
Abstract
Aims/hypothesis
SUR1(ABCC8)−/− mice lacking functional KATP channels are an appropriate model to test the significance of KATP channels in beta-cell function. We examined how this gene deletion interferes with stimulus-secretion coupling. We tested the influence of metabolic inhibition and galanin, whose mode of action is controversial.
Methods
Plasma membrane potential (Vm) and currents were measured with microelectrodes or the patch-clamp technique; cytosolic Ca2+ concentrations ([Ca2+]c) and mitochondrial membrane potential (ΔΨ) were measured using fluorescent dyes.
Results
In contrast to the controls, SUR1−/− beta cells showed electrical activity even at a low glucose concentration. Continuous spike activity was measured with the patch-clamp technique, but with microelectrodes slow oscillations in Vm consisting of bursts of Ca2+-dependent action potentials were detected. [Ca2+]c showed various patterns of oscillations or a sustained increase. Sodium azide did not hyperpolarize SUR1−/− beta cells. The depolarization of ΔΨ evoked by sodium azide was significantly lower in SUR1−/− than SUR1+/+ cells. Galanin transiently decreased action potential frequency and [Ca2+]c in cells from both SUR1−/− and SUR1+/+ mice.
Conclusion/interpretation
The strong dependence of Vm and [Ca2+]c on glucose concentration observed in SUR1+/+ beta cells is disrupted in the knock-out cells. This demonstrates that both parameters oscillate in the absence of functional KATP channels. The lack of effect of metabolic inhibition by sodium azide shows that in SUR1−/− beta cells changes in ATP/ADP no longer link glucose metabolism and Vm. The results with galanin suggest that this peptide affects beta cells independently of KATP currents and thus could contribute to the regulation of beta-cell function in SUR1−/− animals.
Similar content being viewed by others


KATP channels are essential for normal stimulus-secretion coupling in pancreatic beta cells because they link changes in electrical activity that trigger insulin secretion to beta-cell metabolism. Glucose influx into beta cells leads to enhanced glycolytic flux, an increased ATP/ADP ratio, closure of KATP channels, plasma membrane depolarization and opening of voltage-dependent Ca2+ channels. This results in a rise in the cytosolic free Ca2+ concentration, [Ca2+]c, which activates exocytosis of insulin-containing granules [1]. In addition to this well-characterized glucose-dependent triggering pathway, an amplifying pathway has been described [2] which is independent of KATP channels and changes in membrane potential [3, 4]. Animals deficient in beta cell-type KATP channels provide a useful model in which to study the role of these channels in the metabolic and hormonal control of insulin secretion and in the feedback mechanism(s) driving oscillations in beta cells. Two mouse models have been developed which lack functional KATP channels as a consequence of genetic disruption of either the Kir6.2 (KCNJ11) gene, which encodes the K+ ion-selective pore of the channel [5], or the Sur1 (ABCC8) gene which constitutes the neuroendocrine specific sulfonylurea receptor [6, 7]. Surprisingly, random-fed animals exhibit almost normal blood glucose concentrations while glucose-induced insulin secretion is decreased [5, 6]. This contrasts with loss-of-function mutations in human SUR1 and Kir6.2 which cause a recessive form of persistent hyperinsulinaemic hypoglycaemia of infancy (PHHI) characterised by oversecretion of insulin despite severe hypoglycaemia [8, for review see 9]. Thus in the animal models, KATP channel-independent mechanisms must prevent excessive insulin secretion. SUR1−/− mice show a reduced response to incretins, i.e., GLP-1 and GIP [7, 10] as a consequence of their impaired cAMP-induced potentiation of insulin secretion [10] which could contribute to their unexpected normoglycaemia. Moreover, metabolic and/or hormonal signals could contribute additional regulatory pathways. Glucose metabolism, via ATP production, affects not only KATP channel activity, but could alter insulin secretion by influencing the activity of ion pumps, protein phosphorylation, or exocytosis. We tried to evaluate whether in SUR1−/− beta cells a glucose dependence of membrane potential and [Ca2+]c still exists.
The neuropeptide galanin is known to inhibit insulin secretion [11] and thus is a potential counterregulator of excessive insulin secretion. However, the mechanism of this inhibitory effect is controversial because it has been attributed to both KATP channel-dependent, via activation and hyperpolarization, and KATP channel-independent pathways [11, 12, 13, 14, 15, 16]. We have compared the effects of galanin and a metabolic inhibitor, NaN3, on stimulus-secretion coupling in beta cells of SUR1 knock-out versus wildtype mice in a search for additional regulatory pathways.
Material and methods
Cell and islet preparation
Experiments were done on islets or single pancreatic beta cells isolated from fed SUR1−/−, NMRI, or C57Bl/6 mice killed by cervical dislocation or CO2. The principles of laboratory animal care were followed (NIH publication No. 85-23, revised 1985) and experiments were carried out according German laws. For measurements of cell membrane potential with microelectrodes, a piece of pancreas was fixed in a perifusion chamber and islets were microdissected by hand. Other experiments were done on islet cells isolated by collagenase digestion. Cells were dispersed in Ca2+-free medium and cultured up to 4 days in RPMI 1640 medium (11.1 mmol/l glucose) supplemented with 10% fetal calf serum, 100 U/ml penicillin and 100 µg/ml streptomycin [17].
Solutions and chemicals
Cell membrane potential and KATP current patch-clamp recordings were done at 32°C with amphotericin B (250 µg/ml) in the pipette solution which contained (in mmol/l): 10 KCl, 10 NaCl, 70 K2SO4, 4 MgCl2, 2 CaCl2, 10 EGTA, 20 HEPES, pH 7.15 adjusted with KOH. The bath solution contained (in mmol/l): 140 NaCl, 5 KCl, 1.2 MgCl2, 2.5 CaCl2, 0.5 glucose, 10 HEPES, pH 7.4 adjusted with NaOH. This bath solution was also used for determination of [Ca2+]c and ΔΨ at 37°C at the indicated glucose concentrations.
For cell membrane potential measurements with intracellular microelectrodes the extracellular fluid contained (in mmol/l): 120 NaCl, 5 KCl, 2.5 CaCl2, 1.2 MgCl2, 24 NaHCO3, 15 glucose and was gassed with 95% O2 and 5% CO2 to maintain a pH of 7.4 at 37°C.
Fura-2AM and rhodamine 123 were obtained from Molecular Probes (Eugene, Ore., USA). RPMI1640 medium, penicillin/streptomycin and glutamine were from GIBCO/BRL (Karlsruhe, Germany). Galanin was from Bachem (Heidelberg, Germany). All other chemicals were purchased from Sigma (Deisenhofen, Germany) and Merck (Darmstadt, Germany) in the purest form available.
Patch-clamp recordings
Patch pipettes were pulled from borosilicate glass capillaries (Clark, Pangbourne, UK) and had resistances between 3 and 5 MΩ when filled with pipette solution. Membrane currents and potentials were recorded with an EPC-9 patch-clamp amplifier and software “Pulse” (HEKA, Lambrecht, Germany) in the voltage-clamp or current-clamp mode, respectively. Whole-cell KATP currents were measured at a holding potential of −70 mV and during 300 ms voltage pulses to −80 mV and −60 mV at 15-s intervals. Under these experimental conditions the currents measured with control NMRI beta cells are due almost entirely to KATP channels and are blocked by tolbutamide [18].
Membrane potential measurements
Vm was determined using high resistance microelectrodes [19]. Beta cells from SUR1+/+ mice were identified by their characteristic membrane potential oscillations. Beta cells from SUR1−/− islets cells were discriminated from α-cells by their lack of Na+ action potentials and their contrary glucose dependence of electrical activity [20].
Measurement of [Ca2+]c
The cytosolic free Ca2+ concentration ([Ca2+]c) was measured in single cells or small clusters by the fura-2 method according to [21] using equipment and software from TILL photonics (Gräfelfing, Germany). Cells were considered to be beta cells when [Ca2+]c was not decreased by 15 mmol/l glucose as described for α-cells [22]. The cells were loaded with fura-2AM (5 µmol/l) for 30 min at 37°C. Intracellular fura-2 was excited alternately at 340 nm or 380 nm by means of an oscillating diffraction grating. The excitation light was then directed through the objective (PlanNeofluar40x objective, Zeiss, Stuttgart, Germany) by means of a glass fiber light guide and a dichroic mirror. The emitted light was filtered (LP515 nm) and measured by a digital camera. The ratio of the emitted light intensity at 340 nm/380 nm excitation was used to calculate [Ca2+]c using an in vitro calibration with fura-2 K+-salt.
Measurement of ΔΨ
The mitochondrial membrane potential (ΔΨ) was measured using the same equipment as for [Ca2+]c. Rhodamine 123 (Rh123) fluorescence was excited at 480 nm and the intensity of the emitted light (LP515 nm) was measured and given in arbitrary units (a.u.) (12-bit grey values of the CCD camera). Cells were loaded with Rh123 (10 µg/ml) for 10 min at 37°C. An increase of Rh123 fluorescence corresponds to a decrease in mitochondrial membrane potential [23, 24].
Presentation of results
Electrophysiological experiments, [Ca2+]c and ΔΨ are illustrated by recordings representative of the indicated number of experiments carried out with different cells. At least three different cell preparations have been used for each series of experiments. If possible the means ± SEM are given in the text for the indicated number of experiments. For evaluation of electrical activity in microelectrode experiments the last 4 to 6 min prior to changes in glucose concentration were analysed. The frequency of Ca2+ action potentials was calculated by the number of action potentials during the first 5 s of each burst phase. The statistical significance of differences between means was assessed by a one sample t test or Student’s t test for paired values when two samples were compared. Multiple comparisons were made by ANOVA followed by Student-Newman-Keuls test. A p value of less than 0.05 was considered significant.
Results
Comparison of membrane potential and [Ca2+]c in cells from SUR1+/+ and SUR1−/− mice
Figure 1 shows the effect of glucose stimulation on the plasma membrane potential and whole-cell KATP currents of beta cells from SUR1+/+ mice (Fig. 1a) versus SUR1−/− (Fig. 1b) mice measured with the perforated-patch technique. The traces illustrate the membrane potential recorded in the current-clamp mode. At the intervals indicated by VC the amplifier was switched to the voltage-clamp mode to register currents, illustrated at the right side, during voltage pulses from −70 mV to −60 mV. In SUR1+/+ mice, with functional KATP channels, the plasma membrane potential changed in response to an increase in glucose concentration (Fig. 1a). On average, the plasma membrane potential was −72±2 mV at a glucose concentration of 0.5 mmol/l (n=9). Increasing the glucose concentration to 15 mmo/l led to depolarization of the plasma membrane potential and the occurrence of Ca2+-dependent action potentials. The plateau potential (potential from which the spikes start) was −46±2 mV (n=9, p≤0.001). The KATP current amplitude was 10.6±1.7 pA in the presence of 0.5 mmol/l glucose and 2.0±0.4 pA after switching to 15 mmol/l glucose (n=9, p≤0.001). In all isolated SUR1−/− beta cells the membrane potential was persistently depolarized in 0.5 mmol/l glucose and continuous Ca2+ action potentials were observed. This pattern did not change with increasing glucose concentration (Fig. 1b). The plateau potential was −38±2 mV (n=12) at the low glucose concentration and −40±1 at 15 mmol/l glucose (n=14). No current similar to the KATP current was detectable in voltage-clamp mode, i.e., 1.8±0.3 pA leak current at 0.5 mmol/l glucose (n=10) and 1.1±0.2 pA at 15 mmol/l glucose (n=10) (compare VC3 and VC4 with VC2).

Comparison of the glucose dependence of membrane potential and current in SUR1+/+ (a) and SUR1−/− beta cells (b) measured in the current-clamp mode with the perforated-patch technique to maintain normal cell metabolism. The change in glucose concentration from 0.5 mmol/l (0.5G) to 15 mmol/l (15G) is indicated by the horizontal bars. At the intervals indicated by VC the amplifier was switched to the voltage-clamp mode to record the current at −70 mV (continuous trace) and during 300 ms voltage steps to −60 mV and −80 mV applied every 15 s (upper and lower deflections). The currents measured during the pulses to −60 mV, marked VC1 to VC4, are illustrated at the right side of the figure (c). VC1 and VC3: 0.5 mmol/l glucose, VC2 and VC4: 15 mmol/l glucose, VC1 and VC2: SUR1+/+ beta cell, VC3 and VC4: SUR1−/− beta cells. In contrast to SUR1+/+ beta cells the SUR1−/− beta cells showed electrical activity at the low glucose concentration and the corresponding leak current was small. The recordings are representative of nine (SUR1+/+) and fourteen (SUR1−/−) experiments
Changes in membrane potential normally control the free cytosolic Ca2+ concentration ([Ca2+]c) which triggers insulin secretion in beta cells. Figure 2 illustrates the effects of different glucose concentrations on [Ca2+]c in isolated beta cells or small cell clusters from SUR1+/+ (Fig. 2a) and SUR1−/− (Fig. 2b–e) mice. As described previously [24, 25], control cells respond to an increase of the glucose concentration from 0.5 mmol/l to 15 mmol/l with an initial decrease (due to an activation of Ca2+ pumps) followed by a longer period with increased [Ca2+]c and finally slow oscillations of [Ca2+]c (Fig. 2a). On average [Ca2+]c was 81±3 nmol/l (n=13) at a glucose concentration of 0.5 mmol/l in control cells, decreasing to 57±4 nmol/l after switching to 15 mmol/l glucose (p≤0.001), then abruptly rising to a peak value of 406±23 nmol/l (n=13, p≤0.001). Cells from SUR1−/− mice exhibited variable patterns of [Ca2+]c, but importantly the direct correlation between glucose concentration and [Ca2+]c was lost (Fig. 2). The only consistent response of [Ca2+]c to changes in glucose was a drop upon shifting from 0.5 mmol/l to 15 mmol/l (see arrowheads in Fig. 2b,c,e). A spontaneous rise in [Ca2+]c in SUR1−/−, but not wildtype beta cells, often occurred in the presence of 0.5 mmol/l glucose (n=11, see Fig. 2b). In four of eleven cells increased glucose produced spontaneous oscillations in [Ca2+]c, in the remaining cells the increase was sustained without oscillations. In three cells or cell clusters the behavior was similar to that observed in wildtype cells (Fig. 2c). In three other cells [Ca2+]c was oscillating in 0.5 mmol/l glucose and increased to a sustained level after changing the glucose concentration to 3 mmol/l and 7 mmol/l. These cells sustained the higher level of [Ca2+]c upon returning the glucose concentration to 0.5 mmol/l (Fig. 2d). In another series [Ca2+]c was increased, but not oscillating, in 0.5 mmol/l glucose (Fig. 2e, n=6). In these cells [Ca2+]c was 213±9 nmol/l in the presence of 0.5 mmol/l glucose. Raising the glucose concentration to 15 mmol/l evoked a transient drop in [Ca2+]c to 89±9 nmol/l (p≤0.001) corresponding to the initial decrease observed in Fig. 2b, c.

Comparison of the glucose dependence of [Ca2+]c in SUR1+/+ (a) and SUR1−/− beta cells (b–e). (a) In beta cells from SUR1+/+ mice increasing the glucose concentration from 0.5 mmol/l to 15 mmol/l produced an initial decrease in [Ca2+]c followed by a rapid increase at the beginning of the first phase and finally a second phase with oscillations. This recording is representative of thirteen experiments with similar results. (b–e) In SUR1−/− beta cells the pattern of the [Ca2+]c response to glucose was quite variable. (b) In eleven cells [Ca2+]c rose spontaneously in low glucose (0.5 mmol/l) and oscillated in four cells. (c) In three cells [Ca2+]c was low at 0.5 mmol/l glucose and increasing glucose produced a result similar to that seen in control cells. (d) In three other cells [Ca2+]c was oscillating in the presence of 0.5 mmol/l glucose and increased to a sustained level when glucose was increased and remained increased when glucose was lowered to 0.5 mmol/l. (e) In six cells, a sustained increase was observed at 0.5 mmol/l glucose and increasing the concentration of glucose produced a transient reduction in [Ca2+]c
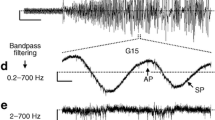
It is commonly accepted that the oscillations in [Ca2+]c in wildtype beta cells are linked to glucose metabolism via oscillations in electrical activity [26]. The persistent Ca2+-dependent action potentials with variable [Ca2+]c observed here raise the question of whether [Ca2+]c and membrane potential remain coupled in SUR1−/− beta cells. The patch-clamp experiments done with single isolated beta cells suggest the strong coupling between changes in membrane potential and [Ca2+]c observed in cells with functional KATP channels could be abrogated in SUR1−/− beta cells. To test this point we measured the cell membrane potential in intact islets using intracellular microelectrodes since oscillatory activity is difficult to detect in single beta cells [27, 28, 29]. Surprisingly, the membrane potential of SUR1−/− beta cells oscillated under these conditions in the presence of 15 mmol/l glucose (n=8, Fig. 3b, c) resembling recordings from wildtype cells in intact islets (Fig. 3a). In contrast to wildtype cells the oscillations persisted in 0.5 mmol/l (n=9, Fig. 3b), 25 mmol/l (n=8, Fig. 3c, d), and 40 mmol/l glucose (n=4, Fig. 3d).

Recording of plasma membrane potential using intracellular microelectrodes on intact islets. (a) SUR1+/+ islets from C57Bl/6 mice in the presence of 0.5 mmol/l, 15 mmol/l and 25 mmol/l glucose. (b–d) Continuous oscillations in SUR1−/− islets perifused with 0.5 mmol/l, 15 mmol/l, 25 mmol/l and 40 mmol/l glucose. Record (d) is the direct continuation of (c). The recordings are representative of five (a), nine (b), eight (c) and four (d) experiments
Measurements with intracellular microelectrodes on intact islets clearly show oscillations in the absence of KATP channels. To exclude the possibility that the different ionic compositions of the bath solutions contribute to the conflicting result, additional patch-clamp experiments were done with the bicarbonate buffer used for intracellular recording. In this series of experiments the membrane potential did not oscillate and was persistently depolarized at 0.5 mmol/l (−38±3 mV, n=4) and 15 mmol/l glucose (−43±2 mV; n=7; not shown).
The microelectrode experiments showed that SUR1−/− beta-cell activity could be modulated by glucose (Fig. 3 and Table 1): The reduction of glucose concentration from 15 to 0.5 mmol/l led to a decrease in the frequency of Ca2+-dependent action potentials whereas the fraction of plateau phase (FOPP; percentage of time with spike activity) and burst duration were increased (Fig. 3b, n=9). Increasing the glucose concentration from 15 to 25 mmol/l or 40 mmol/l did not change the FOPP, but did increase the frequency of Ca2+-dependent action potentials (n=5 with 25 mmol/l glucose and n=4 with 40 mmol/l glucose). The influence of glucose concentration on the electrical activity in SUR1−/− islets is summarized in Table 1.
The membrane potential oscillations of SUR1−/− beta cells were not influenced by 100 µmol/l tolbutamide (n=9) or 100 µmol/l diazoxide (n=7) (Fig. 4).

Recordings of plasma membrane potential using intracellular microelectrodes on intact SUR1−/− islets in the presence of (a) 100 µmol/l tolbutamide or (b) 100 µmol/l diazoxide. The recordings are representative of nine (a) or seven (b) experiments
Effects of metabolic inhibition on SUR1+/+ and SUR1−/− beta cells
Figure 5 compares the changes in beta-cell membrane potential and current induced by the metabolic inhibitor, NaN3. When applied in the presence of 15 mmol/l glucose NaN3 markedly increased KATP currents in SUR1+/+ beta cells, hyperpolarizing their plasma membranes, and suppressing spike activity (Fig. 5a). The plateau potential was −52±4 mV in the presence of 15 mmol/l glucose and decreased to −76±1 mV after addition of NaN3 (n=4, p≤0.002). The KATP current was essentially zero in 15 mmol/l glucose (VC2), but increased to 12.0±4.4 pA during treatment with NaN3 (VC1; n=4, p≤0.01). NaN3 did not influence the remaining leak currents or plateau potential in SUR1−/− beta cells, but did partially inhibit spike activity (Fig. 5b). The plateau potential was −42±2 mV in 15 mmol/l glucose and −34±3 mV after addition of NaN3 (n=5). The corresponding leak current amplitudes were 0.9±0.2 pA (VC3) and 1.0±0.3 pA (VC4), respectively (n=3). The inhibitory effect of NaN3 in SUR1−/− beta cells is unlikely to be mediated by glucose metabolism as it also appeared in the presence of 0.5 mmol/l glucose (n=4).

Effects of NaN3 on beta-cell membrane potential and currents. (a) SUR1+/+ and (b) SUR1−/− beta cells. Recordings were made using the perforated patch mode on beta cells in 15 mmol/l glucose. In control beta cells 5 mmol/l NaN3 hyperpolarised the membrane potential and increased the KATP current amplitude (a). This recording is representative of four experiments. In SUR1−/− beta cells 5 mmol/l NaN3 did not alter the membrane potential (n=5) or current (n=3), but suppressed spike activity (b). The currents measured during voltage pulses to −60 mV (VC1 to VC4) are illustrated in (c). VC1 and VC3: 15 mmol/l glucose and 5 mmol/l NaN3, VC2 and VC4: 15 mmol/l glucose, VC1 and VC2: SUR1+/+ beta cell, VC3 and VC4: SUR1−/− beta cell
Ca2+ homeostasis and ATP production are tightly coupled in normal beta cells raising the question of how the loss of glucose-dependent regulation of [Ca2+]c in isolated SUR1−/− beta cells influences their metabolism. The mitochondrial membrane potential, ΔΨ, reflects ATP production and thus changes in beta-cell metabolism in response to glucose. Increasing the glucose concentration from 0.5 mmol/l to 15 mmol/l induced a clear hyperpolarization of ΔΨ in both SUR1+/+ (Fig. 6a) and SUR1−/− beta-cells (Fig. 6b) followed by a smaller depolarization. The hyperpolarization is caused by an increase in ATP production while the depolarization (arrow heads in Fig. 6) is a consequence of Ca2+ influx [24]. The addition of NaN3 in 15 mmol/l glucose depolarized ΔΨ in both cell types. The glucose-evoked hyperpolarization was larger in wildtype (647±49 a.u., n=21, or 48±3% compared to maximal depolarization achieved with 5 mmol/l NaN3, respectively, n=6) than knock-out beta cells (365±74 a.u., n=13; p≤0.002, or 26±2% compared to maximal depolarization by 5 mmol/l NaN3, respectively, n=12; p≤0.0001). The result suggests that the mitochondria of SUR1−/− beta cells do not hyperpolarize to the same extend as in wildtype beta cells and implies a lower rate of ATP production. This might, at least in part, be due to the higher [Ca2+]c in the knock-out beta cells which is expected to induce a depolarizing current in the inner mitochondrial membrane [24]. However, hyperpolarization of SUR1−/− beta cell mitochondria was not increased by removing extracellular Ca2+ (n=14), thus additional factors contribute to this difference.

Effects of glucose concentration and NaN3 on mitochondrial membrane potential (ΔΨ). (a) SUR1+/+ (b) and SUR1−/− beta cells. Increasing glucose from 0.5 to 15 mmol/l hyperpolarised ΔΨ in both cell types. This is indicated by the decrease in fluorescence (a.u.=arbitrary units) followed by a small increase (arrowheads) due to Ca2+ influx into cytosol and mitochondria. In 15 mmol/l glucose, NaN3 depolarized ΔΨ. The effects of glucose and NaN3 were smaller in SUR1−/− versus SUR1+/+ beta cells. These recordings are representative of twenty-one (glucose) and six (NaN3) experiments with SUR1+/+ beta cells and thirteen (glucose) and twelve (NaN3) experiments with SUR1−/− beta cells
Effects of galanin on SUR1+/+ and SUR1−/− beta cells
In addition to metabolic signals, beta-cell activity is regulated by hormones and neuropeptides. The inhibitory effect of galanin on insulin secretion has been suggested to require activation of KATP channels in various insulin-secreting cell lines [12, 30]. We tested the ability of this neuropeptide to hyperpolarize SUR1+/+ vs SUR1−/− beta cells and affect [Ca2+]c. Galanin slightly hyperpolarized the membrane potential transiently suppressing spike activity in both SUR1+/+ (Fig. 7a) and SUR1−/− beta cells (Fig. 7b) (−10±3 mV, n=9 vs −7±1 mV, n=5, respectively). The galanin-induced current is too small to be detected under these conditions even in the absence of KATP currents. The average membrane conductance in SUR1+/+ beta cells amounted to 0.20±0.04 nS in the presence of 15 mmol/l glucose and 0.31±0.07 nS after addition of 50 nmol/l galanin (n=9). In SUR1−/− beta cells the average conductance was 0.09±0.03 nS before and 0.11±0.02 nS after treatment with galanin (n=3). As the beta-cell membrane resistance is high, even small changes in current are sufficient to evoke membrane hyperpolarization. Galanin lowered [Ca2+]c in both cell types (Fig. 8a–d), but consistent with the incomplete suppression of spike activity the effect was transient. In SUR1+/+ beta cells the first peak after increasing the glucose concentration from 0.5 mmol/l to 15 mmol/l reached 407±21 nmol/l (n=16). Application of galanin led to a rapid, transient decrease to 96±8 nmol/l (n=16; p≤0.0001) followed by [Ca2+]c oscillations and/or a steady sustained level. The galanin induced changes in [Ca2+]c in SUR1−/− beta cells were similar; before addition, [Ca2+]c was 276±21 nmol/l (n=9) in the presence of 15 mmol/l glucose, then dropped to 105±19 nmol/l (n=9; p≤0.0001) after application before oscillations resumed. To ensure the lowering of [Ca2+]c was not coincidental with the end of a phase with increased [Ca2+]c, galanin was applied immediately after the beginning of the first rise in [Ca2+]c observed with 15 mmol/l glucose in 3 out of the 16 experiments with SUR1+/+ cells (Fig. 8b) and three out of the nine experiments with SUR1−/− beta cells (Fig. 8d).

Effects of galanin on beta-cell membrane potential and currents. (a) SUR1+/+ (b) and SUR1−/− beta cells. Galanin (50 nmol/l) slightly hyperpolarized the membrane potential and transiently suppressed electrical activity in both cell types. The currents measured during voltage pulses to −60 mV (VC1 and VC2) in the presence of galanin are illustrated in (c). These recordings are representative of nine experiments with SUR1+/+ beta cells and five experiments with SUR1−/− beta cells

Effects of galanin on beta-cell [Ca2+]c. (a, b) SUR1+/+ and (c, d) SUR1−/− beta cells. 50 nmol/l galanin transiently decreased [Ca2+]c and subsequently induced oscillations in SUR1+/+ as well as SUR1−/− beta cells in 15 mmol/l glucose. In b and d galanin was added immediately after the first rise of [Ca2+]c in 15 mmol/l glucose to show that the effect of galanin is independent of the point of application. The recordings are representative of thirteen (a) and three (b) experiments for SUR1+/+ and six (c) and three (d) experiments for SUR1−/− beta cells
Discussion
In SUR1 knock-out mice the beta-cell membrane potential, measured using the whole-cell configuration of the patch-clamp technique, was depolarized with continuous spike activity occurring in all cells independent of the glucose concentration. This is in agreement with the occurrence of spontaneous, Ca2+-dependent action potentials observed in all beta cells lacking functional KATP channels, i.e., beta cells from Kir6.2 [5] and SUR1 knock-out mice [6, 7] or beta cells from patients with PHHI (persistent hyperinsulinaemic hypoglycaemia of infancy) [8], a disorder which can be caused by the loss of functional KATP channels [8]. The depolarized membrane potential, Ca2+-dependent action potentials, and increased [Ca2+]c suggest that insulin secretion should be increased and result in hypoglycaemia. This is the case in patients with PHHI, but not in SUR1−/− or Kir6.2−/− mice which are mildly glucose intolerant, but not hypoglycaemic [5, 6, 7]. Thus rodents are able to compensate for the lack of functional KATP channels.
The [Ca2+]c oscillations that occur in some SUR1−/− beta cells were unexpected for two reasons: First, the generally accepted assumption is that changes in beta-cell membrane potential govern changes in [Ca2+]c [26] and thus insulin secretion [31]. Second, KATP channels have been considered to play a key role in the oscillatory activity of beta cells [24, 32, 33, 34]. For example, it has been reported recently that a rise in [Ca2+]c lowers ATP production evoking an increase in the KATP currents which act as a negative feedback to reduce Ca2+ influx by hyperpolarizing the beta cell [35]. In normal beta cells oscillations in [Ca2+]c are thought to be necessary for pulsatile insulin secretion [36]. Whether this applies to SUR1−/− beta cells remains to be verified. Our patch-clamp experiments could be interpreted to suggest that [Ca2+]c oscillations in isolated SUR1−/− beta cells are regulated independently of membrane potential. However, it is often difficult to detect membrane potential oscillations when patch-clamping single or small clusters of beta cells [27, 28, 29], although they invariably occur in experiments with intact islets [37], when electrically coupled cells, surrounded by nerve endings and blood vessels, are impaled by intracellular microelectrodes. In short, microelectrode impalements are a better reflection of the situation in vivo than patch-clamp experiments. Of interest, it has been reported [38] that the pattern of [Ca2+]c oscillations observed in single beta cells can differ markedly from those recorded with intact islets. Our data show that the membrane potential of SUR1−/− beta cells can oscillate in the absence of functional KATP channels. This points to the existence of a second underlying oscillator able to substitute in SUR1−/− mice for the one in WT beta cells involving KATP channels. Several lines of evidence suggest that this secondary oscillator may not be operative in normal mouse beta cells. First, it is almost impossible to obtain continuous oscillations of membrane potential or [Ca2+]c in wildtype islets comparable to the behaviour of SUR1−/− islets when KATP channels are inhibited by tolbutamide and the perifusion time with glucose-free medium is sufficient to exclude the influence of glucose metabolism [39]. Second, oscillations are not observed when KATP channels are opened with diazoxide and cells are depolarized by increasing the external K+ concentration [40, 41]. Recently, it has been shown that oscillatory activity in wildtype beta cells involves activation of a low-conductance Ca2+-dependent K+ current (IK,slow) [42, 43].
While both membrane potential and [Ca2+]c clearly oscillate in SUR1−/− beta cells in islets, determining whether membrane potential and [Ca2+]c are as tightly coupled as in control beta cells [26] will require simultaneous measurement of Vm and [Ca2+]c. The experiments with galanin support the view that changes in [Ca2+]c in SUR1−/− beta cells, like SUR1+/+ beta cells, are governed by changes in membrane potential since galanin-induced hyperpolarization caused a decrease in [Ca2+]c in both. Patch-clamp recordings from isolated beta cells from PHHI patients exhibit Ca2+-dependent spikes or action potentials equivalent to those observed in SUR1−/− beta cells. We are unaware of any intracellular microelectrode studies on PHHI islets. One plausible explanation for the severity of the disturbance in insulin secretion in PHHI patients versus the knock-out mouse models is that PHHI beta cells are persistently depolarized in islets. Corroboration for this speculation will require measurement of the beta-cell membrane potential in PHHI islets using intracellular microelectrodes.The discrepancy in glucose homeostasis between animal models lacking functional beta-cell KATP channels and PHHI patients raises interesting questions about the mechanisms involved in normalising insulin secretion in the knockout animals. We used the metabolic inhibitor NaN3 to test the possibility that metabolic factors influence beta-cell activity and thus insulin secretion at a site distinct from KATP channels. NaN3 increased the KATP current amplitude and thus hyperpolarized the plasma membrane potential of beta cells from wildtype mice (Fig. 5, [44]), an effect attributed to reduced ATP production and diminished ATP/ADP. Remarkably, NaN3 influenced the electrical activity of SUR1−/− beta cells by decreasing spike activity without concomitant membrane hyperpolarization. We recently showed that NaN3 inhibits L-type Ca2+ currents in SUR1+/+ beta cells using the standard whole-cell configuration [44]. This implies the reduction of Ca2+ action potentials in SUR1−/− beta cells could result from a direct effect of NaN3 on the L-type Ca2+ channel. We cannot exclude that inhibition of mitochondrial metabolism has additional effects on L-type Ca2+ channel activity as suggested by [45] in experiments with oligomycin . The data imply that the membrane hyperpolarization induced by decreasing the metabolic rate in SUR1+/+ beta cells is mediated by openings of KATP channels, and that decreased ATP/ADP alone does not influence the membrane potential in SUR1−/− beta cells. Activation of other K+ channels by adrenaline or neuropeptides, e.g. somatostatin or galanin, known to reduce glucose-induced insulin secretion, are candidates for suppression of the hyperinsulinaemia observed in PHHI and could contribute to glucose homeostasis in KATP-channel deficient mice. Somatostatin is used to treat PHHI patients [46] where it is suggested to activate a K+ channel distinct from the KATP channel. Galanin has been reported to lower insulin secretion via a direct effect on exocytosis [13, 16] and has been suggested to increase the K+ permeability of beta cells [11, 15]. One report [15] shows that clonidine induces a K+ current insensitive to sulphonylureas in mouse beta cells. For galanin a direct proof of the nature of the K+ channel underlying membrane hyperpolarisation is still lacking. Direct activation of KATP channels has been described for various tumour cell lines [12, 30], but these results may not directly transfer to the normal situation. Our experiments indicate that there is no significant difference in the action of galanin on SUR1+/+ compared with SUR1−/− beta cells and exclude the possibility that galanin evokes membrane hyperpolarisation and lowering of [Ca2+]c via KATP channels.
The mitochondrial membrane potential, ΔΨ, governs ATP production and thus reflects cellular metabolic status. The glucose-induced hyperpolarization and NaN3-induced depolarization of ΔΨ are markedly lower in beta cells from SUR1−/− mice implying reduced ATP synthesis in the knock-out beta cells. This suggests that [Ca2+]c may not be strongly coupled to glucose metabolism in SUR1−/− beta cells. Changes in [Ca2+]c are known to influence [Ca2+]m, the mitochondrial Ca2+ concentration [47, 48] and persistent increased [Ca2+]c in the presence of a low glucose could provoke Ca2+ influx into mitochondria and result in their depolarization. This would increase the open probability of the mitochondrial transition pore (mPTP) [49] and would explain the decreased hyperpolarization of ΔΨ in SUR1−/− beta cells. This hypothesis is not fully supported by our experiments with Ca2+-free medium, therefore additional factors appear to contribute to impaired mitochondrial function.
In conclusion, the new insights gained from these experiments with KATP-channel deficient mice are: (i) Oscillations in plasma membrane potential and [Ca2+]c can occur despite the loss of channels presumed to play a key role in normal beta-cell oscillations; (ii) the oscillations of Vm and [Ca2+]c in SUR1−/− beta cells are not driven by glucose metabolism, but changes in [Ca2+]c seem to be coupled to Vm; (iii) the loss of KATP channels affects mitochondrial metabolism; (iv) the action of galanin does not require KATP channels. Consequently, this neuropeptide is able to influence membrane potential and [Ca2+]c in SUR1−/− beta cells and could constitute a KATP channel independent contribution to the maintenance of glucose homeostasis in these cells.
Abbreviations
- Kir6.2:
-
inward rectifying K+ channel type 6.2
- SUR1:
-
sulfonylurea receptor type1
- ΔΨ:
-
mitochondrial membrane potential
- Vm:
-
plasmamembrane potential
- [Ca2+]c :
-
cytosolic free calcium concentration
References
Ashcroft F, Rorsman P (1989) Electrophysiology of the pancreatic β-cell. Prog Biophys Mol Biol 54:87–143
Henquin JC (2000) Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49:1751–1760
Gembal M, Gilon P, Henquin J-C (1992) Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse β-cells. J Clin Invest 89:1288–1295
Sato Y, Aizawa T, Komatsu M, Okada N, Yamada T (1992) Dual functional role of membrane depolarization/Ca2+ influx in rat pancreatic β-cell. Diabetes 41:438–443
Miki T, Nagashima K, Tashiro F et al. (1998) Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc Natl Acad Sci USA 95:10402–10406
Seghers V, Nakazaki M, DeMayo F, Aguilar-Bryan L, Bryan J (2000) SUR1 knockout mice A model for KATP channel-independent regulation of insulin secretion. J Biol Chem 275:9270–9277
Shiota C, Larsson O, Shelton KD et al. (2002) Sulfonylurea receptor type 1 knock-out mice have intact feeding-stimulated insulin secretion despite marked impairment in their response to glucose. J Biol Chem 277:37176–37183
Kane C, Shepherd RM, Squires PE et al. (1996) Loss of functional KATP channels in pancreatic β-cells causes persistent hyperinsulinemic hypoglycemia of infancy. Nat Med 2:1344–1347
Aguilar-Bryan L, Bryan J (1999) ATP-sensitive potassium channels, sulfonylurea receptors, and persistent hyperinsulinemic hypoglycaemia of infancy. Diabetes Rev 4:336–346
Nakazaki M, Crane A, Hu M, Seghers V, Ullrich S, Aguilar-Bryan L, Bryan J (2002) cAMP-activated protein kinase-independent potentiation of insulin secretion by cAMP is impaired inSUR1 null islets. Diabetes 51:3440–3449
Drews G, Debuyser A, Nenquin M, Henquin JC (1990) Galanin and epinephrine act on distinct receptors to inhibit insulin release by the same mechanisms including an increase in K+ permeability of the B-cell membrane. Endocrinology 126:1646–1653
Dunne MJ, Bullett MJ, Li GD, Wollheim CB, Petersen OH (1989) Galanin activates nucleotide-dependent K+ channels in insulin-secreting cells via a pertussis toxin-sensitive G-Protein. EMBO J 8:413–420
Ullrich S, Wollheim CB (1989) Galanin inhibits insulin secretion by direct interference with exocytosis. FEBS Lett 247:401–404
Homaidan FR, Sharp GW, Nowak LM (1991) Galanin inhibits a dyhydropyridine-sensitive Ca2+ current in the RINm5f cell line. Proc Natl Acad Sci USA 88:8744–8748
Rorsman P, Bokvist K, Ämmälä C, Arkhammar P, Berggren P-O, Larsson O, Wåhlander K (1991) Activation by adrenaline of a low-conductance G protein-dependent K+ channel in mouse pancreatic B cells. Nature 349:77–79
Renström E, Ding W-G, Bokvist K, Rorsman P (1996) Neurotransmitter-induced inhibition of exocytosis in insulin-secreting β-cells by activation of calcineurin. Neuron 17:513–522
Plant TD (1988) Properties and calcium-dependent inactivation of calcium currents in cultured mouse pancreatic B-cells. J Physiol 404:731–747
Trube G, Rorsman P, Ohno-Shosaku T (1986) Opposite effects of tolbutamide and diazoxide on the ATP-dependent K+ channel in mouse pancreatic beta-cells. Pflugers Arch Eur J Physiol 407:493–499
Meissner HP, Schmelz H (1974) Membrane potential of beta-cells in pancreatic islets. Pflugers Arch Eur J Physiol 351:195–206
Barg S, Galvanovskis J, Göpel SO, Rorsman P, Eliasson L (2000) Tight coupling between electrical activity and exocytosis in mouse glucagon-secreting α-cells. Diabetes 49:1500–1510
Grynkiewicz G, Poenie M, Tsien RY (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260:3440–3450
Nadal A, Quesada I, Soria B (1999) Homologous and heterologous asynchronicity between identified α-, β- and δ-cells within intact islets of Langerhans in the mouse. J Physiol 517:85–93
Duchen MR, Smith PA, Ashcroft FM (1993) Substrate-dependent changes in mitochondrial function, intracellular free calcium concentration and membrane channels in pancreatic β-cells. Biochem J 294:35–42
Krippeit-Drews P, Düfer M, Drews G (2000) Parallel oscillations of intracellular calcium activity and mitochondrial membrane potential in mouse pancreatic B-cells. Biochem Biophys Res Commun 267:179–183
Grapengiesser E, Gylfe E, Hellman B (1988) Dual effect of glucose on cytoplasmic Ca2+ in single pancreatic β-cells. Biochem Biophys Res Commun 150:419–425
Santos RM, Rosario LM, Nadal A, Garcia-Sancho J, Soria B, Valdeolmillos M (1991) Widespread synchronous [Ca2+]i oscillations due to bursting electrical activity in single pancreatic islets. Pflugers Arch Eur J Physiol 418:417–422
Smith PA, Ashcroft FM, Rorsman P (1990) Simultaneous recordings of glucose dependent electrical activity and ATP-regulated K+-currents in isolated mouse pancreatic β-cells. FEBS Letters 261:167–190
Ämmälä C, Larsson O, Berggren P-O, Bokvist K, Juntti-Berggren L, Kindmark H, Rorsman P (1991) Inositol trisphosphate-dependent periodic activation of a Ca2+-activated K+ conductance in glucose-stimulated pancreatic β-cells. Nature 353:849–852
Kinard TA, Vries G de, Sherman A, Satin LS (1999) Modulation of the bursting properties of single mouse pancreatic β-cells by artificial conductances. Biophys J 76:1423–1435
Weille J de, Schmid-Antomarchi J, Fosset M, Lazdunski M (1988) ATP-sensitive K+ channels that are blocked by hypoglycemia-inducing sulfonylureas in insulin-secreting cells are activated by galanin, a hyperglycemia-inducing hormone. Proc Natl Acad Sci USA 85:1312–1316
Gilon P, Shepherd RM, Henquin JC (1993) Oscillations of secretion driven by oscillations of cytoplasmic Ca2+ as evidences in single pancreatic islets. J Biol Chem 268:22265–22268
Tornheim K (1997) Are metabolic oscillations responsible for normal oscillatory insulin secretion? Diabetes 46:1375–1380
Detimary P, Gilon P, Henquin JC (1998) Interplay between cytoplasmic Ca2+ and the ATP/ADP ratio: a feedback control mechanism in mouse pancreatic islets. Biochem J 333:269–274
Kindmark H, Köhler M, Brown G, Bränström R, Larsson O, Berggren PO (2001) Glucose-induced oscillations in cytoplasmic free Ca2+ concentration precede oscillations in mitochondrial membrane potential in the pancreatic beta-cell. J Biol Chem 276:34530–34536
Rolland JF, Henquin JC, Gilon P (2002) Feedback control of the ATP-sensitive K+ current by cytosolic Ca2+ contributes to oscillations of the membrane potential in pancreatic β-cells. Diabetes 51:376–384
Kjems LL, Ravier MA, Jonas JC, Henquin JC (2002) Do oscillations of insulin secretion occur in the absence of cytoplasmic Ca2+ oscillations in β-cells? Diabetes 51:S177–S182
Drews G, Krämer C, Düfer M, Krippeit-Drews P (2000) Contrasting effects of alloxan on islets and single mouse pancreatic β-cells. Biochem J 352:389–397
Gilon P, Ravier MA, Jonas JC, Henquin JC (2002) Control mechanisms of the oscillations of insulin secretion in vitro and in vivo. Diabetes 51:S144–S151
Henquin J-C (1998) A minimum of fuel is necessary for tolbutamide to mimic the effect of glucose on electrical activity in pancreatic B-cells. Endocrinology 139:993–998
Gilon P, Henquin J-C (1992) Influence of membrane potential changes on cytoplasmic Ca2+ concentration in an electrically excitable cell, the insulin-secreting pancreatic B-cell. J Biol Chem 267:20173–20720
Sato Y, Anello M, Henquin JC (1999) Glucose regulation of insulin secretion independent of the opening or closure of adenosine triphosphate-sensitive K+ channels in beta cells. Endocrinology 140:2252–2257
Göpel SO, Kanno T, Barg S, Eliasson L, Galvanovskis J, Renström E, Rorsman P (1999) Activation of Ca2+-dependent K+ channels contributes to rhythmic firing of action potentials in mouse pancreatic β-cells. J Gen Physiol 114:759–769
Goforth PB, Bertram R, Khan FA, Zhang M, Sherman A, Satin LS (2002) Calcium-activated K+ channels of mouse β-cells are controlled by both store and cytoplasmic Ca2+: experimental and theoretical studies. J Gen Physiol 120:301–322
Düfer M, Krippeit-Drews P, Drews G (2002) Inhibition of mitochondrial function affects cellular Ca2+ handling in pancreatic B-cells. Pflugers Arch Eur J Physiol 444:236–243
Smith PA, Rorsman P, Ashcroft F (1989) Modulation of dihydropyridine-sensitive Ca2+ channels by glucose metabolism in mouse pancreatic β-cells. Nature 342:550–553
Kane C, Lindley KJ, Johnson PR et al. (1997) Therapy for persistent hyperinsulinemic hypoglycemia of infancy. Understanding the responsiveness of beta cells to diazoxide and somatostatin. J Clin Invest 100:1888–1893
Kennedy ED, Rizzuto R, Theler JM et al. (1996) Glucose-stimulated insulin secretion correlates with changes in mitochondrial and cytosolic Ca2+ in aequorin-expressing INS-1 cells. J Clin Invest 98:2524–2538
Meachler P, Kennedy ED, Pozzan T, Wollheim CB (1997) Mitochondrial activation directly triggers the exocytosis of insulin in permeabilized pancreatic β-cells. EMBO J 16:3833–3841
Ichas F, Mazat J-P (1998) From calcium signalling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim Biophys Acta 1366:33–50
Acknowledgements
This work was supported by grants from the Deutsche Forschungsgemeinschaft (Dr225/6-1) and the U.S. National Institutes of Health (DK52771).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Düfer, M., Haspel, D., Krippeit-Drews, P. et al. Oscillations of membrane potential and cytosolic Ca2+ concentration in SUR1−/− beta cells. Diabetologia 47, 488–498 (2004). https://doi.org/10.1007/s00125-004-1348-0
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-004-1348-0