
Abstract
Aims/hypothesis
Diabetes is known to reduce survival after myocardial infarction. Our aim was to examine whether diabetes is associated with enhanced cardiomyocyte apoptosis and thus interferes with the post-infarction remodelling process in myocardium in rat.
Methods
Four weeks after intravenous streptozotocin (diabetic groups) or citrate buffer (controls) injection, myocardial infarction was produced by ligation of left descending coronary artery. Level of cardiomyocyte apoptosis was quantified by TUNEL and caspase-3 methods. Collagen volume fraction and connective tissue growth factor were determined under microscope. Left ventricular dimensions were evaluated by echocardiography and planimetry.
Results
The number of apoptotic cardiomyocytes was equally high in diabetic and non-diabetic rats after 1 week from infarction. At 12 weeks after infarction the number of apoptotic cells was higher in the diabetic as compared to non-diabetic rats both in the border zone of infarction and in non-infarcted area. Correspondingly, left ventricular end diastolic diameter, relative cardiac weight, connective tissue growth factor-expression and fibrosis were increased in diabetic compared with non-diabetic rats with myocardial infarction.
Conclusion/interpretation
Sustained cardiomyocyte apoptosis, left ventricular enlargement, increased cardiac fibrosis and enhanced profibrogenic connective tissue growth factor expression were detected after myocardial infarction in experimental diabetes. Apoptotic myocyte loss could be an important mechanism contributing to progressive dilatation of the heart and poor prognosis after myocardial infarction in diabetes.
Similar content being viewed by others


Diabetic patients have about a two-fold increase in a risk of short- and long-term mortality after myocardial infarction (MI). Even after adjustment for the infarct size, and the extent of coronary disease, diabetes is independently associated with reduced survival conferring a similar risk of mortality as a previous myocardial infarction [1]. The mechanisms mediating adverse effects of diabetes include neurohormonal and metabolic factors as well as changes in coagulation factors. Together with more extensive coronary artery disease, this leads to higher risk of new MI [2].
We and others have shown that cardiomyocyte apoptosis remains increased several months post-MI, correlating with ventricular enlargement [3, 4]. As recent studies have shown that diabetes could be associated with enhanced susceptibility to apoptosis [5], we wanted to study in an experimental rat MI model, whether cardiomyocyte apoptosis is increased in diabetic compared to non-diabetic animals after MI, and whether it would relate to cardiac remodelling.
Materials and methods
Study design
Male Wistar rats were randomized to receive either streptozotocin (STZ) 45 mg/kg (Sigma Chemical, St Louis, Mo., USA; diabetic group) or citrate buffer (non-diabetic control group) intravenously by the tail vein. Blood glucose was determined with Glucocard II Super test meter 3 days after the injection and at 2-week intervals thereafter throughout the study under non-fasting conditions. Animals with a blood glucose concentration greater than 20 mmol/l were included in the diabetic group. Long-acting insulin (Insulin Humutard Ultra, Lilly, Fegersheim, France), 4 IU per day s.c. was administered to diabetic animals to prevent ketoacidosis and to promote weight gain without rendering the rats euglycaemic. The Helsinki University animals ethics committee and the Provincial State Office of Southern Finland approved the procedures. Four weeks after the STZ or citrate buffer injections the animals underwent either sham or MI operation (n=7–11 in each group). MI was produced by ligation of the left anterior descending coronary artery as described [3]. The rats were killed 1, 4 and 12 weeks after the operation, the hearts were weighed, embedded in paraffin, and cut into sections for histology, planimetry, and assessment of apoptosis and connective tissue growth factor (CTGF).
Histology and infarct size
The presence of signs of acute MI (eosinophilia, karyolysis, and leucocyte infiltration) or collagen scars compatible with MI was verified by examination of Van Gieson stained transverse left ventricular (LV) sections. Infarct size was determined planimetrically [3].
Apoptosis
Apoptotic cardiomyocytes were detected with terminal deoxynucleotide transferase-mediated ddUTP nick end-labelling (TUNEL) assay which labels DNA strand breaks formed during the final stages of apoptosis. The assay was standardized as described [6]. Caspase-3 is a specialized cysteine-dependent protease being directly involved in cleavage of structural elements in both the cytoplasm and nucleus of cells causing the morphological features as well as internucleosomal DNA fragmentation in apoptotic cells. Activation of caspase-3 was detected immunohistochemically with an antibody recognizing the active form of caspase-3 (Asp175, Cell Signaling Technologies, Beverly, Mass., USA) and visualized with Vectastain ABC Elite Kit (Vector Laboratories, Burlingame, Calif., USA) [7]. The percentages of TUNEL-positive and caspase-3-positive myocytes were calculated separately in the border zone of infarction and in the remote non-infarcted myocardium in the transverse LV section that showed maximal infarct size [3].
Cardiac dimensions and myocardial fibrosis
All animals underwent echocardiography under medetomidine 0.25 mg/kg (Domitor, Orion Pharma, Espoo, Finland) sedation before the operation, at 1 and 4 weeks. Two-dimensional echocardiographical measurements were carried out using a 7–10 MHz phased array sector probe. LV end-diastolic diameter (LVEDD) was determined from parasternal projections [8]. Interstitial collagen volume fraction was determined with picrosirius red method from four different slices of Direct Red 80 (Fluka Chemika, Switzerland) stained left ventricular sections under polarized light. Areas of connective tissue network and myocytes were quantified by semi-automated computer-based analysis system [9]. Connective tissue growth factor (CTGF) was determined immunohistochemically with an anti-CTGF antibody (Abcam, Cambridge, UK) as a primary antibody. Each sample was scored from 0 to 4 according to the CTGF intensity in stained sections [10]. Cardiac weights are presented relative to rat body weight (mg/g). Brain natriuretic peptide (BNP) mRNA expression was determined both from the border zone of MI and from the remote area of the LV as described earlier [11]. Results are presented as means±SEM. Data were analyzed using analysis of variance (ANOVA) followed by Neuman-Keul’s test. A p value of less than 0.05 was considered statistically significant.
Results
The size of the myocardial infarction was similar in diabetic and non-diabetic groups (Table 1). Blood glucose concentration in diabetic rats was 28.1±0.7 mmol/l and 5.6±0.3 mmol/l in non-diabetic rats. Body weight did not differ between the MI groups. Relative heart weight was significantly higher in diabetic rats in comparison with non-diabetic rats (p<0.05).
Apoptosis in myocardial tissue samples was detected by TUNEL and caspase methods (Fig. 1, Fig. 2). The percentage of apoptotic cardiomyocytes in sham operated control rats was low throughout the experiment as judged by both TUNEL (0.004–0.008±0.001%) and caspase-3 (0.001–0.009±0.006%) methods. No differences were found in the level of apoptosis between diabetic and non-diabetic sham-operated rats. The number of apoptotic cells increased after the infarction both in the border zone and remote area in both groups as compared to the sham-operated rats (Fig. 2, p<0.001). The highest levels of myocardial apoptosis in border zone were dectected 1 week after the infarction. The number of apoptotic cells in border zone decreased in a more gradual manner after MI in diabetic rats and the difference was statistically significant at time-point 12 weeks . Similarly, in the remote areas the percentage of the apoptotic cells remained higher in diabetic as compared to non-diabetic rats(p<0.01 by TUNEL and p<0.001 by caspase-3) (Fig. 2).

Time-scale of myocardial apoptosis in diabetic and non-diabetic rats 1 to 12 weeks after myocardial infarction in border zone (a) and in remote area (b) measured by TUNEL and caspase-3 methods. *p<0.05, **p<0.01, ***p<0.001 in comparison with 1-week levels

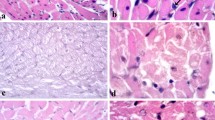
Cardiomyocyte apoptosis in diabetic vs non-diabetic rats measured by TUNEL and caspase-3 methods at 12 weeks. (A) and (C) show apoptosis in the border zone of MI measured by TUNEL and caspase-3 methods, respectively. (B) and (D) show the amount of apoptosis in the remote area of LV measured by the same methods. (E) shows cardiomyocyte in border zone of MI demonstrating an intense local immunoreactivity of the activated caspase-3 (arrow, Ea), the nucleus of the same cell seen in an adjacent tissue section is TUNEL positive (arrow, Eb) and contains condensed chromatin as shown by DAPI-staining (arrow, Ec). Magnification ×400
Left ventricular dimensions were measured by echocardiography (at baseline, 1 week and 4 weeks after MI) and by planimetry 12 weeks after MI (Table 2). Before the operation LVEDD was 8.2±0.1 mm in diabetic rats and 8.3±0.1 mm in non-diabetic animals (p=0.57). Four weeks after coronary ligation LVEDD (10.0±0.2 vs 9.6±0.2 mm) was significantly larger in diabetic compared with non-diabetic MI rats (p<0.05). Larger cardiac dimensions were also detected in sham-operated diabetic compared with non-diabetic animals (LVEDD 8.8±0.1 mm vs 8.4±0.1 mm, respectively, p<0.05) at 4 weeks. In accordance with the echocardioraphical results, planimetry from transverse left ventricular sections 12 weeks after MI showed a trend towards larger left ventricular areas in diabetic animals as compared to non-diabetic animals. Myocardial collagen content and CTGF score were measured from stained myocardial tissue samples (Fig. 3A–D). Both collagen volume fraction and CTGF score were higher in diabetic as compared to non-diabetic MI rats (p<0.05 and p<0.01 respectively).

Myocardial fibrosis and connective tissue growth factor (CTGF) stainings 12 weeks after myocardial infarction in diabetic and non-diabetic rats. (A) shows picrosirius red stained left venticular sections under polarized light from non-diabetic (Aa) and diabetic (Ab) rats. (B) depicts myocardial tissue samples stained by anti-CTGF antibody in non-diabetic (Ba) and diabetic (Bb) rats. (C) and (D) show quantitated collagen volume fraction (CVF) and CTGF score, respectively
BNP mRNA expression in the remote zone of myocardial infarction showed a gradual upregulation between 1 and 12 weeks after the infarction (Fig. 4). After 12 weeks, remote zone BNP mRNA levels were higher in diabetic animals in comparison with non-diabetic animals (p<0.05). Border zone BNP mRNA expression was highly upregulated 1 week after MI in both groups. Although, border zone BNP mRNA levels decreased from 1 to 12 weeks in both the diabetic and non-diabetic groups, the levels remained significantly higher in diabetic animals as compared to non-diabetic animals (p<0.05).

Myocardial BNP mRNA expression in remote area (a) and border zone (b) after myocardial infarction in diabetic (black bars) and non-diabetic rats (open bars). Results are shown relative to GAPDH and 1 week vehicle (remote zone)
Discussion
The finding in this study is that experimental diabetes leads to prolonged cardiomyocyte apoptosis after MI. The enhanced apoptosis in diabetic rats with MI takes place in parallel with increased LV enlargement, hypertrophy and fibrosis, and increased expression of pro-fibrogenic and pro-apoptotic CTGF.
Myocardial remodelling after MI is characterized by compensatory hypertrophy of myocytes, ventricular dilatation and increased interstitial collagen deposition and fibrosis. The mechanisms behind the development of heart failure are not fully understood but continuous apoptotic loss of myocytes is suggested to be involved [12]. In experimental models apoptosis has been shown to continue at an elevated level after MI correlating to ventricular enlargement and severity of LV dysfunction [3, 13]. In our study in diabetic rats, the number of apoptotic cardiomyocytes remained high both in the border zone of infarction and in the remote non-infarcted area up to 12 weeks after coronary ligation compared to non-diabetic MI rats. Parallel to sustained activation of apoptosis, diabetic MI rats developed larger LVEDD, increased relative heart weight and higher amount of cardiac fibrosis compared to non-diabetic rats with MI.
Recently, hyperglycaemia was shown to induce apoptosis in STZ-induced diabetes in rats and mice. Exposure to high levels of glucose has been shown to activate apoptosis in cardiac myocytes and myoblasts in vitro [14, 15]. In our study, hyperglycaemia alone was not a sufficient stimulus to promote myocyte apoptosis as no differences in the amount of apoptosis were found between diabetic and non-diabetic sham-operated rats. In contrast to another study [14], STZ-diabetic rats in our experiment received insulin daily to keep the blood glucose level between 20–30 mmol/l. Moreover, possible effect of acute STZ-toxicity on apoptosis was avoided by timing the coronary ligation or sham operation 4 weeks after the STZ-injections.
Diabetes is known to predispose to development of cardiac structural and functional changes. In agreement with previous reports on diabetic cardiomyopathy in STZ-induced diabetes [16, 17], sham-operated diabetic rats in our study had larger LVEDD and higher relative heart weight than non-diabetic controls. However, sustained myocyte apoptosis in diabetic rats was observed only after they had been subjected to MI. This is in line with a study [5], which reported that insulin treatment inhibits cardiac apoptosis in diabetic mice. In contrast to a recent paper reporting increased acute myocardial infarct size caused by hyperglycaemia in ischaemia-reperfusion model [18], we did not find any difference in MI size between diabetic and non-diabetic animals in our experimental setting. Thus, the enhanced apoptosis cannot be attributed to larger infarcts in diabetic rats. In another recent work [19], it was shown that in mice subjected to coronary ligation, survival, LV function as well as fibrosis were affected by the presence of diabetes and the outcome was clearly worse in diabetic compared with non-diabetic mice. Taken together, these observations point to diabetes per se as the adverse prognostic factor.
The results of our work supported by another study [19] also suggest that this effect is mediated at least in part by enhanced apoptosis after myocardial infarction. Using a modified DNA ladder technique, it was shown [19] that apoptosis seemed to be increased in the diabetic compared with non-diabetic remote (non-infarcted) myocardium at 2 weeks after a massive MI. Using the quantitative TUNEL technique and demonstration of caspase-3 activation, we were able to show that the apoptotic signal remained increased in the diabetic compared with non-diabetic animals during prolonged (12 weeks) convalescence after moderate infarction. Moreover, apoptosis was localised in the cardiomyocytes of both the remote and the border zone areas. It is possible that relevant apoptosis occurs also in other cell types (including endothelial cells, fibroblasts or even inflammatory leukocytes), but detecting such phenomena in cardiac tissue samples with any degree of accuracy remains technically very demanding. Therefore, we have to limit our discussion to cardiomyocyte apoptosis which nevertheless is highly relevant to post-MI remodelling.
The hallmarks of cardiac remodelling after MI are dilatation, hypertrophy and fibrosis. The fact that apoptosis remained high also in the peri-infarct border zones of the diabetic animals points to the role of this anatomical area in the progressive dilatation of the left ventricle after MI. The key signal to increased apoptosis as well as hypertrophy in the border zone is myocardial stretch. Indeed, 12 weeks after MI brain natriuretic peptide (BNP) mRNA was higher in diabetic compared with non-diabetic animals both in the border and remote zones. We also have shown that the expression of CTGF, a fibrogenic protein induced by TGFβ1 and high glucose [20, 21], was upregulated in the hearts of diabetic MI rats. CTGF has been shown to induce apoptosis via downregulation of the anti-apoptotic genes [22]. In contrast, the replacement fibrosis after cardiomyocyte loss could be amplified by the increased expression of fibrogenic factors such as CTGF. There was no difference in the infarct size and the level of fibrosis between the MI groups at 4 weeks (data not shown). Therefore, we can speculate that increased and prolonged cardiomyocyte apoptosis might precede the development of myocardial fibrosis, another hallmark of the remodelling process.
In conclusion, sustained cardiomyocyte apoptosis is associated with LV enlargement and increased cardiac fibrosis after MI in experimental diabetes. Apoptotic myocyte loss could be an important mechanism contributing to progressive dilatation of the heart and poor prognosis after MI in diabetes.
Abbreviations
- STZ:
-
streptozotozin
- MI:
-
myocardial infarction
- CTGF:
-
connective tissue growth factor
- LV:
-
left ventricular
- LVEDD:
-
LV end-diastolic diameter
- BNP:
-
B-type natriuretic peptide
References
Mukamal KJ, Nesto RW, Cohen MC, et al. (2001) Impact of diabetes on long-term survival after acute myocardial infarction: Comparability of risk with prior myocardial infarction. Diabetes Care 24:1422–1427
Paulson DJ (1997) The diabetic heart is more sensitive to ischemic injury. Cardiovasc Res 34:104–112
Palojoki E, Saraste A, Eriksson A, et al. (2001) Cardiomyocyte apoptosis and ventricular remodeling after myocardial infarction in rats. Am J Physiol 280:H2726–H2731
Cheng W, Kajstura J, Nitahara JA, et al. (1996) Programmed myocyte cell death affects the viable myocardium after infarction in rats. Exp Cell Res 226:316–327
Cai L, Li W, Wang G, Guo L, Jiang Y, Kang YJ (2002) Hyperglycemia-induced apoptosis in mouse myocardium. Mitochondrial cytochrome c-mediated caspase-3 activation pathway. Diabetes 51:1938–1948
Saraste A, Pulkki K, Kallajoki M, Henriksen K, Parvinen M, Voipio-Pulkki L-M (1997) Apoptosis in human acute myocardial infarction. Circulation 95:320–323
Saraste A, Pulkki K (2000) Morphologic and biochemical hallmarks of apoptosis. Cardiovasc Res 45:528–537
Thomas WP, Gaber CE, Jacobs GJ (1993) Recommendations for standard in transthoracic two-dimensional echocardiography in the dog and cat. J Vet Intern Med 7:247–252
Brooks WW, Bing OHL, Robinson KG, Slawsky MT, Chaletsky DM, Conrad CH (1997) Effect of angiotensin-converting enzyme inhibition on myocardial fibrosis and function in hypertrophied and failing myocardium from spontaneous hypertensive rat. Circulation 96:4002–4010
Finckenberg P, Inkinen K, Ahonen J, et al. (2003) Angiotensin II induces connective tissue growth factor gene expression via calcineurin-dependent pathways. Am J Pathol 163:355–366
Bäcklund T, Palojoki E, Saraste A, et al. (2003) Effect of vasopeptidase inhibitor omapatrilat on cardiomyocyte apoptosis and ventricular remodeling in rat myocardial infarction. Cardiovasc Res 57:727–737
Saraste A, Pulkki K, Kallajoki M, et al. (1999) Cardiomyocyte apoptosis and progression of heart failure to transplantation. Eur J Clin Invest 29:380–386
Sam F, Sawyer DB, Chang DL-F, et al. (2000) Progressive left ventricular remodeling and apoptosis late after myocardial infarction in mouse heart. Am J Physiol 279:H422–H428
Fiordaliso F, Li B, Latini R, et al. (2000) Myocyte death in streptozotocin-induced diabetes in rats is angiotensin II-dependent. Lab Invest 80:513–527
Fiordaliso F, Leri A, Cesselli D, et al. (2001) Hyperglycemia activates p53 and p53-regulated genes leading to myocyte cell death. Diabetes 50:2363–2375
Miric G, Dallemagne C, Endre Z, Margolin S, Taylor SM, Brown L (2001) Reversal of cardiac and renal fibrosis by pirfenidone and spironolactone in streptozotocin-diabetic rats. Br J Pharmacol 133:687–694
Riva E, Andreoni G, Bianchi R, et al. (1998) Changes in diastolic function and collagen content in normotensive and hypertensive rats with long-term streptozotocin-induced diabetes. Pharmacol Res 37:233–240
Marfella R, D’Amico M, Di Filippo C, et al. (2002) Myocardial infarction in diabetic rats: role of hyperglycemia on infarct size and early expression of hypoxia-inducible factor 1. Diabetologia 45:1172–1181
Shiomi T, Tsutsui H, Ikeuchi M, et al. (2003) Streptozotocin-induced hyperglycemia exacerbates left ventricular remodeling and failure after experimental myocardial infarction. J Am Coll Cardiol 42:165–172
Duncan MR, Frazier KS, Abramson S, et al. (1999) Connective tissue growth factor mediates transforming growth factor beta-induced collagen synthesis: downregulation by cAMP. FASEB J 13:1774–17
Way KJ, Isshiki K, Suzuma K, et al. (2002) Expression of connective tissue growth factor is increased in injured myocardium associated with protein kinase C [beta]2 activation and diabetes. Diabetes 51:2709–2718
Hishikawa K, Oemar BS, Nakaki T (2001) Static pressure regulates connective tissue growth factor expression in human mesangial cells. J Biol Chem 276:16797–16816
Acknowledgements
We thank Prof. M. S. Nieminen for his valuable comments during the writing of the manuscript as well as for contributing to the echocardiography studies and Dr. T. Grönholm for her help conducting the experiment. We acknowledge M. Louhilainen, T. Suvanto, T. Riihimäki and R. Hatakka for excellent technical assistance. This study was supported by the Research Grants of Helsinki University Hospital and the Sigrid Juselius Foundation. M.L. and E.P. received personal grants from Paavo Nurmi Foundation and T.B. from von Frenckell’s Foundation. We also wish to thank Dr. P. Virkkunen for the access to echocardiographic facilities in the Department of Radiology, Helsinki University Hospital.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bäcklund, T., Palojoki, E., Saraste, A. et al. Sustained cardiomyocyte apoptosis and left ventricular remodelling after myocardial infarction in experimental diabetes. Diabetologia 47, 325–330 (2004). https://doi.org/10.1007/s00125-003-1311-5
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-003-1311-5