
Abstract
Aims/hypothesis
Although matrix metalloproteinase-9 (MMP-9) is specifically induced and apoptosis of endothelial cells is evidenced in diabetes mellitus, the mechanism of endocardial endothelial dysfunction in diabetes mellitus is not clear. The increase in MMP-9 activity is associated with endocardial endothelial apoptosis and dysfunction in diabetes mellitus.
Methods
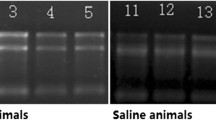
Diabetes was created by injecting 65 mg/kg alloxan in tail vein of MMP-9 knockout (–/–) and wild-type (WT, C57BL/J6) mice. At 8 weeks mice were grouped: (i) WT+saline; (ii) WT+alloxan; (iii) MMP+saline; (iv) MMP+alloxan. The MMP-9 genotype was determined by observing single PCR product of different mobility than the PCR product from wild-type in blood from tail vein.
Results
MMP-9 activity, measured by zymography, increased in plasma and in the left ventricle of alloxan-induced diabetic wild-type mice. The concentrations of cardiac inhibitor of metalloproteinase, that blocks MMP-9 activity, were decreased in diabetic MMP-9 knockouts as well as in wild-type mice. Diabetes induced apoptosis, detected by TUNEL assays, in wild-type but not in MMP-9 knockouts. Endocardial endothelial function was severely impaired in diabetic wild-type mice compared with normoglycaemic animals, while non-diabetic MMP-9 knockout mice showed partial endocardial endothelial dysfunction which was not further exacerbated by the developments of diabetes.
Conclusion/interpretation
The results suggest an association between increased MMP-9 activity and endocardial endothelial apoptosis in diabetic mice, while genetic ablation of MMP-9 correlated with amelioration of endocardial endothelial dysfunction and apoptosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Interstitial and perivascular fibrosis contribute to diabetic cardiomyopathy. Despite important strides made toward the understanding of mechanism of fibrosis, the role of extracellular matrix (ECM) composition and concentration in matrix accumulation is not clear. Sixteen percent of the myocardium is capillaries, including lumen and endothelium [1]. Capillary endothelium, strategically located between the superfusing luminal blood and the underlying cardiac muscle, plays an important role in controlling the myocardial performance [2, 3, 4, 5, 6, 7]. In fact evidence suggests that the frequency of apoptosis of capillary microvessel cells predicts the development of the histologic lesions in diabetes [8]. The capillary surface area is reduced and tissue thickness from capillaries to myocytes is increased in the left ventricle (LV) of diabetic rats [9]. In diabetes mellitus alteration in the ultrastructure leads to cardiovascular dysfunction [10, 11, 12, 13]. Structural pathological manifestations in diabetic cardiomyopathy are reversed by insulin treatment in rats [14]. The composition and concentration of collagen and elastin in the basement membrane of capillary endothelium contribute to the structural alterations in the heart leading to accumulation of oxidized-matrix between endothelium and myocytes. Abnormal collagen glycation, and chamber tissue stiffness, affecting diastolic function have appeared to be a major factor in impaired glucose tolerance in diabetes mellitus [15, 16]. Alteration in LV diastolic filling is also associated with reciprocal changes in the LV collagen gene [17], and accumulation of myocardial collagen in insulin-resistant syndrome [18]. There is enhanced ECM production in rat heart endothelial cells [19], and the degree of MMP activity is excessive in diabetes mellitus [20]. The elastinolytic proteinases are upregulated in the basement membrane of microvessels of diabetes [21]. MMP-2 and -9 degrade elastin [22], and MMP-2 also degrades ultrastructure collagen [23]. Because elastin turnover is remarkably lower than collagen [24], elastin and ultrastructural collagen are replaced by oxidatively modified stiffer collagen, leading to a greater distance between endothelium and myocyte, and impairing eeNO diffusion from endothelium to myocyte. Endothelial cell density is decreased, ECM is increased, and MMP is activated in diabetes mellitus. The mechanism of EE dysfunction, and matrix accumulation and increased tissue thickness between endothelium and muscle in diabetes mellitus is not clear. We hypothesize that MMP-9 instigates unabated proteolysis and leads to endothelial apoptosis and dysfunction.
Materials and methods
MMP-9 knockout and diabetes mouse model
MMP-9 was inactivated using homologous recombination in C57BL/6J mouse embryonic stem cells by disrupting the coding sequence of MMP-9 gene by Neo gene as described [25]. MMP-9 knockout (–/–) mice were bred. At 8 weeks male mice were selected and genotyping was carried out using PCR on tail vein blood. To generate wild-type litermate homozygous MMP-9-deficient and WT C57BL/6J mice were cross-bred. MMP-9 PCR was carried out using sense oligo primer (5′-CCA GTT TCC ATT CAT CTT CC-3′); and an antisense primer (5′-ACA GTA GTG GCC GTA GAA GG-3′) [gene code MN 004994]. PCR products were analysed on 1.2% agarose gel and stained by ethidium bromide. The phenotype was determined using gelatin zymography on tail vein blood. Male WT and MMP-9 (–/–) at age 8 to 10 weeks, weighing 25 to 30 g were divided into the following four study groups: (i) WT; (ii) WT + Alloxan (A); (iii) MMP; (iv) MMP + A. Diabetes was induced by a single dose of 65 mg/kg of alloxan injected through tail vein. Animal room temperature was maintained between 22°C and 24°C. A 12-h light to dark cycle was maintained by artificial illumination. In accordance with the National Institute of Health Guidelines for animal research, all animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Mississippi Medical Center, Jackson, USA. The animals were fed standard chow and water ad libitum. To determine whether treatment with alloxan caused any change in food and water intake, food and water were measured every 2 days during the treatment period. After 8 weeks of alloxan treatment physiological and biochemical parameters were measured.
Glucose
Blood was collected in heparinized tubes. The plasma was separated and glucose concentrations were measured using Bio-Rad glucose measurement kit.
Haemodynamic parameters
Mice were anesthetized with tribromoethanol (100 mg/kg i.p.). This drug had minimal effects on cardiovascular function in mice [26]. The aortic blood pressure, heart rate (HR), and systolic and diastolic blood pressure (SBP, DBP) were measured by a PE-10 catheter in aorta through the right common carotid artery [27]. After arterial pressure measurements, the catheter was advanced to LV and LVP, EDP and dP/dt were measured. The catheter was connected to a pressure transducer (Micro-Med, Louisville, Ky., USA) positioned at the heart level. Pulsatile arterial pressure signal was analyzed by a computer using customized software (Micro-Med). To determine plasma glucose and to ensure mouse to mouse variation, 0.5 ml blood was collected from each mouse by the same catheter after the measurements of vital parameters.
Urinary protein
To collect the urine since mouse urine had large quantities of proteins (primary mouse major urinary protein, MUP) and male mice had much higher urinary MUP than female mice, male mice were kept in 24-h metabolic cages for several days of acclimatization to reduce separation effects, prior to haemodynamic measurements. MUP was measured by Bio-Rad dye binding assay [28].
Tissue processing
Under deep anaesthesia, the mice were killed. The hearts were arrested in diastole by injecting 0.2 ml/100 g body weight of a 20% KCl solution. Hearts and kidneys were excised. The LV and RV were separated. LV tissue homogenates were prepared. Bio-Rad dye binding assay was applied to estimate total protein concentration in the tissue extracts according to the method of Bradford [28]. Sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE) was done with and without reduction as described [29].
MMP activity
Plasma and LV MMP activity were measured using zymography [30]. To determine MMP-2 and -9 activity, the gelatin substrate gel zymography containing 1% gelatin in 8% SDS-PAGE was carried out [30]. Plasma and LV tissue homogenates were loaded onto the gel under the identical condition of total protein. The scanned band intensity was normalized with beta actin.
CIMP and α-actin
The concentrations of CIMP and actin were measured by western blot analysis using mouse monoclonal anti-TIMP-4 antibody (Chemicon, Richmond, Calif., USA). The specificity of antibodies was established by immunoprecipitating the antigen before loading onto the gel, by antibody-conjugated agarose beads (Upstate Biotechnology, Lake Placid, N.Y., USA). To determine whether total protein loaded onto the gel was identical, α-actin western blots were done by using anti-actin antibody (Sigma Chem, St. Louis, Mo., USA). The alkaline phosphatase conjugated secondary antibody was used as a detection system. Bands on blots were scanned by Bio-Rad GS-700 densitometer.
Preparation of acetylcholine, nitroprusside, and alloxan solutions
The concentrations of acetylcholine, nitroprusside, and alloxan were based on weight measurements. All dilutions from stock solutions in phosphate-buffered saline were made before the experiment. Phosphate-buffered saline was used as vehicle control.
Histology, collagen and elastin contents
Paraffin embedded 10 micron tissue sections were stained with trichrome of collagenous matrix. The area stained bluish for collagen was scanned and measured as an arbitrary unit (AU)/cm2. The amounts of collagen and elastin were measured by estimating hydroxyproline and desmosine/isodesmosine, respectively, as described [31].
TUNEL and immuno-labelling
To determine whether diabetes caused capillary endothelial and endocardial injury, the serial tissue sections were labelled with transferase deoxyuridine nick end labelling (TUNEL) according to the instructions of the manufacturer (Oncogene Research Products, Fluorescein-FragEL cat# QIA39), for identification of nicked DNA. Apoptosis was indexed by counting TUNEL positive cells per cm2. Endothelial cells were characterized using FITC-labelled CD31 (PECAM-1) antibody (Sigma) and counted per cm2 in a fixed grid. Randomly selected five grids per tissue were analysed. The numbers of cells were normalized with the cells in control tissue and the percentage of endothelial cells was reported.
Endocardial endothelial function
The determination of endothelial function in isolated papillary muscle preparation did not demonstrate what happened in the entire transmyocardial wall. To determine cardiac function, Langendorff preparation was used. However, this did not differentiate the specific contribution due to regional ischaemia, hypertrophy, stunning, and/or hibernation of myocytes in myocardial wall. Rather it gave global contractile response to cardiotonic agents. Furthermore, it did not separate the effects of LV from RV. We have compared the data obtained from cardiac rings prepared from hypertensive rats [31], and found similar pressure-volume curves as obtained by Langendorff preparation. In addition, the cardiac ring preparation separated the effect of LV from RV. To determine the specific regional differences in contractile function, the rings can be prepared to include or to exclude the homogenous or inhomogeneous regions of the transmural myocardial wall [31, 32, 33, 34]. The "deli" shaped LV rings were mounted in a tissue myobath. One of the two mounted wires was connected to a force transducer. The ring was stretched and brought to resting tension at which the 20 mmol/l CaCl2 was added. At the maximum CaCl2 contraction, acetylcholine (endothelial-dependent) or nitroprusside (endothelial-independent) was added. To minimize the differences due to orientation of cardiac muscle, the rings were rotated 90° and contraction was measured. The average of two contractions was recorded. The percentage of relaxation was calculated based on 100% contraction to 20 mmol/l CaCl2. The dose-response curves were generated. To avoid ischaemia, oxygen at 20 psi was continuously bubbled through the myobath. This pressure was sufficient for penetration of cardiotonic agent [32]. The experiments were completed within 40 min. During that time there was minimal injury from ischaemia, under these conditions [32].
Statistical analysis
Values were given as means±SEM from n=6 in each group. Differences between groups were evaluated by using a two-way ANOVA, followed by the Bonferroni post hoc test [35], focusing on the respective effects of –/– and diabetes. The results of the four comparisons by Bonferroni test are reported as: *, +/+A vs –/–A; **, +/+A vs +/+; #, –/–A vs –/–; and ##, +/+ vs –/–. A p value of less than 0.05 was considered significant.
Results
Diabetes induced MMP-9
To determine whether selective induction of MMP-9 in diabetes mellitus causes EE dysfunction, MMP-9 gene ablation model was used. The phenotype of MMP-9 –/– mouse is shown in Fig. 1. There was no induction of MMP-9 in diabetes-induced MMP-9 –/– mice. However, the creation of diabetes in wild-type mice selectively increased MMP-9 activity in plasma (Fig. 2) as well as in the left ventricle (Fig. 3). The significance of increased MMP-2 activity could imply its role in compensatory remodelling in diabetes, and therefore, MMP-2 could be a survival factor.

Genotype analysis of MMP-9 (–/–) mice: Representative PCR analysis of MMP-9, lanes 1–3 MMP-9 (–/–); lane 4 wild-type control mice. PCR was carried out using forward primer, CCAGTTTCCATTCATCTTCC; reverse primer ACAGTAGTGGCCGTAGAAGG; PCR product 479 bp, TA 60°C; accession code NM004994 of MMP-9 gene. Mr represents molecular weight marker

Phenotype analysis and induction of MMP-9 by alloxan-induced diabetes mellitus (DM): Plasma MMP activity was measured in MMP-9 (–/–) and wild-type mice at 8 weeks after injection of alloxan as compared with untreated mice. The plasma MMP activity was measured by zymography. Lane 1, –/–; lane 2, –/– treated with alloxan; lane 3, wild-type (+/+) treated with alloxan; and lane 4, wild-type control. The n=6 was used in each experimental group

Left ventricular concentrations of MMP activity: A Zymographic analysis. Lane 1, –/–; lane 2, –/– treated with alloxan; lane 3, wild-type (+/+) treated with alloxan; and lane 4, wild-type control. The 25 µg total protein was loaded onto each lane. To demonstrate that other proteins were not altered, corresponding actin controls are shown. B Histographic concentrations of MMP-2 and −9 lytic activity (arbitrary scan unit, AU). The scanned band intensity is reported as means ± SEM from n=6 in each group. *, +/+A vs –/–A; **, +/+A vs +/+; and #, –/–A vs –/–. A p<0.05 was considered significant
Diabetes decreased CIMP
To determine whether increased MMP-9 activity was associated with decreased CIMP, the concentrations of CIMP were measured. CIMP was decreased both in the wild-type and in MMP-9 –/– mice after diabetes was induced (Fig. 4), suggesting unabated proteolysis in diabetes mellitus.

Left ventricular concentrations of CIMP: A Western blot analysis. Lane 1, –/–; lane 2, –/– treated with alloxan; lane 3, wild-type (+/+) treated with alloxan; and lane 4, wild-type control. The 25 µg total protein was loaded onto each lane. The corresponding actin controls are shown. B Histographic demonstration of the levels of CIMP (arbitrary scan unit, AU). The scanned band intensity is reported as means ± SEM from n=6 in each group. **, +/+A vs +/+; #, –/–A vs –/–; and ##, +/+ vs –/–. A p<0.05 was considered significant
Matrix-deposition
To determine whether increased MMP-9 activity and decreased CIMP concentrations were associated with accumulation of collagen matrix and myocyte hypertrophy, the hearts were stained with trichrome for collagenous material. The results suggest an increase in matrix accumulation in diabetes-induced wild-type mice. In addition, there was significant hypertrophy in diabetic mice (Fig. 5). There were basal concentrations of fibrosis in MMP-9 –/– mice; however, the creation of diabetes tended to increase the concentrations of fibrosis in MMP-9 –/– mice, but not significantly (Table 1, Fig. 5).

Histological staining in the endocardium by trichrome: A, wild-type; B, wild-type + alloxan; C, MMP-9 –/–; and D, MMP-9 –/– mice treated with alloxan. Arrows indicate sites of fibrosis. All sections are at 100× magnification. Arrows indicate the sites of fibrosis
Haemodynamic parameters in diabetes
The concentrations of increased glucose were associated with increased renal injury as measured by increased concentrations of MUP (Table 1). Blood glucose concentrations were low in WT non-diabetic mice. This could suggest a reduction in glucose concentrations, in part, due to the treatment of anaesthesia before the plasma of WT mice was collected. The increased MUP was correlated with increased LV weight, i.e. LVH and hypertension. There were significant diastolic and systolic dysfunctions in diabetic mice (Table 1). These results suggest that increased glucose toxicity induced overall cardiac injury and dysfunction.
Decreased EE cell density in diabetes
To determine whether the increase in cardiac muscle size associated with decreased capillary cell density, the hearts were stained with PECAM-1/CD31 antibody and TUNEL positive cells were identified. There were inhomogeneous TUNEL-positive cells in diabetes mellitus hearts, and there was a focal decrease in endocardial endothelial cell density in diabetic wild-type mice compared with the control mice (**p<0.001, Fig. 6). The number of endothelial cells were lower in MMP-9 –/– mice as compared with wild-type mice. Diabetes did not decrease further the number of endothelial cells in MMP-9 –/–, as in WT mice. The number of TUNEL positive cells were higher in MMP-9 –/– as compared with wild-type mice. The diabetes induced apoptosis in WT mice but had no effect on the number of endothelial cells in MMP-9 –/– mice (Fig. 7), suggesting an association of TUNEL positivity with MMP-9 activity.

Representative endothelial antigen (CD31/PECAM-1) staining in the endocardium: Labelling of deparaffinized fixed tissue sections of the heart from diabetes (B) and control (A) mice; ×40 magnification. Arrows indicate capillaries including endothelium. C Histographic presentation of the percentage of endothelial cells. Wild-type without any treatment is 100%. *, +/+A vs –/–A; and **, +/+A vs +/+. A p<0.05 was considered significant

Representative TdT-mediated dUTP nick-end labelling (TUNEL)-positive labelling in the endocardium of diabetes (B) as compared with wild-type control (A) mice; ×40 magnification. Arrows indicate sites of TUNEL positive cells. C Relative counts of apoptotic nuclei. *, +/+A vs –/–A; and **, +/+A vs +/+. A p<0.05 was considered significant
EE dysfunction in diabetes
To determine whether decreased EE-cell density was associated with decreased EE function, the EE function of MMP-9 –/– diabetic mice was measured. The response to acetylcholine was attenuated in EE of diabetic WT mice. Although acetylcholine-response was low in MMP-9 –/–, diabetes had no further attenuation of acetylcholine response in MMP-9 –/– mice (Fig. 8B). The response to nitroprusside was similar in both WT mice with or without diabetes (Fig. 8C). These results suggest that diabetes induces EE dysfunction in wild-type and has no effect in MMP-9 –/– mice. There were no effect in muscle.

Endocardial endothelial response to acetylcholine (ACH): LV rings from WT sham, WT+diabetes mellitus (DM), Sham –/–, and DM –/–, groups of mice were precontracted with 20 mmol/l CaCl2. Different doses of ACH were added to the ring in a tissue myobath. The relaxation to ACH was estimated as the percentage remaining CaCl2 contraction. Data are an average of at least six mice. A Typical contractile response to CaCl2 and relaxation to 109, 10-8, 107, and 10-6 mol/l ACH of a wild-type mice. B ACH dose-response curves. C Nitroprusside (Npr) dose-response curves. **, +/+A vs +/+; and ##, +/+ vs –/–. A p<0.05 was considered significant
Discussion
Our results suggest that increased MMP-9 activity was associated with endothelial dysfunction and apoptosis in diabetes mellitus. Studies have shown an increase in the concentrations of constitutively-expressed matrix metalloproteinase-2 (MMP-2, gelatinase A) in the compensatory phase, and robust induction of MMP-9 (gelatinase B) in the decompensatory phase of congestive heart failure (CHF) [45]. Others have shown apoptosis in CHF [38]. Evidence suggests that fibrosis is generated by inflammatory reaction, necrosis and apoptosis [36], and decreased MMP-9 activity in MMP-9 –/– mice is associated with echocardiographic wall abnormalities [37]. During diabetes generalized MMP activation [20], specifically the induction of MMP-9 has been observed [39]. It is a paradox that increased MMP activity and decreased concentrations of CIMP by fibrosis, co-exist in diabetes. This can be explained by the scenario that MMP-1, and -2 degrade interstitial collagen [23]. The MMP-1 and -2 are inhibited by TIMP-1; however, the activity of MMP-9 is not inhibited by TIMP-1. Previous studies have suggested increased concentrations of TIMP-1 in diabetes [41]. CIMP inhibits MMP-9 [34], but the concentrations of CIMP are decreased in diabetes. MMP-2 and -9 degrade elastin [22] as well as cell adhesive molecules more efficiently than other MMPs. Because the turnover of collagen is faster than elastin, oxidatively-modified collagen (glycated-collagen) is deposited faster than elastin or any other ultrastructural matrix proteins. Consequently, this constitutes fibrosis. In addition, the elastin-derived peptides, i.e. the signal from outside of the cell to inside of the cell, lead to alterations in the cytoskeletal structure of the cell [42].
ECM degradation is required for opening up binding sites on integrin (ECM-receptor) during compensatory remodelling, and MMP-2 plays an important role; however, unabated degradation leads to complete cell-ECM disconnect and cell apoptosis [40]. We suggest that activation of MMP-9 was one of the causes of apoptosis in diabetic cardiomyopathy. This leads to cardiac fibrosis. The histological changes in the myocardium were consistent with the deposition of oxidized and glycated fibrotic collagen in diabetes mellitus [15, 16]. The endothelial function is impaired suggesting that MMP-9 can also play other important roles. In addition, the expression of the cardiac inhibitor of MMPs, TIMP-4 is decreased in both the diabetic WT and knockout mouse, suggesting that concentration of this inhibitor can also be a more important factor regulating ECM turnover. There is a tendency to increase fibrosis, and others have shown compensatory increase in MMP-3 and -13 in this model [37], suggesting elastin degradation and collagen accumulation.
Metabolic parameters, e.g. MAP, SP, DP are significantly affected by knockout of MMP-9. The decreased MMP-9 activity has been associated with hypertension [43] and increased MMP-9 activity has been coordinated with decreased blood pressure [44]. This was consistent with the possibility that MMP-9 degraded elastin more efficiently than oxidized collagen. This led to deposition of matrix and increased vascular stiffness and resistance. Our results demonstrate an increase in blood pressure in MMP-9 –/– mice compared with wild-type (Table 1). The levels of blood pressure were similar between WT+A and MMP –/– treated with alloxan. These results suggest an additive effect of MMP-9 deficiency and diabetes on blood pressure. There were increased concentrations of MUP in MMP-9 –/– mice than wild-type. These results could suggest microvascular injury in diabetes. The effect of MMP-9 in heart was different in the sense that LVP was high in MMP-9 –/– than the control mice. Diabetes increased MMP-9 activity in plasma and LV of WT mice. In vitro cell culture derived from MMP-9 –/– hearts in the presence of high or low glucose concentration could provide a more clear picture of the role of MMP-9 in preventing the loss of endothelial cells. Whilst ablation of MMP-9 activity contributes to decreases in apoptosis, decreased TIMP-4 could also play an important role. Cell culture experiments using TIMP-4 antibody could provide important information regarding the relative contribution of each. Although previous studies have shown that most of the MMPs in the heart are in latent forms, including the MMP-9, and can be activated by oxidative stress [30]. Our study suggests a direct involvement of MMP-9 in endothelial cell apoptosis by an increase in the redox and proteolytic stress during diabetes. This study, however, did not measure the concentrations of redox components generated in diabetes in the presence and absence of MMP-9.
Abbreviations
- A:
-
Alloxan
- CIMP:
-
cardiac inhibitor of metalloproteinase
- ECM:
-
extracellular matrix
- EE:
-
endocardial endothelial
- eNOS:
-
endothelial nitric oxide synthase
- LV:
-
left ventricle
- MMP:
-
matrix metalloproteinase
- MUP:
-
mouse urinary protein
- NO:
-
nitric oxide
- PCR:
-
polymerase chain reaction
- TIMP:
-
tissue inhibitor of metalloproteinase
- TUNEL:
-
terminal uridine deoxynucleotide neck-end labeling
- WT:
-
wild-type
References
Hoppeler H, Kayar SR (1988) Capillary and oxidative capacity of muscles. News in Physiol Sci 3:113–116
Roberts JT, Wearn JT (1941) Quantitative changes in the capillary-muscle relationship in human hearts during normal growth and hypertrophy. Am Heart J 617–633
Hayden MR, Tyagi SC (2000) Remodeling of the endocrine pancreas: The central role of amylin and insulin resistance. Southern Med J 93:24–28
Hayden MR, Tyagi SC (2001) "A" is for amylin and Amyloid in type 2 diabetes mellitus. J Pancreas 2:124–139
Henderson AH, Lewis MJ, Shah AM, Smith JA (1992) Endothelium, endocardium, and cardiac contraction. Cardiovasc Res 26:305–308
Smith JA, Shah AM, Fort S, Lewis MJ (1992) The influence of endocardial endothelium on myocardial contraction. Trends Pharmacol Sci 13:113–116
Mebazaa A, Wetzel R, Cherian M, Abraham M (1995) Comparison between endothelial and great vessel endothelial cells: morphology, growth, and prostaglandin release. Am J Physiol 268:H250–H259
Kern TS, Tang J, Mizutani M et al. (2000) Response of capillary cell death to aminoguanidine predicts the development of retinopathy: comparison of diabetes and galactosemia. Invest Ophthalmol 41:3972–3978
Warley A, Powell JM, Skepper JN (1995) Capillary surface area is reduced and tissue thickness from capillaries to myocytes is increased in LV of STZ-diabetic rats. Diabetologia 38:413–421
Di Bello V, Giampietro O, Matteucci E et al. (1996) Ultrasonic video-densitometric analysis in type I diabetic myocardium. Coron Artey Dis 7:895–901
Sokolov EI, Zaichikova OS, Tsyplenkova VG (1998) Ultrastructure of the myocardium in patients with cardiac pathology complicated by diabetes mellitus. Arkh Patol 60:49–54
Dhalla NS, Prierce GN, Innes IR, Beamish RE (1985) Pathogenesis of cardiac dysfunction in diabetes mellitus. Can J Cardiol 14:263–281
Abe T, Ohga Y, Tabayashi N et al. (2002) LV diastolic dysfunction in type 2 DM model rats. Am J Physiol 282:H138–H148
Thompson EW (1998) Structural manifestations of diabetic cardiomyopathy in the rat and its reversal by insulin treatment. Am J Anat 182:270–282
Avendano GF, Agarwal RK, Bashey RI (1999) Effects of glucose intolerance on myocardial function and collagen-lonked glycation. Diabetes 48:1443–1447
Turk Z, Misur I, Turk N, Benko B (1999) Rat tissue collagen modified by advanced glycation: co-relation with duration of diabetes and glycemic control. Clin Chem Lab Med 37:813–820
Katayama S, Abe M, Negishi K et al (1994) Reciprocal changes in LV collagen alpha 1 chain gene expression between type I and IV in spontaneously diabetic rats. Diabetes Res Clin Pract 26:163–169
Mizushige K, Yao L, Noma T et al. (2000) Alteration in LV diastolic filling and accumulation of mycardial collagen at insulin-restant prediabetic stage of a type 2 diabetic rat model. Circulation 101:899–907
He Q, Spiro MJ (1995) Isolation of rat heart endothelial cells and pericytes: evaluation of their role in the formation of ECM components. J Mol Cell Cardiol 27:1173–1183
Ryan ME, Usman A et al. (2001) Excessive matrix metalloproteinase activity in diabetes: inhibition by tetracycline analogues with zinc reactivity. Curr Med Chem 8:305–316
Kwan CY, Wang RR, Beazley JS, Lee RM (1988) Alterations of elastin and elastase-like activities in aortas of diabetic rats. Biochim Biophys Acta 967:322–325
Senior RM, Griffin GL, Eliszar CJ, Shapiro SD, Goldberg GI, Welgus HG (1991) Human 92- and 72- kilodalton type IV collagenases are elastases. J Biol Chem 266:7870–7875
Aimes RT, Quigley JP (1995) MMP-2 is an interstitial collagenase. J Biol Chem 270:5872–5876
Rucklidge GJ, Milne G, McGaw BA, Milne E, Robins SP (1992) Turnover rates of different collagen types measured by isotope ratio mass spectrometry. Biochim Biophys Acta 11:1156–1157
Vu TH, Shipley JM, Bergers G et al. (1998) MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 93:411–422
Papaioannou VE, Fox JG (1993) Efficacy of tribromoethanol anesthesia in mice. Lab Anim Sci 43:189–192
Miller AD, Tyagi SC (2002) Mutation in collagen gene induces cardiomyopathy in transgenic mice. J Cell Biochem 85:259–267
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Tyagi SC, Ratajska A, Weber KT (1993) Myocardial matrix metalloproteinases: localization and activation. Mol Cell Biochem 126:49–59
Mujumdar VS, Smiley LM, Tyagi SC (2001) Activation of matrix metalloproteinase dilates and decreases cardiac tensile strength. Int J Cardiol 79:277–286
Tyagi SC, Smiley LM, Mujumdar VS (1999) Homocyst(e)ine impairs endocardial endothelial function. Can J Physiol Pharmacol 77:950–957
Miller A, Mujumdar V, Palmer L, Bower JD, Tyagi SC (2002) Reversal of endocardial endothelial dysfunction by folic acid in homocysteinemic hypertensive rats. Am J Hypertens 15:157–163
Cox MJ, Sood HS, Hunt MJ et al. (2002) Apoptosis in the left ventricle of chronic volume overload causes endocardial endothelial dysfunction in rats. Am J Physiol (Heart & Circulatory) 282:H1197–H1205
Tarone RE (1990) A modified Bonferroni method for discrete data. Biometrics 46:515
Tyagi SC (1997) Dynamic ECM remodeling in the heart failure: cardiac hypertrophy, dilatation and fibrosis. Pathophysiology 4:227–234
Ducharme A, Frantz S, Aikawa M et al. (2000) Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargment and collagen accumulation after experimental myocardial infarction. J Clin Invest 106:55–62
Frustaci A, Kajstura J, Chimenti C et al. (2000) Myocardial cell death in human diabetes. Circ Res 87:1123–1132
Uemura S, Matsushita H, Li W et al. (2001) Diabetes mellitus enhances vascular matrix metalloproteinase activity:role of oxidative stress. Circ Res 88:1291–1298
Meredith JE, Fazeli B, Schwartz MA (1993) The ECM as a cell survival factor. Mol Biol Cell 4:953–961
Shankland SJ, Ly H, Thai K, Scholey JW (1996) Glomerular expression of TIMP-1 in normal and diabetic rats. J Am Soc Nephrol 7:97–104
Guliano RL, Haskill S (1993) Signal trasduction from the ECM. J Cell Biol 120:577–585
Viellard-Baron A, Frisdal E, Eddahibi S et al. (2000) Inhibition of MMP by lung TIMP-1 gene transfer or doxycycline aggravates pulmonary hypertension in rats. Circ Res 87:418–425
Li-Saw-Hee FL, Edumnds E, Blann AD (2000) MMP-9 and TIMP-1 levels in essential hypertension. Relationship to left ventriclular mass and anti-hypertensive therapy. Int J Cardiol 75:43–47
Mujumdar VS, Tyagi SC (1999) Temporal regulation of extracellular matrix components in transition from compensatory hypertrophy to decompensatory heart failure. J Hypertens 17:261–270
Acknowledgements
This work was supported in part by NIH grants GM-48595, HL-71010, and HL-74185.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Camp, T.M., Tyagi, S.C., Senior, R.M. et al. Gelatinase B(MMP-9) an apoptotic factor in diabetic transgenic mice. Diabetologia 46, 1438–1445 (2003). https://doi.org/10.1007/s00125-003-1200-y
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-003-1200-y