
Abstract
Aims/hypothesis
We investigated the effect of cerivastatin, a statin, on the development of diabetic nephropathy in spontaneously hypertensive rats (SHR) with streptozotocin-induced diabetes.
Methods
Diabetic SHR were given standard chow or chow containing cerivastatin at a dose of 0.1 mg/kg or 1.0 mg/kg for 12 weeks. Effects of cerivastatin on urinary albumin excretion, mesangial expansion, glomerular macrophage infiltration, and the number of anionic sites on the glomerular basement membrane (GBM) were assessed.
Results
Cerivastatin did not affect the blood glucose concentration, blood pressure or serum cholesterol concentration in diabetic SHR. However, cerivastatin treatment caused a dose-dependent decrease of albuminuria and hyperfiltration. At 1.0 mg/kg, cerivastatin inhibited the diabetes-induced expansion of mesangial and tuft areas on histological examination of the kidneys, as well as the loss of anionic sites from the GBM evaluated with polyetyleneimine and the intraglomerular infiltration of ED1-positive macrophages evaluated by immunohistochemistry. Whole-kidney expression of mRNA for MCP-1 and TGF-β, estimated by the real-time quantitative RT-PCR, was increased (both 2.6-fold) in untreated diabetic SHR at 12 weeks. Cerivastatin treatment (1.0 mg/kg) inhibited the up-regulated expression of MCP-1 and TGF-β mRNA (decreased to 48% and 34%, respectively) in diabetic SHR.
Conclusion/interpretation
In this hypertensive model of diabetic nephropathy, cerivastatin decreased albuminuria through suppression of glomerular hyperfiltration, mesangial expansion, and the loss of charge barrier independently of a cholesterol-lowering effect. These preventive effects could be at least partly due to inhibition of macrophage recruitment and activation, and inhibition of TGF-β overexpression.
Similar content being viewed by others


Diabetic nephropathy is the leading cause of end-stage renal disease and the most frequent cause of mortality in patients with diabetes. Hypertension frequently co-exists in patients with Type 2 diabetes and could be another risk factor that accelerates cardiovascular and renal complications [1]. The pathogenesis of diabetic nephropathy is multifactorial, including accumulation of extracellular matrix (ECM) that results in mesangial expansion and thickening of the glomerular basement membrane (GBM) [2]. However, the upstream abnormalities that lead to these morphological changes are not clear. Evidence has supported the hypothesis that macrophages and macrophage products play an important role not only in inflammatory glomerular disease, but also in the pathogenesis of glomerulosclerosis. The glomeular infiltration of macrophages is also a characteristic finding in diabetic patients [3], and is observed before the onset of ECM expansion and glomerulosclerosis [4]. Macrophages that migrate into the glomeruli could cause structural damage through the release of proinflammatory and profibrotic cytokines. We previously showed that TNF-α enhanced the expression and production of monocyte chemoattractant protein-1 (MCP-1), a specific chemoattractant for monocytes, in human mesangial cells [5]. MCP-1 is thought to be involved in recruiting and activating macrophages in the glomeruli in proliferative glomerular diseases [6]. We have obtained in vivo evidence showing that administration of cerivastatin, or 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase inhibitor (or statin), could inhibit lipopolysaccharide (LPS)-induced overproduction of inflammatory mediators by macrophages and prevent LPS-induced death [7]. To investigate whether statins can prevent the progression of diabetic nephropathy and/or hypertensive nephrosclerosis by suppressing the activation and recruitment of macrophages, we assessed the effect of cerivastatin on the development of albuminuria in a hypertensive model of diabetic nephropathy, i.e., spontaneously hypertensive rats (SHR) with streptozotocin (STZ)-induced diabetes.
Materials and methods
Animal model and experimental design
Male spontaneously hypertensive rats (Funabashi Farm, Chiba, Japan), weighing 220 to 240 g and 10 to 12 weeks old, were made diabetic by a single intravenous injection of STZ (50 mg/kg, Sigma, St Louis, Mo., USA), as described previously [8]. The blood glucose concentration was measured 48 h after injection, and it was confirmed that the blood glucose concentration of all diabetic rats was over 16.5 mmol/l before starting this study. After the induction of diabetes, the diabetic SHR were divided into the following three groups: (a) rats given normal chow (Oriental Yeast, Tokyo, Japan) (STZ, n=16), (b) rats given chow mixed with cerivastatin sodium (donated by Bayer Pharmaceutical, Osaka, Japan) at a dose of 0.1 mg/kg (STZ+0.1Ceriva, n=5), and (c) rats given chow mixed with cerivastatin sodium at a dose of 1.0 mg/kg (STZ+1.0Ceriva, n=16). Non-diabetic SHR treated with or without cerivastatin was used as a control group. Three groups of non-diabetic SHR were also investigated: (d) a control group (n=9), (e) a 0.1Ceriva group (n=5), and (f) a 1.0Ceriva group (n=6). The rats were allowed unrestricted access to water and to the standard or mixed chow. All animal procedures were in accordance with the standards set forth in the Guidelines for the Care and Use of Laboratory Animals at the Takara-machi campus of Kanazawa University.
Blood pressure measurement
Systolic blood pressure (SBP) was assessed by tail-cuff plethysmography (BP-98A, Softron, Tokyo, Japan) in conscious pre-warmed rats before and 2, 4, 8 and 12 weeks after the start of cerivastatin treatment.
Blood sampling and analysis
Blood samples were obtained from the tail vein before and 2, 4, 8 and 12 weeks after the creation of diabetes. Blood glucose was measured by the glucose-oxidase method using Glucocard (Aventis Pharma, Tokyo, Japan). The blood samples were centrifuged, and serum was frozen at −70°C for subsequent measurement of the serum creatinine (Cr), total cholesterol (TC), HDL-cholesterol (HDL-C) concentrations by quantitative colorimetric assay (BML, Tokyo, Japan).
Urinary albumin excretion
Urinary albumin excretion (mg/24 h) was assessed before and 2, 4, 8 and 12 weeks after the start of treatment. After each rat was placed in an individual metabolic cage to collect urine over 24 h, their urine samples were collected and frozen at −70°C for subsequent analysis of albumin and Cr concentrations. Urinary albumin concentrations were measured by enzyme-linked immunoabsorbent assay using an anti-rat albumin antibody (Nephrat, Exocell, Philadelphia, Pa., USA). Urinary Cr concentrations were measured by a quantitative colorimetric assay (BML, Tokyo, Japan). Creatinine clearance (Ccr, ml·min−1 kg−1) was calculated as urinary Cr urine volume serum Cr−1 body weight−1.
Morphological analysis—mesangial expansion
After 12 weeks of cerivastatin administration, the animals were killed and their kidneys were fixed in 10% buffered formalin and embedded in paraffin. Light microscopy was done by using periodic acid-Schiff (PAS)-stained 6-µm serial sections containing 40 to 60 glomeruli. The glomerular cross-sectional area (Ag), mesangial area (Am), and tuft area (At) were measured in 30 glomerular profiles per rat by using NIH image 1.58 software. The glomerular volume (Vg) was then calculated as: Vg=β/k [Ag]3/2, where β=1.38 is the size distribution coefficient and k=1.1 is the shape coefficient for glomeruli idealized as a sphere [9]. All measurements were done in a masked fashion.
Morphological analysis—macrophage infiltration into glomeruli
Kidney specimens were incubated with mouse anti-rat monocyte/macrophage monoclonal antibody ED1 (Serotec, Oxford, UK) for 1 h at room temperature. This was followed by application of the immunoperoxidase technique using an ABC kit (Vector Laboratories, Burlingame, Calif., USA). Peroxidase activity was identified by reaction with 3′,3′-diaminobenzidine (Sigma Chemical, St Louis, Mo., USA). Each incubation was followed by three washes in phosphate-buffered saline. The average number of ED1-positive cells per glomerulus was calculated by counting the cells in 30 glomeruli per kidney.
Morphological analysis—anionic sites on the lamina rara externa
To investigate the effects of cerivastatin on the anionic charge of the GBM, we evaluated the number of anionic sites on the lamina rara externa (LRE) of the GBM, as described previously [8]. Anionic sites were identified as particles that showed intensive staining with polyetyleneimine. The number of anionic sites per 1000 nm of LRE was calculated on electron micrographs (an average of 10 random visual fields) in each rat at a final magnification of 100 000×. Counting of anionic sites was done by two observers who were blinded to the treatment groups.
Analysis of mRNA for MCP-1 and TGF-β
Total RNA was extracted from each kidney by acid guanidium thiocyanate-phenol-chloroform method, followed by synthesis of cDNA as described previously [10]. Real-time quantitative PCR was done for MCP-1 and transforming growth factor-β1 (TGF-β1) using the ABI Prism 7700 Sequence Detection System (Applied Biosystems, Foster City, Calif., USA). To control for variation in the amount of DNA available for PCR in the different samples, gene expressions of the target sequence were normalized in relation to the expression of an endogenous control, 18S ribosomal RNA (18S rRNA TaqMan Control Reagent Kit; Applied Biosystems). The sets of primers and TaqMan probes were designed with a primer design software Primer Express version 1.5 (Applied Biosystems). The forward primers were 5′-TCTCTTCCTCCACCACTATGCA-3′ for rat MCP-1, and 5′-TTCCTGGCGTTACCTTGGT-3′ for rat TGF-β1. The reverse primers were 5′-GGCTGAGACAGCACGTGGAT-3′ for MCP-1, and 5′-GCCACTGCCGGACAACT-3′ for TGF-β1. The TaqMan probes were 5′-TCACGCTTCTGGGCCTGTTGTTCA-3′ for MCP-1, and 5′-TACGCCTGAGTGGCTGTCTTTTGA-3′ for TGF-β1. The PCR conditions were one cycle of 50°C for 2 min, 95°C for 10 min, followed by 50 cycles of 95°C for 15 s and 58°C for 1 min.
Statistical analysis
All results are shown as the mean or the mean ± SEM. Data were analysed by one-way analysis of variance. A p value of less than 0.05 was considered statistically significant. All calculations were done with Statview version 5.0 software for Macintosh (Abacus Concepts, Berkley, Calif., USA)
Results
Metabolic and biochemical parameters
Body weight, SBP, the blood glucose concentration and the serum TC concentration at each time point in each group are summarized (Table 1). Cerivastatin administration did not affect the health of the rats, including the food intake and body weight. The mean body weights of the groups were similar at the time of STZ injection. At each subsequent time point studied, body weight was lower in diabetic SHR (STZ, STZ+0.1Ceriva and STZ+1.0Ceriva) than in non-diabetic SHR (Control, 0.1Ceriva and 1.0Ceriva). Cerivastatin did not affect the weights of the diabetic SHR.
Mean SBP values were similar in all groups at all times. Administration of cerivastatin did not affect the SBP in diabetic SHR (Table 1).
The blood glucose concentration did not differ among the groups at the beginning of the study. STZ-injected SHR showed an increase of blood glucose concentrations at 2 weeks after injection, whereas no differences were found among the diabetic SHR groups. The blood glucose concentrations of diabetic SHR treated with cerivastatin did not differ from those of untreated diabetic SHR at all times (Table 1).
Induction of diabetes did not affect the serum TC concentration, while it increased HDL-C concentration in diabetic SHR. Cerivastatin had no influence on the serum TC and HDL-C concentrations in diabetic SHR (Table 1). Further, cerivastatin administration did not affect TC and HDL-C concentrations in non-diabetic SHR.
Urinary albumin excretion is shown in Fig. 1A. In untreated diabetic SHR, albuminuria gradually increased from 4 to 12 weeks after the onset of diabetes. There were no statistically significant changes of albuminuria among non-diabetic SHR at any point. Accordingly, albuminuria was higher in untreated diabetic SHR than in non-diabetic SHR after 8 weeks (p<0.01). When compared with untreated diabetic SHR, albuminuria was lower from weeks 8 to 12 in rats given high-dose cerivastatin (1.0 mg/kg) and it was also lower at 12 weeks in rats given low-dose cerivastatin (0.1 mg/kg). Thus, the suppressive effect of cerivastatin on albuminuria in diabetic SHR was dose-dependent after 12 weeks of treatment.

Time course of changes in urinary albumin excretion (A) and creatinine clearance (Ccr, B) in control rats (●), untreated diabetic SHR (○), and diabetic SHR treated with 0.1 mg/kg (△) or 1.0 mg/kg (□) of cerivastatin. Each value represents the mean ± SEM (* p<0.01 vs control; † p<0.05, †† p<0.01 vs untreated diabetic SHR)
Ccr increased gradually in untreated diabetic SHR during the 12-week study period (Fig. 1B). Ccr was higher in untreated diabetic SHR than in the non-diabetic SHR after 8 weeks (p<0.01), suggesting that hyperfiltration contributes to the development of albuminuria in diabetic SHR. Ccr was lower at 12 weeks in rats given high-dose cerivastatin when compared with untreated diabetic SHR.
Mesangial expansion
Table 2 shows the morphologic parameters for each group. In untreated diabetic SHR, the glomerular tuft and mesangial area were increased at 12 weeks when compared with the values in non-diabetic SHR (p<0.01). There was a trend for an increase of glomerular volume in untreated diabetic SHR compared with non-diabetic SHR. Cerivastatin treatment ameliorated the increase of both tuft area and mesangial area.
Glomerular macrophages
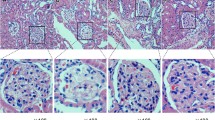
ED1-positive cells (representing monocytes/macrophages) infiltrated the glomeruli of untreated diabetic SHR at 12 weeks (Figs 2A,B, 3). ED1-positive cell infiltration of the periglomerular and tubulointerstitial areas was also observed in untreated diabetic SHR (Fig. 2B). ED1-positive cell infiltration into the glomeruli was suppressed by 1.0 mg/kg of cerivastatin at 12 weeks of treatment (p<0.001 vs STZ; Figs 2B,C, 3). Cerivastatin also inhibited the infiltration of ED1-positive cells into the tubulointerstitial areas.

Immunohistochemical staining for ED1-positive cells in glomeruli from a control rat (A) a diabetic SHR (B), and a diabetic SHR treated with 1.0 mg/kg of cerivastatin (C) at 12 weeks. Positive staining was observed in the glomerulus (arrows) and in the periglomerular region (arrowhead). Sections are counterstained with hematoxylin (magnification ×200)

Number of ED1-positive cells per glomeruli in control rats (Control), diabetic SHR (STZ), and diabetic SHR treated with 1.0 mg/kg of cerivastatin (STZ+1.0Ceriva) at 12 weeks. Each value represents the mean ± SEM (* p<0.0001, ** p<0.001 vs control; † p<0.001 vs STZ, n=6 per control; n=7 per STZ; n=9 per STZ+1.0Ceriva group)
Expression of MCP-1 and TGF-β mRNA
To clarify the mechanism by which cerivastatin inhibited macrophage recruitment into the glomeruli of diabetic SHR, the whole-kidney expression of MCP-1 mRNA was estimated by quantitative RT-PCR. MCP-1 mRNA expression was increased by 2.6-fold in the kidney of diabetic SHR when compared with non-diabetic SHR at 12 weeks (p<0.05; Fig. 4A). Cerivastatin treatment (1.0 mg/kg) completely inhibited the up-regulated renal expression of MCP-1 mRNA to 48% in diabetic SHR (p<0.05 vs STZ).

Effects of cerivastatin on monocyte chemoattractant protein-1 (MCP-1) (A) and transforming growth factor-β (TGF-β) (B) mRNA expression in the whole-kidney estimated by the real-time quantitative RT-PCR in diabetic SHR at 12 weeks. MCP-1 and TGF-β mRNA expressions were normalized with 18S ribosomal RNA mRNA. Each value represents the mean ± SEM. * p<0.05 vs control rats (Control); † p<0.05 vs diabetic SHR (STZ), n=4 per control; n=7 per STZ, diabetic SHR treated with 1.0 mg/kg of cerivastatin (STZ+1.0Ceriva group)
To better understand the mechanism by which cerivastatin was able to inhibit mesangial expansion in diabetic SHR, the whole-kidney expression of TGF-β was estimated. TGF-β mRNA expression was increased by 2.6-fold in untreated diabetic SHR when compared with non-diabetic SHR at 12 weeks (p<0.05; Fig. 4B), whereas cerivastatin treatment completely suppressed the increase of TGF-β mRNA expression to 34% in diabetic SHR (p<0.05 vs STZ).
Anionic sites on the LRE
Representative electron micro anionic sites on the LRE of the GBM are shown in Fig. 5. Control SHR showed a regular distribution of anionic sites (Fig. 5A). In untreated diabetic SHR, anionic sites were sparse and irregular compared with those in non-diabetic SHR (Fig. 5B). On the other hand, anionic sites were abundant and regularly arranged in diabetic SHR treated with cerivastatin, findings similar to those in control SHR (Fig. 5C). To evaluate the changes of anionic sites quantitatively, we counted the number of sites per 1000 nm of LRE (Fig. 6). In untreated diabetic SHR, the number of anionic sites was decreased when compared with that in non-diabetic SHR (p<0.001). However, no difference in the number of sites was found between diabetic SHR treated with cerivastatin and control SHR. Thus, cerivastatin treatment prevented the loss of anionic sites.

Electron micrographs of the glomerular basement membrane in a control rat (A), and diabetic SHR without (B) or with cerivastatin treatment (1.0 mg/kg) (C) at 12 weeks. Arrows show anionic sites on the lamina rara externa in the glomerular basement membrane (magnification ×100 000)

The number of anionic sites per 1000 nm of the lamina rara externa in control rats (Control), and diabetic SHR without (STZ) or with cerivastatin treatment (1.0 mg/kg) (STZ+1.0Ceriva) at 12 weeks. Each value represents the mean ± SEM of five experiments (* p<0.001 versus control; † p<0.001 vs STZ; n=5)
Discussion
Diabetic nephropathy is a serious complication and is the most common cause of renal failure requiring haemodialysis in Japanese patients with diabetes. Microalbuminuria in diabetic patients predicts the onset of proteinuria, as well as an increased risk of death and cardiovascular events [11]. Therefore, it is important to appropriately treat patients with microalbuminuria as soon as they are identified. Large-scale clinical trials have indicated that strict glycaemic and blood pressure control is required to prevent the development of diabetic nephropathy [1, 12]. In the case of pharmacological approaches, only angiotensin-converting enzyme inhibitors and angiotensin receptor blockers have been suggested to ameliorate albuminuria in diabetic patients [1, 13]. Although a recent meta-analysis showed that statins can decrease proteinuria and preserve glomerular filtration in patients with chronic renal disease [14], little is known about its mechanism involved.
In our study, cerivastatin treatment decreased albuminuria dose-dependently without modifying serum TC, HDL-C, blood glucose, or blood pressure. In particular, even the high-dose of cerivastatin did not affect the serum lipid concentrations but markedly improved the diabetes-induced albuminuria. Renoprotective effect of cerivastatin was through the lipid-independent actions as reported previously [15, 16]. Because cerivastatin did not ameliorate albuminuria in non-diabetic SHR, it seemed to prevent diabetes-specific changes related to the development of nephropathy. The mechanism of diabetic nephropathy is multifactorial. The features associated with disease progression are glomerular hypertrophy [17], thickening of the GBM and mesangial expansion (which lead to proteinuria), and eventual loss of glomerular filtration and glomerulosclerosis. Not only Ccr, which might be an unreliable index of glomerular filtration rate, but also tuft area increased in diabetic SHR, indicating that hyperfiltration occurred in the diabetic state in our model. We showed that cerivastatin treatment could inhibit both an increase of Ccr and expansion of the tuft area in diabetic SHR. Thus, cerivastatin inhibited hyperfiltration in diabetic SHR. These results indicate that the inhibitory effect of cerivastatin on albuminuria could be partly due to the improvement of renal haemodynamics.
As reported previously [3, 18], an increase of glomerular macrophage was observed in diabetic SHR. Secondary to this macrophage infiltration, renal MCP-1 expression was up-regulated in diabetic SHR. The mechanisms leading to the up-regulation of MCP-1 in various types of glomerular injury, including diabetic nephropathy, have yet to be fully understood. However, human and rodent mesangial cells can synthesize MCP-1 in response to several factors that are thought to be involved in glomerular injury, such as interleukin-1, TNF-α, and low-density lipoprotein [19]. In the case of human mesangial cells, high concentration of glucose as well as glycated albumin have been reported to promote MCP-1 production [20, 21]. Thus, diabetic nephropathy could be considered to be an inflammatory process that is characterized by the up-regulation of MCP-1 and infiltration of macrophages into glomeruli. In this study, cerivastatin suppressed up-regulation of MCP-1 expression in diabetic SHR kidneys and also inhibited the glomerular infiltration of macrophages. Taken together with our previous observation that cerivastatin inhibited the LPS-induced production of proinflammatory cytokines by rat macrophages [7], these findings suggest that statins might possess anti-inflammatory properties. In fact, a study has shown that statins can reduce the proliferation and activation of macrophages in atherosclerotic plaques [22]. The promoter region of the human MCP-1 gene contains putative consensus binding sites for NF-κB [23]. Furthermore, farnesylated p21 Ras (a low molecular weight cell membrane G protein) and growth factor receptor complex seem to activate NF-κB [24]. By inhibiting mevalonic acid synthesis, statins could also inhibit the synthesis of isoprenoid intermediates of the cholesterol biosynthesis pathway, particularly farnesyl pyrophosphate and geranylgeranylpyrophosphate [25]. These intermediates serves as lipid attachments for the post-translational modification of several important proteins, including cellular membrane G proteins, Ras, Rho, and Rac [26], whose proper membrane localization and function are dependent on isoprenylation. Thus, cerivastatin might reduce the expression of MCP-1 mRNA through inhibiting the production of proinflammatory cytokines by macrophages, as well as by inhibiting NF-κB activation.
Both mesangial expansion and GBM thickening in diabetic nephropathy are associated with TGF-β, a fibrogenic factor that is synthesized by activated macrophages or constitutively produced by glomerular cells. TGF-β stimulates matrix synthesis and inhibits matrix degradation [27]. Many studies have shown the increased expression of TGF-β in the kidneys of diabetic animals and patients [28], which leads to mesangial matrix expansion and GBM thickening [29]. Inhibition of TGF-β activity by specific neutralyzing antibodies was reported to reduce collagen synthesis and expression of the type IV collagen and fibronectin genes in the renal cortex [30] and the glomeruli of diabetic rats [31]. In the present study, TGF-β mRNA expression was up-regulated in the kidney of diabetic SHR. As mesangial deposition of ECM increased and proteinuria progressed, the expression of TGF-β mRNA expression was markedly up-regulated, while cerivastaitn ameliorated TGF-β mRNA expression in the kidneys of diabetic SHR. These data suggest that cerivastatin inhibits mesangial expansion by controlling renal overexpression of TGF-β mRNA in diabetic rats, possibly by preventing the activation of protein kinase C and mitogen-activated protein kinase. Both pathways have been shown to affect the expression of ECM proteins [32]. Because MCP-1 has been reported to mediate collagen deposition due to TGF-β in experimental glomerulonephritis [33], the up-regulation of MCP-1 and TGF-β could be a common pathway involved in the development of diabetic nephropathy. We immunohistologically confirmed that MCP-1 and TGF-β protein was indeed expressed in glomeuli (particularly in mesangial cells) in diabetic SHR as reported previously [20, 21, 28]. Diabetic state promotes TGF-β production not only by accumulating macrophages but also by mesangial cells themselves. Cerivastatin suppressed up-regulation of MCP-1 expression in diabetic glomeruli with a consecutive decrease of macrophage infiltration, which led to a decrease of TGF-β expression and/or directly suppressed the overexpression of TGF-β in diabretic glomeruli. Thus, the inhibitory effects of cerivastatin on both MCP-1 and TGF-β expression might play an additive role in preventing mesangial expansion.
As we observed previously [8], the number of anionic sites on the GBM was decreased in diabetic SHR in the present study. These sites consist of glycosaminoglycans, which are rich in heparan sulphate proteoglycan (HSPG) [34]. HSPG molecules on the GBM are synthesized by mesangial cells as well as by glomerular epithelial cells. The anionic sites regulate the transudation of circulating macromolecules across the GBM as a charge-selective filtration barrier, so their loss leads to an increase in the permeation of anionic proteins such as ferritin and albumin [35, 36]. It is reported that hyperglycaemia decreases production and sulphation of HSPG by reducing the activity of glucosaminyl N-deacetylase, a key enzyme in HSPG synthesis [37]. Moreover, oxidative stress has been suggested to cause loss of the charge barrier in isolated kidneys [38]. In this study, cerivastatin inhibited diabetes-induced loss of anionic sites on the GBM. Thus, cerivastatin could inhibit the loss of anionic sites by improving synthesis or sulphation of HSPG. The mechanism by which cerivastatin inhibits the loss of charge barrier should be examined, especially from the view of the effect of a statin on oxidative stress [39].
An HMG CoA reductase inhibitor, cerivastatin, prevented the development of nephropathy in diabetic SHR. Cerivastatin decreased albuminuria by suppressing glomerular hyperfiltration, mesangial expansion, and loss of the charge barrier independently of a cholesterol-lowering effect. Diabetes-induced up-regulation of MCP-1 and TGF-β mRNA in the kidney was also inhibited by cerivastatin, along with the reduction of glomerular macrophage infiltration and mesangial expansion. Statins could have a novel therapeutic value for slowing or preventing the progression of diabetic nephropathy.
Abbreviations
- SHR:
-
Spontaneously hypertensive rats
- MCP-1:
-
monocyte chemoattractant protein-1
- TGF-β:
-
transforming growth factor-β
- ECM:
-
extracellular matrix
- GBM:
-
glomerular basement membrane
- LRE:
-
lamina rara externa
- HMG CoA:
-
3-hydroxy-3-methylglutaryl coenzyme A
- LPS:
-
lipopolysaccharide
- STZ:
-
streptozotocin
- TC:
-
total cholesterol
- Ccr:
-
creatinine clearance
- NF-κΒ:
-
nuclear factor-κB
- HSPG:
-
heparan sulphate proteoglycan
References
UK Prospective Diabetes Study Group (1998) Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes. UKPDS 38. BMJ 317:703–713
Mauer SM (1994) Structural-functional correlations of diabetic nephropathy. Kidney Int 45:612–622
Furuta T, Saito T, Ootaka T et al. (1993) The role of macrophages in diabetic glomerulosclerosis. Am J Kidney Dis 21:480–485
Klahr S, Schreiner G, Ichikawa I (1988) The progression of renal disease. N Engl J Med 318:1657–1666
Ohta MY, Nagai Y, Takamura T, Nohara E, Kobayashi K (2000) Inhibitory effect of troglitazone on tumor necrosis factor alpha-induced expression of monocyte chmoattractant protein-1 in human mesangial cells. Metabolism 49:163–166
Oppenheim JJ, Zachariae CO, Mukaida N, Matsushima K (1991) Properties of the novel proinflammatory supergene ''intercrine'' cytokine family. Annu Rev Immunol 9:617–648
Ando H, Takamura T, Ota T, Nagai Y, Kobayashi K (2000) Cerivastatin improves survival of mice with lipopolysaccharide-induced sepsis. J Pharmacol Exp Ther 294:1043–1046
Yamashita H, Nagai Y, Takamura T, Nohara E, Kobayashi K (2002) Thiazolidinedione derivatives ameliorate albuminuria in streptozotocin-induced diabetic spontaneous hypertensive rat. Metabolism 51:403–408
Weibel ER (1979) Stereological methods: practical methods for biological morphometry. Academic Press, London, pp 162–203
Takamura T, Nohara E, Nagai Y, Kobayashi K (2001) Stage-specific effects of a thiazolidinedione on proliferation, differentiation and PPARγ mRNA expression in 3T3-L1 adipocytes. Eur J Pharmacol 422:23–29
Mogensen CE (1984) Microalbuminuria predicts clinical proteinuria and mortality in maturity-onset diabetes. N Engl J Med 310:356–360
UK Prospective Diabetes Study Group (1998) Intensive blood glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes: UKPDS 33. Lancet 352:837–853
Parving HH, Lehnert H, Brochner-Mortensen J, Gomis R, Anderson S, Arner P, Irbesartan in Patients with Type 2 Diabetes and Microalbuminuria Study Group (2001) The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med 345:870–878
Fried LF, Orchard TJ, Kasiske BL (2001) Effect of lipid reduction on the progression of renal disease: a meta-analysis. Kidney Int 59:260–269
Park JK, Muller DN, Mervaala EM et al. (2000) Cerivastatin prevents angiotensin II-induced renal injury independent of blood pressure- and cholesterol-lowering effects. Kidney Int 58:1420–1430
Ni W, Egashira K, Kataoka C et al. (2001) Antiinflammatory and antiarteriosclerotic actions of HMG-CoA reductase inhibitors in a rat model of chronic inhibition of nitric oxide synthesis. Circ Res 89:415–421
Hostetter TH, Troy JC, Brenner BM (1981) Glomerular hemodynamics in experimental diabetes mellitus. Kidney Int 19:410–415
Young BA, Johnson RJ, Alpers CE et al. (1995) Cellular events in the evolution of experimental diabetic nephropathy. Kidney Int 47:935–944
Rovin BH, Tan LC (1992) LDL stimulates mesangial fibronectin and chemoattractant expression. Kidney Int 43:218–225
Ihm CG, Park JK, Hong SP et al. (1998) A high glucose concentration stimulates the expression of monocyte chemotactic peptide 1 in human mesangial cells. Nephron 79:33–37
Banba N, Nakamura T, Matsumura M, Kuroda H, Hattori Y, Kasai K (2000) Possible relation of monocyte chemoattractant protein-1 with diabetic nephropathy. Kidney Int 58:684–690
Aikawa M, Rabkin E, Sugiyama S et al. (2001) An HMG CoA reductase inhibitor, cerivastatin, suppresses growth of macrophage expressing matrix metalloproteinases, and tissue factor in vivo and in vitro. Circulation 103:276–283
Ueda A, Okuda K, Ohno S et al. (1994) NF-kappa B and Sp1 regulate transcription of the human monocyte chemoattractant protein-1 gene. J Immunol 153:2052–2063
O'Donnell MP, Kasiske BL, Kim Y, Atluru D, Keane WF (1993) The mevalonate pathway: importance in mesangial cell biology and glomerular disease. Miner Electrolyte Metab 19:173–179
Goldstein JL, Brown MS (1990) Regulation of the mevalonate pathway. Nature 343:425–430
Maltese WA (1990) Posttranslational modification of proteins by isoprenoids in mammalian cells. FASEB J 4:3319–3328
Border WA, Noble NA (1994) Transforming growth factor β in tissue fibrosis. N Engl J Med 331:1286–1292
Yamamoto T, Nakamura T, Noble NA, Ruoslahti E, Border WA (1993) Expression of transforming growth factor-β is elevated in human and experimental diabetic nephropathy. Proc Natl Acad Sci USA 90:1814–1818
Sharma K, Ziyadeh FN (1995) Hyperglycemia and diabetic kidney disease: the case for transforming growth factor-β as a key mediator. Diabetes 44:1139–1146
Sharma K, Jin Y, Guo J, Ziyadeh FN (1996). Neutralization of TGF-β by anti- TGF-β antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes 45:522–530
Bollineni JS, Reddi AS (1996) Transforming growth factor-β1 enhances glomerular collagen synthesis in diabetic rats. Diabetes 42:1673–1677
Haneda M, Araki S, Togawa M, Sugimoto T, Isono M, Kikkawa R (1997) Mitogen-activated protein kinase cascade is activated in glomeruli of diabetic rats and glomerular mesangial cells cultured under high glucose conditions. Diabetes 46:847–853
Schneider A, Panzer U, Zahner G et al. (1999) Monocyte chemoattractant protein-1 mediates collagen deposition in experimental glomerulonephritis by transforming growth factor-β. Kidney Int 56:135–144
Kanwar YS, Farquhar MG (1979) Isolation of glycosaminoglycans (heparan sulfate) from glomerular basement membranes. Proc Natl Acad Sci USA 76:4493–4497
Kanwar YS, Linker A, Farquhar MG (1980) Increased permeability of the glomerular basement membrane to ferritin after removal of glycosaminoglycans (heparan sulfate) by enzyme digestion. J Cell Biol 86:688–693
Rosenzweig LJ, Kanwar YS (1982) Removal of sulfated (heparan sulfate) or nonsulfated (hyaluronic acid) glycosaminoglycans results in increased permeability of the glomerular basement membrane to125I-bovine serum albumin. Lab Invest 47:177–184
Kofoed-Enevoldsen A (1992) Inhibition of glomerular glucosaminyl N-deacetylase in diabetic rats. Kidney Int 41:763–767
Kashihara N, Watanabe Y, Makino H, Wallner EI, Kanwar YS (1992) Selective decreased de novo synthesis of glomerular proteoglycans under the influence of reactive oxygen species. Proc Natl Acad Sci USA 89:6309–6313
Moriyama T, Kawada N, Nagatoya K et al. (2001) Fluvastatin suppress oxidative stress and fibrosis in the interstitium of mouse kidneys with unilateral ureteral obstruction. Kidney Int 58:2095–2103
Acknowledgements
We are grateful to A. Nakano, M. Kaida and A. Katayama for technical assistance, and Dr. T. Wada for technical suggestion.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ota, T., Takamura, T., Ando, H. et al. Preventive effect of cerivastatin on diabetic nephropathy through suppression of glomerular macrophage recruitment in a rat model. Diabetologia 46, 843–851 (2003). https://doi.org/10.1007/s00125-003-1099-3
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-003-1099-3