
Abstract
Aims/hypothesis
Several studies have shown that maternal diabetes increases the risk of congenital malformations in various organ systems including the neural tube. The present study analysed molecular and morphological changes in the forebrain of embryos from diabetic Albino Swiss mice.
Methods
Maternal diabetes-induced morphological changes in the forebrain were examined histologically. Cell proliferation index was assayed by BrdU labelling. In situ hybridisation and quantitative real-time PCR were used to analyse the expression of genes coding for sonic hedgehog (Shh), Nkx2.1, brain factor-1 (BF-1) and bone morphogenetic protein-4 (Bmp4) that control forebrain patterning.
Results
There were no distinguishable abnormalities in the forebrain of embryos from diabetic pregnancies on embryonic day 0.5. At embryonic day 11.5, embryos of diabetic pregnancies displayed a fusion and thickening of the ventral telencephalic neuroepithelium and a partial absence of the dorsal telencephalon, indicating a severe patterning defect in the dorsoventral axis of the telencephalon. The cell proliferation index was also higher in the ventral telencephalon of these embryos. Molecular analyses indicated that expression of Shh, Nkx2.1 and BF-1 was increased and their expression domains expanded dorsally in the ventral telencephalon in embryos of diabetic mice at embryonic day 11.5. The expression of Bmp4 was reduced in the dorsal forebrain of these embryos. At embryonic day 8.5, only Shh expression was increased.
Conclusions/interpretation
Altered expression of various genes involved in dorsoventral patterning of the forebrain is associated with forebrain malformations in embryos of diabetic mice.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Epidemiological studies and experiments in rodent embryos have shown that there is an increased risk of fetal malformations and spontaneous abortions in diabetic pregnancy [1, 2, 3, 4, 5]. In human epidemiological studies, a direct correlation has been shown between the degree of maternal hyperglycaemia during the first trimester and the incidence and severity of fetal abnormalities [6, 7, 8]. Maternal diabetes-induced malformations have been detected in all major organ systems, including the cardiovascular, gastrointestinal, genito-urinary and neurological systems [9, 10]. Among the more frequent of the latter are neural tube defects such as anencephaly, holoprosencephaly and syntelencephaly [11, 12, 13, 14]. Diabetes-induced embryonic dysmorphogenesis is accompanied by metabolic abnormalities including increased superoxide dismutase activity [15, 16, 17], reduced concentrations of myoinositol and arachidonic acid [18, 19] and inhibition of the pentose phosphate shunt pathway [20]. Moreover, the frequency of embryonic malformations in diabetic pregnancy has been reported to be reduced by dietary supplements of antioxidants such as vitamin E or vitamin C [16, 21, 22, 23] and butylated hydroxytoluene [24], suggesting that oxidative stress is involved in embryonic dysmorphogenesis.
Neural tube defects have been reported to occur after mutations in more than 30 genes in mice [25]. Most of these mutations affect cranial neurulation, which lead to exencephaly and anencephaly. Recently, neural tube defects in embryos of diabetic mice have been associated with reduced expression of Pax3, which is a member of the conserved family of paired box transcription factors and is involved in the development of the midbrain as well as the hindbrain and the caudal neural tube closure [14, 26]. However, little is known about the molecular and cellular mechanisms that regulate patterning of the forebrain in embryos of diabetic mice. The forebrain comprises tissues that are unique within the central nervous system, including the cerebral cortex, eye, thalamus and hypothalamus, and its patterning mechanisms are distinct from those operating in other regions of the central nervous system [27].
The malformations of the forebrain at the centre of the present study, especially malformations in the telencephalic neuroepithelium, have frequently been observed in embryos of diabetic mice, indicating that there could be a defect in patterning of the forebrain. Patterning of the telencephalon is regulated by a distinct set of signalling molecules, which are produced by the ventral and dorsal signalling centres. Candidate signals that regulate dorsoventral patterning of the neural tube include sonic hedgehog (Shh) and bone morphogenetic proteins (BMPs). Shh, a member of the hedgehog family of signalling molecules secreted mainly from two signalling centres, the notochord and the floor plate, induces the differentiation of ventral cell types and inhibits the differentiation of dorsal cell types during early neural tube development [28, 29, 30, 31, 32, 33]. Belonging to the TGF-β superfamily, BMPs oppose Shh influence and promote patterning of the dorsal neural tube [34, 35]. In addition, Shh induces the expression of Nkx2.1 [27, 33], a homeobox transcription factor whose function is required for the formation of the ventral forebrain [36, 37, 38], whereas BMPs antagonise brain factor-1 (BF-1), which is a member of the winged-helix family of transcription factors and is essential for the development of the telencephalon and retina [39]. The present study therefore aimed to explore the molecular mechanisms that contribute to forebrain malformations in embryos of diabetic mice. To do this, we investigated whether the expression of genes coding for Shh, Nkx2.1, BF-1 and Bmp4, which are involved in forebrain patterning, is altered in embryos of diabetic mice.
Materials and methods
Experimental animals
The Albino Swiss mice used in this study were obtained from the Laboratory Animals Centre, Singapore. The mice were housed in clean cages in a temperature-controlled room with a 14-h light and 10-h dark schedule and free access to chow. Type 2 diabetes mellitus was induced in 8-week-old female mice by an intraperitoneal injection of 80 mg/kg streptozotocin (Sigma, St Louis, Mo., USA) dissolved in citrate buffer (0.01 mol/l, pH 4.5) on three successive days. Blood glucose concentrations were measured one week after the streptozotocin injection using a Glucometer Elite (Bayer, Mishawaka, Ind., USA). Female mice with a blood glucose concentration of more than 16 mmol/l were classified as diabetic and mated with age-matched male mice whose blood glucose concentrations were normal (4–6 mmol/l). Noon on the day when a copulation plug was observed was counted as embryonic day (E) 0.5. On E8.5 (9–10 somite stage) or E11.5 (40–42 somite stage), pregnant mice were anaesthetised with 7% chloral hydrate and embryos were collected by Caesarean section. A slight developmental delay of approximately 6 hours was observed in embryos of diabetic mice in comparison to those of control mice. To obtain embryos with the same number of somites, the control and diabetic mice were killed at about a 6-h interval, i.e. at 10 a.m. and at 4 p.m. respectively. All procedures involving animal handling were in accordance with guidelines of the Medical Faculty Ethics Committee (National University of Singapore) and the Principles of laboratory animal care (NIH publication no. 85-23, revised 1985).
Histology and immunohistochemistry
Embryos were fixed overnight in 4% paraformaldehyde in phosphate buffer at 4°C and cryoprotected with 20% sucrose. Cryosections (12–20 µm thick) were mounted on gelatinised slides and stained with Hematoxylin and Eosin. Some of these sections were used to measure the thickness of telencephalic neuroepithelium using an ocular grid in a light microscope (Olympus BX51, Olympus Optical Company, Tokyo, Japan). A total of six sections (two each from three embryos) from each group (normal and diabetic) was used for this morphometric analysis. Student’s t test was used to evaluate statistical significance.
For immunohistochemistry, the cryosections were treated with 1% hydrogen peroxide in methanol to inactivate endogenous peroxidase. The sections were then incubated with the primary antibody, Rabbit TTF-1 (Nkx2.1, diluted 1:500 in PBS) (Biopat Immunotechnologies, Caserta, Italy). Subsequent antibody detection was done using biotinylated anti-rabbit IgG and the ABC kit (Cat No: PK-4000, Vector, Burlingame, Calif., USA). The reaction product was visualised using 3,3′-diaminobenzidine as a substrate and intensified with ammonium nickel sulphate. Tissue sections were finally dehydrated in an ascending series of alcohol and mounted in Permount.
For analysis of cell proliferation, six time-mated pregnant mice from the control and diabetic groups (n=3 from each group) were injected intraperitoneally with BrdU (100 µg/g body weight; Sigma). To overcome the developmental delay observed in embryos of diabetic mice, injections were done at a 6-h interval, i.e. at 8 a.m. and 2 p.m. respectively. Two hours after the injection, embryos (E8.5 and E11.5) were collected by Caesarian section from the pregnant mice, which had been anaesthetised with 7% chloral hydrate. Embryos were then fixed with 4% paraformaldehyde. Cryosections (20 µm thick) at the level of forebrain were collected and processed for immunohistochemical analysis using the anti-BrdU monoclonal antibody (1:100) (Sigma). Photomicrographs were taken with a light microscope (Olympus BX51). For quantitative analysis, BrdU-labelled cells in the telencephalic neuroepithelium were identified and counted under a X20 objective. A total of six sections (two each from three embryos) from each group (normal and diabetic) was examined and the cell counts were averaged and analysed. Student’s t test was used to evaluate statistical significance.
In situ hybridisation
For whole-mount in situ hybridisation, plasmids of Shh (1.3 kb), Bmp4 (1 kb, provided by Dr. L.M. Hogan, Howard Hughes Medical Institute, Nashville, Tenn., USA), Nkx2.1 (2.2 kb, provided by Dr. L.R. Rubenstein, University of California at San Francisco, USA), BF-1 (1.2 kb, provided by Dr. E. Lai, Memorial Sloan-Kettering Cancer Centre, New York, USA) were linearised with the appropriate restriction enzymes. Digoxigenin-labelled antisense mRNA probes were synthesised using an RNA labelling kit (Boehringer, Mannheim, Germany) and RNA polymerase enzyme (Stratagene, La Jolla, Calif., USA). The stained embryos were sectioned transversely with a cryostat.
Quantitation of mRNA levels by real-time PCR
Total RNA was prepared from developing brains of E11.5 embryos of diabetic and non-diabetic mice using RNeasy Mini Kit (Qiagen, Hilden, Germany). Whole embryos were used to extract the total RNA from E8.5 embryos of diabetic and control mice. At least four embryos from four different litters of each group (diabetic and control) were used in this study. For reverse transcription, the reaction mixture containing 2 µg of RNA, 2.5 µmol/l of oligo (dT) primer and 5 units of Molony Murine Leukemia Virus Reverse Transcriptase (Promega, Madison, Wis., USA) in a total volume of 25 µl was incubated for 1 h at 42°C and the reaction was stopped by heating for 5 min at 95°C. For mRNA amplification, an aliquot (0.5 µl) of the reverse transcription products was added to the reaction mixture (20 µl) containing LightCycler-FastStart DNA Master SYBR Green I (Roche Diagnostics, Mannheim, Germany), 0.5 µmol/l of each primer, and 3 mmol/l MgCl2 (Roche). The forward (F) and reverse (R) primers used for amplification of each gene were as follows: (i) mouse Shh: F- 5′-GTG AGG CTG CGA GTG ACC G-3′ and R- 5′-CCT GGT CGT CAG CCG CCA GCA CGC-3′ [40]; (ii) mouse Bmp4: F- 5′-TCC ATC ACG AAG AAC ATC-3′ and R- 5′-TAG TCG TGT GAT GAG GTG-3′ (238 bp; Gene Bank accession No. NM 007554); and (iii) mouse BF-1: F- 5′-CAG CAC TTT GAG TTA CAA CG-3′ and R- 5′-TGG TCT GCG AAG TCA TTG AC-3′ (325 bp; Gene Bank accession No. U36760).
After pre-incubation at 95°C for 10 min, PCR was done with 50 cycles of denaturation at 95°C for 15 s, annealing at 63°C (Shh) or at 58°C (Bmp4) or at 59°C (BF-1) for 10 s, and elongation at 72°C for PCR product size per 25 s. The expression level of specific mRNA transcripts was quantified, expressed as Ct, the cycle number at which the LightCycler System detected the upstroke of the exponential phase of PCR product accumulation, and normalised by the level of glyceraldehyde-3-phosphate dehydrogenase expression in each individual sample [41].
Statistical analysis
Data were analysed by Student’s t test using Microsoft Excel and expressed as means ± SD. A p value of less than 0.05 was considered to be statistically significant.
Results
Morphological changes in the forebrain of embryos of diabetic mice
Our examination of morphological differences in the neural tubes of embryos derived from control and diabetic pregnancies showed that at E8.5 no developmental differences and morphological changes in the neural tubes were distinguishable. Transverse sections also revealed that there were no structural abnormalities in the prospective forebrain of embryos derived from diabetic mice in comparison with prospective forebrains of embryos from control mice (data not shown).
At E11.5, about 12% (±3.2%, n=10) of mouse embryos derived from ten diabetic mothers had neural tube defects, including exencephaly, anencephaly and spina bifida. Only a few embryos (1.3±2.8%, n=10, control vs diabetes: p<0.001) from non-diabetic mice had externally visible neural tube defects. Exencephaly involving the forebrain, midbrain and hindbrain was frequently (75±26% among embryos with neural tube defects, n=10) observed in embryos from diabetic mice (Fig. 1b).

Mouse embryos (E11.5) obtained from normal (a) and diabetic (b) mice. The embryo from a diabetic mouse has an exencephaly extending from the forebrain to hindbrain (arrows). The approximate planes of transverse sections shown here were followed throughout this study. Transverse sections (c, d) of the forebrain at the level of the eyes stained with Hematoxylin and Eosin show obvious morphological defects, including fusion and thickening of the telencephalon, in the embryo from a diabetic mouse. However, the diencephalon derivatives, including the eyes, appear unaltered in the embryo of the diabetic mouse. The red bars indicate the ventral telencephalic wall. Scale bar (a–d): 400 µm. Thickness (diameter in µm) (e) of the ventral telencephalic wall in E11.5 embryos of normal and diabetic mice. Thickness is greatly increased in embryos of diabetic mice. The graph shows mean values ± SD. *p<0.05. e, eye; dc, diencephalon; dt, dorsal telencephalon; fb, forebrain; hb, hindbrain; mb, midbrain; vt, ventral telencephalon
The open neural tube extends from the caudal region of forebrain to hindbrain about the level of otic vesicles. The most rostral region of the forebrain appeared closed (Fig. 1b). The forebrain is a topographically complex structure that comprises the telencephalon and diencephalon. Embryos of diabetic mice showed variable abnormalities in the morphology of the forebrain. Transverse sections through the forebrain of most (80±25%, n=10) of the exencephalic embryos (E11.5) derived from diabetic mice showed that the neuroepithelium of the ventral telencephalon was thickened and both sides of the neuroepithelial wall were fused. As a result, the telencephalic vesicles were absent or reduced in size and volume in comparison to those of normal neural tubes (Fig. 1c,d). Morphometric analysis also showed that the ventral telencephalic wall in embryos of diabetic mice was thickened (Fig. 1e). In addition, the neuroepithelium of the dorsal telencephalon was distorted (Fig. 1d) or absent in some embryos of diabetic mice. However, the structure of the ventral diencephalon and eyes appeared to be normally developed in a majority of the embryos of diabetic mice, although there was a reduction in the volume of the 3rd ventricle.
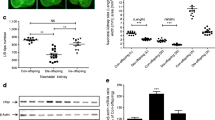
Measurement of the proliferation index within the telencephalic neuroepithelium by BrdU labelling in both embryo groups found that at E8.5 the number of BrdU labelled cells in the prospective forebrain of the embryos of diabetic mice was not different from that of embryos of control mice (Fig. 2a,b,e). At E11.5, the number of BrdU-labelled cells in the ventral telencephalic neuroepithelium was significantly greater (p<0,05) in embryos of diabetic mice than in those of control mice (Fig. 2c–e). Cells in other regions of the forebrain appeared to proliferate normally in embryos of diabetic mice (data not shown).

Transverse sections showing BrdU-labelled cells in the telencephalon of embryos (E8.5 and E11.5) from control and diabetic mice. At E8.5 (a, b) the ratio of BrdU-labelled cells is uniform in the prospective telencephalons of both embryo groups (no distinguishable difference in number of BrdU-cells in the two groups). At E11.5 the BrdU-cells are evenly distributed in the entire telencephalon of the control mouse embryo (c), while the diabetic mouse embryo (d) shows increased proliferation in the ventral telencephalon. Scale bar (a–d): 50 µm. Comparison (e) of percentages of BrdU-labelled cells in the prospective telencephalic (E8.5) and ventral telencephalic (E11.5) neuroepithelium of embryos from control and diabetic mice. Note the increased proliferation in E11.5 embryos of diabetic mice, in contrast with the two groups of embryos at E8.5 (no difference). The graph shows mean values ± SD. *p<0.05. tc, prospective telencephalon; vt, ventral telencephalon
Expression of Shh, Nkx2.1, BF-1 and Bmp4
To investigate whether the forebrain dysmorphogenesis observed in embryos of diabetic mice was accompanied by altered expression pattern of inductive signalling molecules and transcriptional factors, we analysed mRNA expression of Shh, Nkx2.1, BF-1 and Bmp4 in embryos at E8.5 and E11.5.
Shh is initially expressed in the axial meso-endoderm and subsequently in the medial and ventral neural plate. In this study, Shh expression at E8.5 was restricted to the prospective ventral diencephalon in the forebrain (Fig. 3a). In embryos from diabetic pregnancies, the staining intensity and the expression domain of Shh in the diencephalon was increased (Fig. 3b). In E11.5 embryos of control mice, Shh expression was localised to the ventral diencephalon and ventral telencephalon (Fig. 3c). In embryos from diabetic mice, the intensity of Shh expression was stronger and the expression domain of Shh was expanded dorsally in the ventral telencephalon (Fig. 3d).
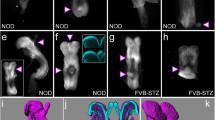
Shh induces ventral telencephalic phenotype, i.e. expression of Nkx2.1, a homeobox gene. In E11.5 embryos of control mice, Nkx2.1 expression was localised in the region of the diencephalon and ventral telencephalon (Fig. 3e), whereas in embryos of diabetic mice, the expression domain in the telencephalon was found to be expanded dorsally and medially (Fig. 3f). Similar results were obtained by immunohistochemical analysis of Nkx2.1 (Fig. 3g,h). However, expression of Nkx2.1 in the prospective forebrain was undetectable in E8.5 embryos of control and diabetic mice (data not shown).

In situ hybridisation for Shh (a–d) and Nkx2.1 (e, f) mRNA expression on transverse sections through the forebrain in embryos of normal and diabetic mice. At E8.5, expression of Shh in the forebrain is restricted to the prospective diencephalon in embryos from control mice (a) , but increased (arrows) in embryos of diabetic mice (b). At E11.5, expression of Shh and Nkx2.1 in embryos from control mice (c, e) is localised to the diencephalon and ventral telencephalon, whereas in embryos of diabetic mice the expression domains of both (d, f) are expanded dorsally in the ventral telencephalon (arrows). Immunohistochemical analysis (g, h) shows that the expression domain of Nkx2.1 is expanded dorsally in the ventral telencephalon in embryos of diabetic mice. Scale bar: (a, b) 100 µm; (c–f) 400 µm; (g, h) 200 µm. dc, diencephalon; e, eye; vt, ventral telencephalon; dt, dorsal telencephalon
BF-1 is required for the development of telencephalic derivatives such as the cerebral hemispheres of the brain. Its expression was localised to the telencephalic neuroepithelium in E8.5 and E11.5 embryos (Fig. 4a,c). At E8.5, embryos of control and diabetic mice displayed no difference in the expression pattern of BF-1 in the prospective telencephalic neuroepithelium (Fig. 4a,b). By E11.5, the expression domain and intensity of BF-1 expression within the telencephalon appeared to be increased while the shape of the telencephalic neuroepithelium was thickened and fused in embryos of diabetic mice (Fig. 4d).
In embryos of control mice, Bmp4 expression in the telencephalic neuroepithelium was detected after cephalic neural tube closure. At E11.5, expression was localised to the area of the dorsomedial telencephalon (Fig. 4e). In embryos of diabetic mice, the Bmp4 expression domain in the telencephalon was absent or reduced (Fig. 4f).

Transverse sections showing mRNA expression of BF-1 and Bmp4 in the telencephalon of embryos from control and diabetic mice. In control embryos BF-1 expression in the forebrain is restricted to the telencephalic neuroepithelium at E8.5 (a) and E11.5 (c). At E8.5, expression of BF-1 in embryos of diabetic mice is unaltered versus controls (b), but stronger at E11.5, with the diabetic mouse embryo (d) displaying a thickened ventral telencephalon. Bmp4 expression in normal embryos at E11.5 (e) is visible in the dorsomedial telencephalon (arrow), but is absent (asterisk) or reduced in some embryos of diabetic mice (f). Scale bar: (a, b) 120 µm; (c-f) 400 µm. dc, diencephalon; dmt, dorso medial telencephalon; dt, dorsal telencephalon; e, eye; III, 3rd ventricle; LV, lateral ventricle; vt, ventral telencephalon
Quantitative analysis of mRNA expression levels of Shh, Bmp4 and BF-1 by real-time PCR
Real-time PCR analysis showed that Shh mRNA expression in E8.5 embryos of diabetic mice and in the developing brains from E11.5 embryos of diabetic mice was significantly increased (p<0.001; Fig. 5a,b). However, just before the cephalic neural tube closure in E8.5 embryos of diabetic mice mRNA expression of BF-1 and Bmp4 was unaltered in comparison with those in control mice (Fig. 5a). At E11.5, the expression level of BF-1 was significantly increased (p<0.001) whereas the Bmp-4 expression level was significantly decreased (p<0.05) in the developing brains of embryos from diabetic mice (Fig. 5b).

Real-time PCR analysis of mRNA expression levels of Shh, Bmp4 and BF-1 in embryos from control and diabetic mice. At E8.5 (a) only Shh expression is significantly increased in embryos of diabetic mice. At E11.5 (b) expression of Shh and BF-1 is significantly greater, whereas Bmp4 expression is significantly lower in brains of embryos from diabetic mice than in embryos from control mice. Graphs show mean values ± SD (n=4). *p<0.05; **p<0.001
Discussion
Neural tube defects are one of the most common malformations associated with diabetic embryopathy [11]. Several genes regulate neurulation, which is the embryonic process in which the neural plate, a specialised region of the ectoderm on the dorsal surface of the embryo, undergoes shaping and folding to form the neural tube. In various experimental conditions mutations in more than 30 genes in the mouse induced neural tube defects, which are mainly marked by a failure of neural tube closure [25, 42]. Recently, decreased expression of Pax-3 was found to be responsible for the development of neural tube defects in the mid-hindbrain regions of embryos from diabetic mice [14, 26]. In the present study, we have characterised the forebrain malformations induced by maternal diabetes, which include a fusion and thickening of the ventral telencephalic neuroepithelium and a partial absence of the dorsal telencephalon and telencephalic vesicles, indicating a severe patterning defect in the dorsoventral axis of the telencephalon. This condition appears similar to the features of syntelencephaly seen in an infant of a diabetic mother [13]. In humans, syntelencephaly, which is characterised by segmental fusion of telencephalic derivatives such as the cerebral hemispheres and other brain structures, has been classified as a distinct variant of holoprosencephaly, the severity of which increases with a smooth posterior to anterior progression of the degree of fusion [43]. While the exact mechanisms of this effect are not known, it is possible that the teratogenicity induced by maternal diabetes alters signalling pathways that regulate forebrain patterning.
Several lines of evidence indicate that Shh is directly responsible for dorsoventral patterning of the forebrain [28, 29, 30].Shh−/− mouse embryos display a loss of ventral neural fates, which include absence of vental forebrain structures and presence of cyclopic eyes that are characteristics of holoprosencephaly in human. Interestingly, in embryos of diabetic mice at E8.5 and E11.5, expression ofShh was found to be up-regulated in the developing brain. In addition, its expression domain in the ventral telencephalon at E11.5 was expanded and the expansion was associated with an increased expression domain of Nkx2.1, which is believed to be the downstream gene of Shh in the ventral telencephalon [33]. Shh organises the dorsoventral axis of the ventral telencephalon by establishing distinct regions of several homeodomain transcription factors, including Nkx2.1[36, 44]. In our study, up-regulation of Shh in embryos of diabetic mice was more evident in the ventral telencephalon, where hyperplasia and fusion of thickened neuroepithelium were seen, than in the diencephalon, indicating that Shh signalling might be regulated by transcription factors expressed locally in the ventral telencephalon. In addition, boundaries of dorsal and ventral fields in the neural plate appeared to be disturbed as the result of Shh up-regulation, and this could interfere with normal folding and proliferation of the neuroepithelium, leading to distortion of the neural tube, as seen here. However, the signalling pathway that regulates expression of Shh and patterning of the forebrain in embryos of diabetic mice has not been identified in its entirety.
Several reports have shown that BMPs, which are expressed in distinct regions of the developing forebrain, regulate patterning of the dorsal neural tube [34, 35]. BMPs seem to act by inhibiting the spread of ventralising signals mediated by Shh [44]. Among BMPs,Bmp4 expression, which was localised to the dorsal medial telencephalon and roof of the telencephalon, has been shown to restrict the proliferation of telencephalic neuroepithelium [34]. In the present study, Bmp4 expression in the developing brain of embryos of diabetic mice at E11.5 was significantly reduced and its expression domain in the dorsomedial telencephalon was absent or reduced. This suggests that the hyperplasia and expansion of expression domains ofShh and its downstream geneNkx2.1 in the ventral telencephalon are associated with the down-regulation of Bmp4 expression observed in embryos of diabetic mice.
It has been shown that expression ofBmp4 regulates BF-1 expression [34]. Mutation of BF-1 in mice leads to the expansion of dorsal cell fate at the expense of ventral cell fate in the telencephalon, suggesting that BF-1 is involved in patterning of the dorsoventral axis of the telencephalon [45]. In embryos of diabetic mice at E11.5, the level of BF-1 mRNA expression was increased and its expression domain within the ventral telencephalon was expanded. It is possible that the altered expression pattern ofBF-1 seen in our study is associated with abnormal dorsoventral patterning of the telencephalon, in which the ventral telencephalon was expanded as revealed by the expression ofShh and Nkx2.1, whereas the dorsal telencephalon was obviously decreased or absent as revealed by Bmp4 expression. AsShh and BF-1 have mitogenic activity for various cell types, the up-regulation ofShh and BF-1 could have contributed to the increased proliferation, thereby resulting in thickening of the ventral telencephalic neuroepithelium in E11.5 embryos of diabetic mice. As BF-1 is required for the down-regulation of Bmp4 in the dorsal telencephalon during forebrain development [45], the reduction or loss of Bmp4 expression in the dorsal telencephalon observed in embryos of diabetic mice could be due to the increased expression of BF-1. In addition, it has been shown that SHH and BMP signalling play reciprocal roles in dorsoventral patterning of the telencephalon [30, 46, 47]. Both decreased SHH signalling and increased BMP signalling have severe effects on growth of the telencephalic vesicles that lead to holoprosencephaly [30, 47, 48].
In the present study, increased expression of Shh and BF-1 and decreased expression of Bmp4 are associated with the expansion and fusion of telencephalic neuroepithelium and loss of the dorsal medial telencephalon. As it was found to be up-regulated as early as E8.5 in embryos of diabetic mice, Shh appears, among the genes examined, to be the up-stream gene regulating various genes that control forebrain patterning. In general, SHH is essential for specification of ventral structures of the entire neural tube and it represses expression of several homeobox genes, notablyPax-3, which is involved in neural tube closure during mesencephalon development [35, 49]. This indicates that the inhibition of Pax3 expression induced by maternal diabetes in the neural tube [14] could be mediated via up-regulation of Shh as observed in the present study.
It has also been shown that overexpression of Pax3 inhibits differentiation of ventral structures as well as inducing dorsalisation of the neural tube, while decreased expression of Pax3 induces ectopic differentiation of ventral structures in the neural tube [49, 50]. It is, therefore, possible that the decreased expression of Pax3 in embryos of diabetic mice inhibits dorsalisation and induces ventralisation in the neural tube, thereby leading to the neural tube anomalies observed by us. However, it is not clear exactly how maternal diabetes alters the expression of genes that are involved in neural tube development.
Although several teratogenic factors such as sorbitol accumulation [51], myo-inositol deficiency [19], arachidonic acid deficiency [18], altered prostaglandin metabolism [52] and increased concentration of 3-deoxyglucosone [53] are altered in embryos of diabetic pregnancy, the mechanisms by which these abnormalities lead to dysmorphogenesis have not been determined. Only recently, it has been shown that oxidative stress induced by maternal diabetes could lead to neural tube defects by impairing the expression of genes that control developmental processes [23]. Moreover, involvement of various other trophic and metabolic factors associated with maternal diabetes in causing neural tube anomalies cannot be ruled out, as multiple genes are also involved in neural tube patterning during embryogenesis.
The present study makes it clear that the signalling molecules that regulate very early events of forebrain patterning during embryogenesis are altered by maternal diabetes. Expression levels of Shh, its downstream gene, Nkx 2.1, and BF-1 in the forebrain, particularly in the ventral telencephalon are induced in embryos of diabetic mice. Shh andBF-1 are known mitogens that could induce hyperplasia in the ventral telencephalon. In addition, the expression ofBmp4 is decreased in the telencephalon, particularly the dorsomedial telencephalon of embryos from diabetic pregnancy and the decrease could be caused by the increased expression of BF-1 as reported previously. We conclude therefore that altered expression of these genes is associated with the forebrain malformations seen in embryos of diabetic mice.
Abbreviations
- Shh :
-
sonic hedgehog
- Bmp4 :
-
bone morphogenetic protein-4
- BF-1 :
-
brain factor-1
- BMPs:
-
bone morphogenetic proteins
- E:
-
embryonic day
References
Mills JL (1982) Malformations in infants of diabetic mothers. Teratology 25:385–394
Hanson U, Persson B, Thunell S (1990) Relationship between haemoglobin A1C in early type 1 (insulin-dependent) diabetic pregnancy and the occurrence of spontaneous abortion and fetal malformation in Sweden. Diabetologia 33:100–104
Kitzmiller JL, Gavin LA, Gin GD, Jovanovic-Peterson L, Main EK, Zigrang WD (1991) Preconception care of diabetes. Glycemic control prevents congenital anomalies. JAMA 265:731–736
Rosenn B, Miodovnik M, Combs CA, Khoury J, Siddiqi TA (1994) Glycemic thresholds for spontaneous abortion and congenital malformations in insulin-dependent diabetes mellitus. Obstet Gynecol 84:515–520
Sakamaki H, Akazawa S, Ishibashi M et al. (1999) Significance of glutathione-dependent antioxidant system in diabetes-induced embryonic malformations. Diabetes 48:1138–1144
Jovanovic L, Druzin M, Peterson CM (1981) Effect of euglycemia on the outcome of pregnancy in insulin-dependent diabetic women as compared with normal control subjects. Am J Med 71:921–927
Miller E, Hare JW, Cloherty JP et al. (1981) Elevated maternal hemoglobin A1c in early pregnancy and major congenital anomalies in infants of diabetic mothers. N Engl J Med 304:1331–1334
Sheridan-Pereira M, Drury MI, Baumgart R, Ua Conaill D, France MW (1983) Haemoglobin A1 in diabetic pregnancy: an evaluation. Ir J Med Sci 152:261–267
Mills JL, Baker L, Goldman AS (1979) Malformations in infants of diabetic mothers occur before the seventh gestational week. Implications for treatment. Diabetes 28:292–293
Becerra JE, Khoury MJ, Cordero JF, Erickson JD (1990) Diabetes mellitus during pregnancy and the risks for specific birth defects: a population-based case-control study. Pediatrics 85:1–9
Kucera J (1971) Rate and type of congenital anomalies among offspring of diabetic women. J Reprod Med 7:73–82
Eriksson UJ (1995) The pathogenesis of congenital malformations in diabetic pregnancy. Diabetes Metab Rev 11:63–82
Robin NH, Ko LM, Heeger S, Muise KL, Judge N, Bangert BA (1996) Syntelencephaly in an infant of a diabetic mother. Am J Med Genet 66:433–437
Phelan SA, Ito M, Loeken MR (1997) Neural tube defects in embryos of diabetic mice: role of the Pax-3 gene and apoptosis. Diabetes 46:1189–1197
Eriksson UJ (1991) Diabetes in pregnancy: effects on post-implantation embryos. Isr J Med Sci 27:478–486
Eriksson UJ, Borg LA (1993) Diabetes and embryonic malformations. Role of substrate-induced free-oxygen radical production for dysmorphogenesis in cultured rat embryos. Diabetes 42:411–419
Cederberg J, Galli J, Luthman H, Eriksson UJ (2000) Increased mRNA levels of Mn-SOD and catalase in embryos of diabetic rats from a malformation-resistant strain. Diabetes 49:101–107
Goldman AS, Baker L, Piddington R, Marx B, Herold R, Egler J (1985) Hyperglycemia-induced teratogenesis is mediated by a functional deficiency of arachidonic acid. Proc Natl Acad Sci USA 82:8227–8231
Hashimoto M, Akazawa S, Akazawa M et al. (1990) Effects of hyperglycaemia on sorbitol and myo-inositol contents of cultured embryos: treatment with aldose reductase inhibitor and myo-inositol supplementation. Diabetologia 33:597–602
Shum L, Sadler TW (1990) Biochemical basis for D,L,-beta-hydroxybutyrate-induced teratogenesis. Teratology 42:553–563
Eriksson UJ, Borg LA (1991) Protection by free oxygen radical scavenging enzymes against glucose-induced embryonic malformations in vitro. Diabetologia 34:325–331
Siman CM, Eriksson UJ (1997) Vitamin C supplementation of the maternal diet reduces the rate of malformation in the offspring of diabetic rats. Diabetologia 40:1416–1424
Chang TI, Horal M, Jain SK, Wang F Patel R, Loeken MR (2003) Oxidant regulation of gene expression and neural tube development: Insights gained from diabetic pregnancy on molecular causes of neural tube defects. Diabetologia 46:538–545
Eriksson UJ, Siman CM (1996) Pregnant diabetic rats fed the antioxidant butylated hydroxytoluene show decreased occurrence of malformations in offspring. Diabetes 45:1497–1502
Juriloff DM, Harris MJ (2000) Mouse models for neural tube closure defects. Hum Mol Genet 9:993–1000
Fine EL, Horal M, Chang TI, Fortin G, Loeken MR (1999) Evidence that elevated glucose causes altered gene expression, apoptosis, and neural tube defects in a mouse model of diabetic pregnancy. Diabetes 48:2454–2462
Shimamura K, Rubenstein LR (1997) Inductive interactions direct early regionalization of the mouse forebrain. Development 124:2709–2718
Echelard Y, Epstein DJ, St Jacques B et al. (1993) Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 75:1417–1430
Belloni E, Muenke M, Roessler E et al. (1996) Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat Genet 14:353–356
Chiang C, Litingtung Y, Lee E et al. (1996) Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383:407–413
Roelink H, Augsburger A, Heemskerk J et al. (1994) Floor plate and motorneuron induction by vhh-1, a vertebrate homolog of hedgehog expressed by the notochord. Cell 76:761–775
Kohtz JD, Baker DP, Corte G, Fishell G (1998) Regionalization within the mammalian telencephalon is mediated by changes in responsiveness to Sonic Hedgehog. Development 125:5079–5089
Ericson J, Muhr J, Jessell TM, Edlund T (1995) Sonic hedgehog: a common signal for ventral patterning along the rostrocaudal axis of the neural tube. Int J Dev Biol 39:809–816
Furuta Y, Piston DW, Hogan BL (1997) Bone morphogenetic proteins (BMPs) as regulators of dorsal forebrain development. Development 124:2203–2212
Liem KF Jr, Tremml G, Roelink H, Jessell TM (1995) Dorsal differentiation of neural plate cells induced by BMP-mediated signals from epidermal ectoderm. Cell 82:969–979
Kimura S, Hara Y, Pineau T et al. (1996) The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev 10:60–69
Qiu M, Shimamura K, Sussel L, Chen S, Rubenstein JL (1998) Control of anteroposterior and dorsoventral domains of Nkx-6.1 gene expression relative to other Nkx genes during vertebrate CNS development. Mech Dev 72:77–88
Sussel L, Marin O, Kimura S, Rubenstein JL (1999) Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development 126:3359–3370
Xuan S, Baptista CA, Balas G, Tao W, Soares VC, Lai E (1995) Winged helix transcription factor BF-1 is essential for the development of the cerebral hemispheres. Neuron 14:1141–1152
Dyer MA, Ferrington SM, Mohn D, Munday JR, Baron, MH (2001) Indian hedgehog activates hematopoiesis and vasculogenesis and can respecify prospective neuroectodermal cell fate in the mouse embryo. Development 128:1717–1730
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408
Copp AJ, Bernfield M (1994) Etiology and pathogenesis of human neural tube defects: insights from mouse models. Curr Opin Pediatr 6:624–631
Cohen MM Jr, Sulik KK (1992) Perspectives on holoprosencephaly: Part II. Central nervous system, craniofacial anatomy, syndrome commentary, diagnostic approach, and experimental studies. J Craniofac Genet Dev Biol 12:196–244
Wilson SW, Rubenstein JL (2000) Induction and dorsoventral patterning of the telencephalon. Neuron 28:641–651
Dou CL, Li S, Lai E (1999) Dual role of brain factor-1 in regulating growth and patterning of the cerebral hemispheres. Cereb Cortex 9:543–550
Pierani A, Brenner-Morton S, Chiang C, Jessell TM (1999) A sonic hedgehog-independent, retinoid-activated pathway of neurogenesis in the ventral spinal cord. Cell 97:903–915
Ohkubo Y, Chiang C, Rubenstein JL (2002) Coordinate regulation and synergistic actions of BMP4, SHH and FGF8 in the rostral prosencephalon regulate morphogenesis of the telencephalic and optic vesicles. Neuroscience 111:1–17
Golden JA, Bracilovic A, McFadden KA, Beesley JS, Rubenstein JL, Grinspan JB (1999) Ectopic bone morphogenetic proteins 5 and 4 in the chicken forebrain lead to cyclopia and holoprosencephaly. Proc Natl Acad Sci USA 96:2439–2444
Goulding MD, Lumsden A, Gruss P (1993) Signals from the notochord and floor plate regulate the region-specific expression of two Pax genes in the developing spinal cord. Development 117:1001–1016
Tremblay P, Pituello F, Gruss P (1996) Inhibition of floor plate differentiation by Pax3: evidence from ectopic expression in transgenic mice. Development 122:2555–2567
Eriksson UJ, Naeser P, Brolin SE (1986) Increased accumulation of sorbitol in offspring of manifest diabetic rats. Diabetes 35:1356–1363
Piddington R, Joyce J, Dhanasekaran P, Baker L (1996) Diabetes mellitus affects prostaglandin E2 levels in mouse embryos during neurulation. Diabetologia 39:915–920
Eriksson UJ, Wentzel P, Minhas HS, Thornalley PJ (1998) Teratogenicity of 3-deoxyglucosone and diabetic embryopathy. Diabetes 47:1960–1966
Acknowledgements
This work was supported by a research grant (R-181-000-038-112) from the National University of Singapore, Singapore. We are grateful to Dr. M.R. Loeken (Joslin Diabetes Centre and Harvard Medical School, Boston, USA) for a critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liao, D.M., Ng, Y.K., Tay, S.S.W. et al. Altered gene expression with abnormal patterning of the telencephalon in embryos of diabetic Albino Swiss mice. Diabetologia 47, 523–531 (2004). https://doi.org/10.1007/s00125-004-1351-5
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-004-1351-5