglitazone fasting, p=0.16; placebo postprandial vs rosiglitazone postprandial, p=0.04). b Plasma triglyceride concentrations (placebo fasting vs rosiglitazone fasting, p=0.86; placebo postprandial vs rosiglitazone postprandial, p<0.01)
Fasting triglyceride concentrations were not altered by rosiglitazone (from 1.92±1.06 to 1.98±1.02 mmol/l, p=0.86) (Fig. 2b). Despite the lack of change in fasting concentrations, rosiglitazone reduced the postprandial rise in triglyceride concentrations compared with placebo by 26% (time-averaged incremental concentration from 855±82 to 632±99 μmol/l, p<0.01) (Fig. 2b). With rosiglitazone treatment, triglyceride concentrations began to decrease after 3 h, whereas during placebo treatment triglyceride concentrations peaked at 4 h before decreasing (Fig. 2b).
RLP-C was measured to give an estimate of the accumulation of remnants of triglyceride-rich lipoproteins after the meal. As would be expected, RLP-C concentrations increased from fasting to postprandial states (on placebo, from 0.38±0.05 to 0.58±0.08 mmol/l [p<0.001]; on rosiglitazone, from 0.46±0.06 to 0.63±0.11 mmol/l [p<0.01]). However, rosiglitazone did not significantly alter fasting or postprandial RLP-C concentrations (p=0.68).
Adipose tissue responses
The net movement of fatty acids from capillaries to the adipocytes, the transcapillary flux, is shown in Fig. 3a. In the fasting state, there was a net release of fatty acids from adipose tissue into the circulation; this was increased by rosiglitazone treatment (p=0.06). Postprandially, there was a net uptake of fatty acids into adipose tissue, and this was not different between treatment and placebo (p=0.59). The fatty acids taken up by adipose tissue are derived from two main sources: from the liberation of fatty acids from the intravascular hydrolysis of circulating triglycerides by LPL, and from the uptake of circulating NEFAs. The rate of action of LPL was unaltered by rosiglitazone treatment (Fig. 3b). Rosiglitazone increased fasting NEFA release by adipose tissue, although this was not statistically significant (+24%, p=0.17) (Fig. 3c). It was associated with an increase in the fasting HSL rate of action from 158±31 to 346±79 nmol/min per 100 g tissue, p=0.01) (Fig. 3d). Postprandial NEFA output and HSL rate of action were unchanged (p=0.15 and p=0.18, respectively) (Fig. 3d).

Subcutaneous abdominal adipose tissue metabolism in response to a mixed meal (given at time 0), after placebo treatment (open circles) and after rosiglitazone treatment (solid circles). a Total flux of fatty acids into and out of subcutaneous abdominal adipose tissue (‘transcapillary flux’), calculated from the V-A difference in NEFA concentrations across the depot and from the A-V difference in triglyceride concentrations across the depot, accounting for tissue blood flow. Negative values represent a net output of fatty acids from the tissue, whilst positive values represent a net uptake of fatty acids into the tissue (placebo fasting vs rosiglitazone fasting, p=0.06; placebo postprandial vs rosiglitazone postprandial, p=0.59). b NEFA output from adipose tissue, calculated from the V-A difference in NEFA concentrations across the depot, accounting for tissue blood flow (placebo fasting vs rosiglitazone fasting, p=0.17; placebo postprandial vs rosiglitazone postprandial, p=0.15). c LPL rate of action in adipose tissue calculated from the removal of triglycerides across the tissue, accounting for tissue blood flow (placebo fasting vs rosiglitazone fasting, p=0.50; placebo postprandial vs rosiglitazone postprandial, p=0.45). d HSL rate of action in adipose tissue calculated from the V-A and A-V differences in glycerol and triglyceride concentrations across the depot, accounting for tissue blood flow (placebo fasting vs rosiglitazone fasting, p=0.01; placebo postprandial vs rosiglitazone postprandial, p=0.18)
To explore the extent to which plasma concentrations of NEFAs were related to the output of NEFAs from subcutaneous abdominal adipose tissue, we plotted the graph of these two variables against one another (mean values are plotted in Fig. 4). Thus, there is no separate time axis on this graph; instead mean values of consecutive time points (−30, 0, 30, 60, 90, 120, 180, 240, 300 and 360 min) are connected with lines. At times −30 and 0 both subcutaneous abdominal adipose tissue and circulating NEFA concentrations were high. After the meal, both the plasma NEFA concentrations and the adipose tissue output of NEFA decreased (Fig. 4). At 120–180 min postprandially, adipose tissue output of NEFAs and plasma NEFA concentrations were both at their minimum (Fig. 4). After 240 min, plasma NEFA concentrations increased as adipose tissue NEFA output increased. The lines joining the consecutive time points formed a circular graph. The points of the circular graph whilst on rosiglitazone were distinct from and displaced downwards relative to the points of the circular graph on placebo. Using regression analysis, we calculated the gradients and slopes of the curve of each individual, comparing rosiglitazone and placebo. Rosiglitazone treatment increased the gradients (p=0.05) and lowered the y intercepts (p<0.001).

Mean plasma NEFA concentrations plotted against mean NEFA output from adipose tissue. Open squares represent values after placebo treatment, and solid squares represent values after rosiglitazone treatment. Solid lines connect values from consecutive time points from 30 to 360 min postprandially, some of which are labelled with arrows. The dotted line represents the mean regression line for the time points on placebo. The dashed line represents the mean regression line for the time points on rosiglitazone. SEMs omitted for clarity (gradients of curve after placebo vs gradients of curve after rosiglitazone, p=0.05; y intercepts of curve after placebo vs y intercepts of curve after rosiglitazone, p<0.001)
Muscle responses
The transcapillary flux of fatty acids into muscle reflects fatty acids from two sources: those derived from LPL lipolysis of triglycerides and those taken up as NEFAs from the circulation. This transcapillary flux, or in other words the net uptake of fatty acids into skeletal muscle, was unaltered by rosiglitazone (Fig. 5a) (fasting, p=0.56; postprandial, p=0.81). However, the relative contributions of fatty acids from circulating NEFAs and from triglyceride lipolysis were altered. Rosiglitazone increased fasting uptake of fatty acids as NEFAs (+560%, p=0.03) (Fig. 5b), but did not significantly decrease fasting triglyceride lipolysis. Postprandially, rosiglitazone did not change NEFA uptake into muscle (p=0.81) (Fig. 5b). The rate of action of muscle LPL was unchanged by rosiglitazone (fasting, p=0.12; postprandial, p=0.88) (Fig. 5c).

Skeletal muscle metabolism in response to a mixed meal (given at time 0) after placebo treatment (open circles) and after rosiglitazone treatment (solid circles). a Total flux of fatty acids into and out of skeletal muscle (‘transcapillary flux’), calculated from the V-A difference in NEFA concentrations across the depot and from the A-V difference in triglyceride concentrations across the depot, accounting for tissue blood flow. Higher values represent increased uptake of fatty acids into muscle (placebo fasting vs rosiglitazone fasting, p=0.56; placebo postprandial vs rosiglitazone postprandial, p=0.81). b NEFA uptake into skeletal muscle, calculated from the A-V difference in NEFA concentrations across the depot, accounting for tissue blood flow (placebo fasting vs rosiglitazone fasting, p=0.03; placebo postprandial vs rosiglitazone postprandial, p=0.81). c LPL rate of action in skeletal muscle, calculated from the removal of triglycerides across the tissue, accounting for tissue blood flow (placebo fasting vs rosiglitazone fasting, p=0.12; placebo postprandial vs rosiglitazone postprandial, p=0.88)
Tissue blood flow
Adipose tissue blood flow was not altered by eating, a finding consistent with previous results in insulin-resistant or obese subjects [45, 46]. However, rosiglitazone therapy increased both fasting and postprandial adipose tissue blood flow relative to placebo by 32% (time-averaged AUC increased from 2.0 to 2.6 ml/min per 100 g of adipose tissue, p=0.03) (Fig. 6a). Forearm blood flow was unaffected by eating (p=0.22) or by rosiglitazone treatment (p=0.20) (Fig. 6b).

Tissue blood flow in response to a mixed meal (given at time 0) after placebo treatment (open circles) and rosiglitazone treatment (solid circles). a Adipose tissue blood flow (placebo vs rosiglitazone, p=0.03). b Forearm muscle blood flow (placebo vs rosiglitazone, p=0.20)
Stable isotope results
To quantify the proportion of the circulating NEFA concentration that was derived from the intravascular lipolysis of circulating triglycerides, we measured concentrations of [1-13C] palmitic acid in the circulation of a representative subset of subjects (n=6).
In this study, [13C]palmitic acid both in the triglyceride fraction (Fig. 7a) and in the NEFA fraction (Fig. 7b) increased postprandially, as would be expected after a meal containing labelled triglycerides. Rosiglitazone did not alter the postprandial rise in [13C]palmitic acid in the triglyceride fraction (p=0.26) (Fig. 7a), but decreased the postprandial rise in [13C]palmitic acid in the NEFA fraction (p=0.04) (Fig. 7b).

Stable isotope concentrations in response to a mixed meal containing 600 mg of [1,1,1-13C]tripalmitin (given at time 0) after placebo treatment (open circles) and rosiglitazone treatment (solid circles). a Concentrations of postprandial [13C]palmitic acid in the triglyceride fraction (p=0.26). b Concentrations of postprandial [13C]palmitic acid in the NEFA fraction (p=0.04)
Gene mRNA expression in muscle and adipose tissue biopsy samples
To supplement the physiological findings of the effects of thiazolidinediones on glucose and fatty acid metabolism, we measured the expression of genes important in these pathways in adipose tissue and skeletal muscle biopsies.
In skeletal muscle, hexokinase II mRNA was increased 2.2-fold (p=0.001) by rosiglitazone and PDK4 was increased 1.5-fold (p=0.04). The expression of other genes involved in glucose metabolism was not significantly altered (Table 2). The expression of genes involved in insulin signalling (IR, IRS-1/2, P85α, p110α and p110β subunits of PI3 kinase) did not change (Table 2). LPL and FABP4 mRNA expression was upregulated, 2.1-fold (p=0.006) and 1.8-fold (p=0.001), respectively. FATP1 was reduced by 30% (p=0.02). There was a statistically significant but modest increase in UCP2 expression of 1.08-fold (p=0.05). Other genes involved in fatty acid metabolism were unchanged.
In adipose tissue the mRNA expression of the known PPARγ target gene, FABP4, increased by 66% (p<0.0001) (Table 3). The expression in adipose tissue of LPL (+36%) and FAT/CD36 (+15%) showed modest increases (Table 3).
Discussion
We set out to explore the association between fatty acid and triglyceride metabolism in response to rosiglitazone treatment in type 2 diabetes. We found that rosiglitazone decreased NEFA and triglyceride concentrations in the late postprandial phase, whilst leaving fasting concentrations unchanged. Mechanisms underlying these observations were explored using arteriovenous and stable isotope techniques.
The postprandial lowering of NEFA concentrations occurred more than 3 h after eating. In theory, this could be a result of either an increased removal or a decreased production of NEFAs. However, plasma NEFA concentrations generally reflect the rate of generation of NEFAs rather than their rate of removal [47, 48]. Here, we quantified the rate of NEFA removal by skeletal muscle and found that this was unchanged by rosiglitazone in the late postprandial period. Other authors have also addressed this issue by quantifying the actual rate of NEFA removal in response to rosiglitazone. They found that rosiglitazone did not change the rate of NEFA removal [49]. Thus, NEFA removal is not likely to be altered by rosiglitazone, implying that the lowered postprandial NEFA concentrations are likely to be secondary to a lower rate of generation of NEFAs.
As PPARγ is highly expressed in adipose tissue [50] and generation of NEFAs only occurs in adipose tissue, this seems to be a likely site of action of rosiglitazone. The adipose tissue depot that is the main source of circulating NEFAs is subcutaneous abdominal adipose tissue [51]. Therefore, we asked whether a reduced output of NEFAs from this depot could account for the lower postprandial NEFA concentrations. Using arteriovenous techniques in combination with measurements of adipose tissue blood flow, we found that rosiglitazone did not affect adipose tissue NEFA release postprandially. When the relationship between plasma NEFA concentrations and subcutaneous adipose tissue NEFA release was explored by regression analysis, the relationship was significantly changed by rosiglitazone. In effect, rosiglitazone lowered the intercept of the regression line towards zero, implying that NEFA generation from sources other than abdominal subcutaneous adipose tissue had been reduced in response to rosiglitazone.
Could different adipose tissue depots respond differently to rosiglitazone? One adipose tissue depot that may behave differently is visceral adipose tissue. A reduction in postprandial NEFA generation from this depot in response to rosiglitazone could account for the observed lowering of postprandial NEFA concentrations and would be consistent with the reduction of visceral adipose tissue mass with rosiglitazone found by other authors [52, 53].
By measuring plasma concentrations of stable isotopes derived from the meal containing [1,1,1-13C]tripalmitin, we were able to identify potential mechanisms underlying the postprandial lowering of plasma NEFA concentrations by rosiglitazone. NEFAs entering the circulation from adipose tissue can be derived from the lipolysis of either intracellular or intravascular triglycerides. The lipolysis of intravascular triglycerides by LPL produces NEFAs which are trapped in adipose tissue or spill over into the systemic circulation. We have previously shown that it is the spillover of NEFAs derived from chylomicron triglycerides that is particularly important in the late postprandial rise in plasma NEFA concentrations [54]. It is this process that we believe is affected by rosiglitazone treatment. [13C]Palmitic acid in the triglyceride fraction is administered in the mixed meal, and so is found in chylomicron particles. The rate of appearance of [13C]palmitic acid in the NEFA fraction decreased, despite no change in the rate of removal of [13C]palmitic acid from the triglyceride fraction, implying that the rate of NEFA spillover from chylomicron triglycerides is lowered by rosiglitazone. Such lowered spillover of NEFAs with rosiglitazone could therefore account for the lowered postprandial NEFA concentrations. However, this mechanism did not seem to be active in subcutaneous abdominal adipose tissue. We suggest that this mechanism operates in visceral adipose tissue in humans and accounts for the observed reduction in plasma NEFA concentrations.
In this study, rosiglitazone lowered triglyceride concentrations in an unusual pattern: late postprandial triglyceride concentrations decreased whilst fasting triglyceride concentrations were unaffected. Such an isolated postprandial reduction has only occasionally been described with any pharmacological agent [55–57]. In rodents, similar fasting-postprandial triglyceride effects have been noted after giving a non-thiazolidinedione PPARγ agonist to male Sprague–Dawley rats [58]. In contrast to rosiglitazone, the other clinically used thiazolidinediones, pioglitazone and troglitazone, reduce both fasting and postprandial triglyceride concentrations. Rosiglitazone, in this and other studies [15–19], does not alter fasting triglyceride concentrations. There is only one previous report of the effect of rosiglitazone on postprandial triglyceride concentrations [17]. As that study was designed primarily to examine glucose metabolism, blood samples were only taken until 180 min postprandially [17]. There was no change in the early postprandial period in plasma triglyceride concentrations in either the present study or that of Raskin et al. [17]. However, ours is the first study to examine triglyceride concentrations during the late postprandial period; we found that rosiglitazone consistently lowered triglyceride concentrations 3 h postprandially.
The postprandial lowering of plasma triglycerides was not simply secondary to an increased rate of removal of triglycerides by the two major tissues expressing LPL, adipose tissue and skeletal muscle: LPL rate of action was unchanged in both of these tissues. Could increased triglyceride removal by the liver account for the postprandial triglyceride reductions? The liver removes triglycerides by whole particle uptake of the remnants of triglyceride-rich lipoproteins. So an increase in the uptake of remnant particles by the liver should be reflected by a decrease in RLP-C concentrations. We found that rosiglitazone did not alter RLP-C concentrations, thus increased triglyceride removal by the liver is unlikely to be the mechanism for the lowered postprandial triglyceride concentrations. An alternative explanation would arise from the decreased postprandial plasma concentrations of NEFAs. This would result in a decreased delivery of NEFAs to the liver, an effect that would be particularly marked if visceral adipose tissue were the main adipose tissue depot affected by rosiglitazone [59]. This in turn would lead to decreased liver fat content, an observation that has been made in rosiglitazone-treated patients [12]. Liver fat would serve as the immediate pool for the synthesis of VLDL triglycerides. Thus a reduction in liver fat might be expected to reduce VLDL triglyceride production by the liver, which would be observed as lower postprandial triglyceride concentrations. Reductions in NEFA concentrations have been previously shown to be associated with lower VLDL triglyceride liver output [60]. A second possible mechanism for lowered postprandial VLDL triglyceride concentrations would be the effects of insulin on VLDL triglyceride secretion. The acute inhibition of VLDL triglyceride secretion by insulin is blunted in type 2 diabetes [61, 62]. It is therefore possible that rosiglitazone restores the effectiveness of insulin in inhibiting the secretion of VLDL triglycerides.
Adipose tissue blood flow is known to be decreased in obesity and type 2 diabetes [45]. Rosiglitazone increased adipose tissue blood flow by a small but consistent amount. This increase in adipose tissue blood flow with rosiglitazone has not been previously described but is consistent with vasodilatory effects attributed to rosiglitazone [63]. Indeed, adipose tissue blood flow is known to be important in the regulation of the tissue’s metabolism [45], and a nutrient-responsive blood flow pattern has been associated with insulin sensitivity [64]. In contrast, muscle blood flow was not changed, suggesting that the effect of rosiglitazone on blood flow was tissue specific.
The activity of HSL, the enzyme responsible for adipose tissue lipolysis, was not decreased but significantly increased in the fasting state, as was the output of NEFAs from adipocytes as reflected in the net transcapillary flux of fatty acids (p=0.06). This finding is, however, entirely consistent with HSL simply responding to circulating insulin concentrations in the fasting state. As hyperinsulinaemia is reduced in the process of insulin sensitisation, HSL activity is consequently upregulated. Thus, adipose tissue may be seen as responding appropriately to ambient insulin concentrations. The postprandial changes in NEFA output and HSL activity in adipose tissue are also consistent with this hypothesis. Adipose tissue NEFA output and HSL rate of action in the postprandial state did not change with rosiglitazone therapy; both were suppressed by insulin after placebo and rosiglitazone treatment. To effectively suppress adipose tissue lipolysis, only a small increment in insulin concentrations above baseline is required [65]. Our data are consistent with those of Mayerson et al. [12] who used a combination of two-step hyperinsulinaemic–euglycaemic clamp and microdialysis techniques to assess in vivo adipose tissue lipolysis [12]. After rosiglitazone treatment, lipolysis was suppressed to a greater extent with a low dose of insulin.
Although the main aim of this study was to assess physiological changes in humans in response to rosiglitazone treatment, the measurement of mRNA expression in biopsy samples allowed the investigation of potential mechanisms underlying the physiological changes observed in skeletal muscle and adipose tissue.
In skeletal muscle, we assessed the expression of genes involved in two pathways important in insulin sensitisation: glucose metabolism and insulin signalling. Rosiglitazone did not alter the expression of genes involved in insulin signalling in muscle, although mRNA concentrations of genes involved in insulin signalling may not directly reflect activity of the pathway. However, our data are consistent with the results of in vitro cultures of muscle cells exposed to rosiglitazone [66] and with the results of protein levels of IRS-1, IRS-2 and GLUT-4 measured in muscle biopsies [67, 68]. The entry of glucose into muscle cells has been shown to be a major controlling step of glucose disposal in type 2 diabetes [69, 70]. Although GLUT-4 mRNA expression was unchanged by thiazolidinedione therapy, this presumably reflects the fact that regulation of GLUT-4 activity occurs mainly at a post-transcriptional level [71]. An early step in skeletal muscle glucose metabolism, hexokinase II, responsible for phosphorylation of glucose on entry into the muscle cell, was induced 2.2-fold by rosiglitazone. This finding suggests a potential mechanism for the observed increased efficiency of forearm muscle glucose uptake with rosiglitazone treatment. Depression of basal skeletal muscle hexokinase II expression in type 2 diabetes was first demonstrated by Vestergaard and colleagues [72], and is supported by the in vivo work of Kelley and colleagues [70]. In healthy people, hexokinase II is induced by insulin to meet the increased demands for glucose handling by the tissue, and this induction fails to occur in type 2 diabetes [73–75]. This is the first study to demonstrate that insulin sensitisation by thiazolidinediones is associated with the normalisation of hexokinase II expression in type 2 diabetes in humans.
In adipose tissue, the expression of genes involved in fatty acid metabolism, such as HSL, FAT/CD36, FAS and SREBP1c, was not substantially altered, although a number of the changes observed were statistically significant. In contrast, FABP4 (aP2), a gene known to be responsive to PPARγ activation [76], was upregulated 1.7-fold in adipose tissue (P<0.0001).
The measurement of mRNA expression in the adipose tissue and skeletal muscle biopsies has limitations. We have demonstrated lower postprandial insulin and NEFA concentrations with rosiglitazone treatment. Thus, it is unclear whether the changes in mRNA concentrations are direct effects of rosiglitazone on skeletal muscle and adipose tissue or whether we are observing the consequences of lowered insulin concentrations. The use of mRNA expression also imposes limitations on the understanding of metabolic processes. For example, LPL is known to be primarily post-transcriptionally regulated and thus its expression does not necessarily correlate with its observed in vivo or ex vivo activity, as demonstrated here.
Conversely, a major strength of this study is that it assesses responses in co-ordinated integrated physiology and metabolism with thiazolidinedione therapy, not simply changes in gene or protein expression: the physiological responses are the final integrated steps of the changes in mRNA and protein concentrations.
We propose that in humans, the main effect of rosiglitazone is not primarily to reduce fasting NEFA concentrations, but instead to reduce postprandial NEFA concentrations. This could be the mechanism of insulin sensitisation by rosiglitazone. This is also the first study to describe that rosiglitazone reduces postprandial triglyceride concentrations. We identify the potential underlying mechanisms for these observations. It is also possible that rosiglitazone has a direct effect on skeletal muscle glucose handling by increasing hexokinase II expression.
Abbreviations
- A-V difference:
-
arteriovenous difference
- FABP4:
-
fatty acid binding protein 4
- FAS:
-
fatty acid synthase
- HSL:
-
hormone-sensitive lipase
- IR:
-
insulin receptor
- LPL:
-
lipoprotein lipase
- PDK4:
-
pyruvate dehydrogenase kinase 4
- Pl kinase:
-
phosphatidyl-inositol-3 kinase
- RLP-C:
-
remnant lipoprotein cholesterol
- V-A difference:
-
venoarterial difference
References
Auwerx J (1999) PPARγ, the ultimate thrifty gene. Diabetologia 42:1033–1049
Way JM, Harrington WW, Brown KK et al (2001) Comprehensive messenger ribonucleic acid profiling reveals that peroxisome proliferator-activated receptor gamma activation has coordinate effects on gene expression in multiple insulin-sensitive tissues. Endocrinology 142:1269–1277
Randle PJ, Garland PB, Hales CN, Newsholme EA (1963) The glucose-fatty acid cycle: its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1:785–789
Boden G (1997) Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes 46:3–10
Lebovitz HE, Banerji MA (2001) Insulin resistance and its treatment by thiazolidinediones. Recent Prog Horm Res 56:265–294
Petersen KF, Krssak M, Inzucchi S, Cline GW, Dufour S, Shulman GI (2000) Mechanism of troglitazone action in type 2 diabetes. Diabetes 49:827–831
Guan HP, Li Y, Jensen MV, Newgard CB, Steppan CM, Lazar MA (2002) A futile metabolic cycle activated in adipocytes by antidiabetic agents. Nat Med 8:1122–1128
Tan GD, Debard C, Tiraby C et al (2003) A “futile cycle” induced by thiazolidinediones in human adipose tissue? Nat Med 9:811–812
Finegood DT, McArthur MD, Kojwang D et al (2001) Beta-cell mass dynamics in Zucker diabetic fatty rats. Rosiglitazone prevents the rise in net cell death. Diabetes 50:1021–1029
Zhou YT, Grayburn P, Karim A et al (2000) Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci U S A 97:1784–1789
Maggs DG, Buchanan TA, Burant CF et al (1998) Metabolic effects of troglitazone monotherapy in type 2 diabetes mellitus. A randomized, double-blind, placebo-controlled trial. Ann Intern Med 128:176–185
Mayerson AB, Hundal RS, Dufour S et al (2002) The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes 51:797–802
Hällsten K, Virtanen KA, Lönnqvist F et al (2002) Rosiglitazone but not metformin enhances insulin- and exercise-stimulated skeletal muscle glucose uptake in patients with newly diagnosed type 2 diabetes. Diabetes 51:3479–3485
Nolan JJ, Jones NP, Patwardhan R, Deacon LF (2000) Rosiglitazone taken once daily provides effective glycaemic control in patients with type 2 diabetes mellitus. Diabet Med 17:287–294
King AB (2000) A comparison in a clinical setting of the efficacy and side effects of three thiazolidinediones. Diabetes Care 23:557
Freed MI, Ratner R, Marcovina SM et al (2002) Effects of rosiglitazone alone and in combination with atorvastatin on the metabolic abnormalities in type 2 diabetes mellitus. Am J Cardiol 90:947–952
Raskin P, Rappaport EB, Cole ST, Yan Y, Patwardhan R, Freed MI (2000) Rosiglitazone short-term monotherapy lowers fasting and post-prandial glucose in patients with type II diabetes. Diabetologia 43:278–284
Wolffenbuttel BH, Gomis R, Squatrito S, Jones NP, Patwardhan RN (2000) Addition of low-dose rosiglitazone to sulphonylurea therapy improves glycaemic control in type 2 diabetic patients. Diabet Med 17:40–47
Fonseca V, Rosenstock J, Patwardhan R, Salzman A (2000) Effect of metformin and rosiglitazone combination therapy in patients with type 2 diabetes mellitus: a randomized controlled trial. JAMA 283:1695–1702
Miyazaki Y, Mahankali A, Matsuda M et al (2001) Improved glycemic control and enhanced insulin sensitivity in type 2 diabetic subjects treated with pioglitazone. Diabetes Care 24:710–719
Bajaj M, Suraamornkul S, Pratipanawatr T et al (2003) Pioglitazone reduces hepatic fat content and augments splanchnic glucose uptake in patients with type 2 diabetes. Diabetes 52:1364–1370
Aronoff S, Rosenblatt S, Braithwaite S, Egan JW, Mathisen AL, Schneider RL (2000) Pioglitazone hydrochloride monotherapy improves glycemic control in the treatment of patients with type 2 diabetes: a 6-month randomized placebo-controlled dose–response study. The Pioglitazone 001 Study Group. Diabetes Care 23:1605–1611
Sakamoto J, Kimura H, Moriyama S et al (2000) Activation of human peroxisome proliferator-activated receptor (PPAR) subtypes by pioglitazone. Biochem Biophys Res Commun 278:704–711
Vu-Dac N, Schoonjans K, Kosykh V et al (1995) Fibrates increase human apolipoprotein A-II expression through activation of the peroxisome proliferator-activated receptor. J Clin Invest 96:741–750
Karpe F (2002) Postprandial lipemia-effect of lipid-lowering drugs. Atheroscler Suppl 3:41–46
Ghazzi MN, Perez JE, Antonucci TK et al (1997) Cardiac and glycemic benefits of troglitazone treatment in NIDDM. The Troglitazone Study Group. Diabetes 46:433–439
Antonucci T, Whitcomb R, McLain R, Lockwood D, Norris RM (1997) Impaired glucose tolerance is normalized by treatment with the thiazolidinedione troglitazone. Diabetes Care 20:188–193
Sunayama S, Watanabe Y, Ohmura H et al (1999) Effects of troglitazone on atherogenic lipoprotein phenotype in coronary patients with insulin resistance. Atherosclerosis 146:187–193
Shirai K, Itoh Y, Sasaki H et al (1999) The effect of insulin sensitizer, troglitazone, on lipoprotein lipase mass in preheparin serum. Diabetes Res Clin Pract 46:35–41
Suter SL, Nolan JJ, Wallace P, Gumbiner B, Olefsky JM (1992) Metabolic effects of new oral hypoglycemic agent CS-045 in NIDDM subjects. Diabetes Care 15:193–203
Minamikawa J, Tanaka S, Yamauchi M, Inoue D, Koshiyama H (1998) Potent inhibitory effect of troglitazone on carotid arterial wall thickness in type 2 diabetes. J Clin Endocrinol Metab 83:1818–1820
Kobayashi J, Nagashima I, Hikita M et al (1999) Effect of troglitazone on plasma lipid metabolism and lipoprotein lipase. Br J Clin Pharmacol 47:433–439
Ranganathan S, Kern PA (1998) Thiazolidinediones inhibit lipoprotein lipase activity in adipocytes. J Biol Chem 273:26117–26122
Rieusset J, Auwerx J, Vidal H (1999) Regulation of gene expression by activation of the peroxisome proliferator-activated receptor gamma with rosiglitazone (BRL 49653) in human adipocytes. Biochem Biophys Res Commun 265:265–271
McTernan PG, Harte AL, Anderson LA et al (2002) Insulin and rosiglitazone regulation of lipolysis and lipogenesis in human adipose tissue in vitro. Diabetes 51:1493–1498
Frayn KN, Coppack SW (2001) Assessment of white adipose tissue metabolism by measurement of arteriovenous differences. Methods Mol Biol 155:269–279
Frayn KN, Coppack SW, Humphreys SM, Whyte PL (1989) Metabolic characteristics of human adipose tissue in vivo. Clin Sci (Lond) 76:509–516
Larsen OA, Lassen NA, Quaade F (1966) Blood flow through human adipose tissue determined with radioactive xenon. Acta Physiol Scand 66:337–345
Rådegran G (1999) Limb and skeletal muscle blood flow measurements at rest and during exercise in human subjects. Proc Nutr Soc 58:887–898
Evans K, Burdge GC, Wootton SA, Clark ML, Frayn KN (2002) Regulation of dietary fatty acid entrapment in subcutaneous adipose tissue and skeletal muscle. Diabetes 51:2684–2690
Karpe F, Boquist S, Tang R, Bond GM, de Faire U, Hamsten A (2001) Remnant lipoproteins are related to intima-media thickness of the carotid artery independently of LDL cholesterol and plasma triglycerides. J Lipid Res 42:17–21
Auboeuf D, Vidal H (1997) The use of the reverse transcription-competitive polymerase chain reaction to investigate the in vivo regulation of gene expression in small tissue samples. Anal Biochem 245:141–148
Evans K, Clark ML, Frayn KN (1999) Effects of an oral and intravenous fat load on adipose tissue and forearm lipid metabolism. Am J Physiol 276:E241–E248
Levy JC, Matthews DR, Hermans MP (1998) Correct homeostasis model assessment (HOMA) evaluation uses the computer program. Diabetes Care 21:2191–2192
Summers LK, Samra JS, Humphreys SM, Morris RJ, Frayn KN (1996) Subcutaneous abdominal adipose tissue blood flow: variation within and between subjects and relationship to obesity. Clin Sci (Lond) 91:679–683
Jansson PA, Larsson A, Lönnroth PN (1998) Relationship between blood pressure, metabolic variables and blood flow in obese subjects with or without non-insulin-dependent diabetes mellitus. Eur J Clin Invest 28:813–818
Issekutz B Jr, Bortz WM, Miller HI, Paul P (1967) Turnover rate of plasma FFA in humans and in dogs. Metabolism 16:1001–1009
Bonadonna RC, Groop LC, Zych K, Shank M, DeFronzo RA (1990) Dose-dependent effect of insulin on plasma free fatty acid turnover and oxidation in humans. Am J Physiol 259:E736–E750
Miyazaki Y, Glass L, Triplitt C et al (2001) Effect of rosiglitazone on glucose and non-esterified fatty acid metabolism in type II diabetic patients. Diabetologia 44:2210–2219
Auboeuf D, Rieusset J, Fajas L et al (1997) Tissue distribution and quantification of the expression of mRNAs of peroxisome proliferator-activated receptors and liver X receptor-alpha in humans: no alteration in adipose tissue of obese and NIDDM patients. Diabetes 46:1319–1327
Guo Z, Hensrud DD, Johnson CM, Jensen MD (1999) Regional postprandial fatty acid metabolism in different obesity phenotypes. Diabetes 48:1586–1592.
Kelly IE, Han TS, Walsh K, Lean ME (1999) Effects of a thiazolidinedione compound on body fat and fat distribution of patients with type 2 diabetes. Diabetes Care 22:288–293
Virtanen KA, Hällsten K, Parkkola R et al (2003) Differential effects of rosiglitazone and metformin on adipose tissue distribution and glucose uptake in type 2 diabetic subjects. Diabetes 52:283–290
Frayn KN, Coppack SW, Fielding BA, Humphreys SM (1995) Coordinated regulation of hormone-sensitive lipase and lipoprotein lipase in human adipose tissue in vivo: implications for the control of fat storage and fat mobilization. Adv Enzyme Regul 35:163–178
Tan KC, Tso AW, Tam SC, Pang RW, Lam KS (2002) Acute effect of orlistat on post-prandial lipaemia and free fatty acids in overweight patients with type 2 diabetes mellitus. Diabet Med 19:944–948
Reitsma JB, Castro Cabezas M, de Bruin TW, Erkelens DW (1994) Relationship between improved postprandial lipemia and low-density lipoprotein metabolism during treatment with tetrahydrolipstatin, a pancreatic lipase inhibitor. Metabolism 43:293–298
Damci T, Yalin S, Balci H et al (2004) Orlistat augments postprandial increases in glucagon-like peptide 1 in obese type 2 diabetic patients. Diabetes Care 27:1077–1080
Laplante M, Sell H, MacNaul KL, Richard D, Berger JP, Deshaies Y (2003) PPAR-gamma activation mediates adipose depot-specific effects on gene expression and lipoprotein lipase activity: mechanisms for modulation of postprandial lipemia and differential adipose accretion. Diabetes 52:291–299
Nielsen S, Guo Z, Johnson CM, Hensrud DD, Jensen MD (2004) Splanchnic lipolysis in human obesity. J Clin Invest 113:1582–1588
Lewis GF, Uffelman KD, Szeto LW, Weller B, Steiner G (1995) Interaction between free fatty acids and insulin in the acute control of very low density lipoprotein production in humans. J Clin Invest 95:158–166
Malmstrom R, Packard CJ, Watson TD et al (1997) Metabolic basis of hypotriglyceridemic effects of insulin in normal men. Arterioscler Thromb Vasc Biol 17:1454–1464
Lewis GF, Uffelman KD, Szeto LW, Steiner G (1993) Effects of acute hyperinsulinemia on VLDL triglyceride and VLDL apoB production in normal weight and obese individuals. Diabetes 42:833–842
Avena R, Mitchell ME, Nylen ES, Curry KM, Sidawy AN (1998) Insulin action enhancement normalizes brachial artery vasoactivity in patients with peripheral vascular disease and occult diabetes. J Vasc Surg 28:1024–1031
Karpe F, Fielding BA, Ilic V, Macdonald IA, Summers LK, Frayn KN (2002) Impaired postprandial adipose tissue blood flow response is related to aspects of insulin sensitivity. Diabetes 51:2467–2473
Campbell PJ, Carlson MG, Hill JO, Nurjhan N (1992) Regulation of free fatty acid metabolism by insulin in humans: role of lipolysis and reesterification. Am J Physiol 263:E1063–E1069
Zierath JR, Ryder JW, Doebber T et al (1998) Role of skeletal muscle in thiazolidinedione insulin sensitizer (PPARγ agonist) action. Endocrinology 139:5034–5041
Kim YB, Ciaraldi TP, Kong A et al (2002) Troglitazone but not metformin restores insulin-stimulated phosphoinositide 3-kinase activity and increases p110beta protein levels in skeletal muscle of type 2 diabetic subjects. Diabetes 51:443–448
Meyer MM, Levin K, Grimmsmann T et al (2002) Troglitazone treatment increases protein kinase B phosphorylation in skeletal muscle of normoglycemic subjects at risk for the development of type 2 diabetes. Diabetes 51:2691–2697
Cline GW, Petersen KF, Krssak M et al (1999) Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N Engl J Med 341:240–246
Kelley DE, Mintun MA, Watkins SC et al (1996) The effect of non-insulin-dependent diabetes mellitus and obesity on glucose transport and phosphorylation in skeletal muscle. J Clin Invest 97:2705–2713
Shepherd PR, Kahn BB (1999) Glucose transporters and insulin action—implications for insulin resistance and diabetes mellitus. N Engl J Med 341:248–257
Vestergaard H, Bjorbaek C, Hansen T, Larsen FS, Granner DK, Pedersen O (1995) Impaired activity and gene expression of hexokinase II in muscle from non-insulin-dependent diabetes mellitus patients. J Clin Invest 96:2639–2645
Pendergrass M, Koval J, Vogt C et al (1998) Insulin-induced hexokinase II expression is reduced in obesity and NIDDM. Diabetes 47:387–394
Kruszynska YT, Mulford MI, Baloga J, Yu JG, Olefsky JM (1998) Regulation of skeletal muscle hexokinase II by insulin in nondiabetic and NIDDM subjects. Diabetes 47:1107–1113
Ducluzeau PH, Perretti N, Laville M et al (2001) Regulation by insulin of gene expression in human skeletal muscle and adipose tissue. Evidence for specific defects in type 2 diabetes. Diabetes 50:1134–1142
Martin G, Schoonjans K, Lefebvre AM, Staels B, Auwerx J (1997) Coordinate regulation of the expression of the fatty acid transport protein and acyl-CoA synthetase genes by PPARα and PPARγ activators. J Biol Chem 272:28210–28217
Acknowledgements
G. Tan was supported by the MRC. We thank J. Levy for providing advice and software to calculate HOMA %S, L. Dennis, V. Ilic and M. Clark for assistance with the studies, and N. Vega and P. Vallier for their work with the mRNA quantification. This study received financial support from GlaxoSmithKline. However, the sponsor had no role in study design, in the collection and interpretation of the data, in the writing of the report, or in the decision to submit the paper for publication.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tan, G.D., Fielding, B.A., Currie, J.M. et al. The effects of rosiglitazone on fatty acid and triglyceride metabolism in type 2 diabetes. Diabetologia 48, 83–95 (2005). https://doi.org/10.1007/s00125-004-1619-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-004-1619-9