
Abstract
Key message
Through substitution mapping strategy, two pairs of closely linked QTLs controlling stigma exsertion rate were dissected from chromosomes 2 and 3 and the four QTLs were fine mapped.
Abstract
Stigma exsertion rate (SER) is an important trait affecting the outcrossing ability of male sterility lines in hybrid rice. This complex trait was controlled by multiple QTLs and affected by environment condition. Here, we dissected, respectively, two pairs of tightly linked QTLs for SER on chromosomes 2 and 3 by substitution mapping. On chromosome 2, two linkage QTLs, qSER-2a and qSER-2b, were located in the region of 1288.0 kb, and were, respectively, delimited to the intervals of 234.9 kb and 214.3 kb. On chromosome 3, two QTLs, qSER-3a and qSER-3b, were detected in the region of 3575.5 kb and were narrowed down to 319.1 kb and 637.3 kb, respectively. The additive effects of four QTLs ranged from 7.9 to 9.0%. The epistatic effect produced by the interaction of qSER-2a and qSER-2b was much greater than that of qSER-3a and qSER-3b. The open reading frames were identified within the maximum intervals of qSER-2a, qSER-2b and qSER-3a, respectively. These results revealed that there are potential QTL clusters for SER in the two regions of chromosome 2 and chromosome 3. Fine mapping of the QTLs laid a foundation for cloning of the genes of SER.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rice is one of the most important staple food crops in the world. In the past decades, great progress has been made in improving rice yield by the utilization of heterosis in hybrid rice (Yuan 2017). The male sterility line (MSL) is a key line of the hybrid rice system. Since the cultivated rice is mainly self-pollination (Virmani and Athwal 1973), improving the outcrossing ability of MSLs is very important for hybrid rice seed production. Stigma exsertion can improve the outcrossing ability of MSLs by catching more pollens from male parents (Marathi and Jena 2015). Therefore, stigma exsertion rate (SER) is an important trait of outcrossing in hybrid rice. MSLs with high SER can produce more hybrid seeds in hybrid rice seed production (Virmani 1994).
In the past two decades, dozens of QTLs responsible for the SER have been identified from rice germplasm resources (Marathi and Jena 2015; Liu et al. 2019; Tan et al. 2020). Because of high SER, MSLs or their maintainer lines were usually used to detect QTLs for SER. From the cytoplasmic male sterile (CMS) maintainer line XieqingzaoB, qSE7 was located on chromosome 7 (Zhang et al. 2018), and qSE11 was mapped on chromosome 11 (Rahman et al. 2017). From the CMS maintainer line II-32B, qSER-7 was identified on chromosome 7 (Liu et al. 2019). The wild rice is the important germplasm resources with strong outcrossing ability (Marathi et al. 2015; Marathi and Jena 2015). Many QTLs for SER were identified from O. rufipogon (Xiong et al. 1999; Li et al. 2001; Uga et al. 2003a; Huang et al. 2012; Bakti and Tanaka 2019; Zou et al. 2020), O. longistaminata (Li et al. 2010), O. glumaepatula (Tan et al. 2020), O. barthii and O. meridionalis (Zou et al. 2020). Many of the QTLs for SER identified from different mapping populations and different mapping methods were found to be position overlapping in rice genome (Tan et al. 2020). The most common and most powerful QTLs for SER were frequently detected on the short arm of chromosome 3. In the region of about 12.0–17.0 Mb of chromosome 3, seven QTLs for SER, qES3 (Yamamtot et al. 2003; Miyata et al. 2007), PES-3 (Yue et al. 2009), qSPE3 (Feng et al. 2010), qSSE3 (Li et al. 2014), qSERb3-1 and qSERm3-1 (Zou et al. 2020), and qSER-3a (Tan et al. 2020) were detected. GS3, a gene controlling seed length (Fan et al. 2006) with pleiotropic effects on stigma length and stigma exsertion (Takano-Kai et al. 2011; Zhou et al. 2017a), were also located in the region. In the region of about 3.0–5.4 Mb of chromosome 2, five QTLs for SER, qPES-2 (Li et al. 2003), qPES-2 (Deng et al. 2010), qPES2.2 (Li et al. 2017), and qSERr2-1 and qSERm2-1 (Zou et al. 2020) were located. Since only one QTL was usually detected in an overlapping region in each study, it is unclear how many QTLs for SER exist in these regions. In addition, although a large number of QTLs for SER have been reported in the rice genome, few QTLs were located within the 500 kb interval and none of them has been cloned (Marathi and Jena 2015; Liu et al. 2019; Tan et al. 2020).
In recent two decades, substitution mapping has become a powerful tool for QTL detection of complex traits instead of traditional genetic mapping. Compared with primary mapping populations, such as F2, recombinant inbred lines (RILs), doubled haploid (DH) lines, the secondary mapping populations, such as chromosomal segment substitution lines (CSSLs), single-segment substitution lines (SSSLs), and near-isogenic lines (NILs), segregate only in target genes (segments) in the same genetic background (Eshed and Zamir 1995; Wang et al. 2012; Tan et al. 2020). For substitution mapping of QTLs in rice genome, we constructed an SSSL library (Zhang et al. 2004; Xi et al. 2006; He et al. 2017; Zhao et al. 2019; Zhang 2019). The SSSL library was widely used to detect QTLs in rice genome (Wang et al. 2012; Zhang et al. 2012; Zhao et al. 2016; Zhou et al. 2017b; Sui et al. 2019). Recently, we detected seven QTLs for SER on five chromosomes using a set of SSSLs derived from O. glumaepatula (Tan et al. 2020). In the present study, we used a set of SSSLs carrying substitution segments of IR66897B (IB), a CMS maintainer line, to fine map QTLs for SER. By substitution mapping, two QTLs for SER were detected in 1288.0 kb region of chromosome 2, and other two QTLs for SER were located in 3575.5 kb region of chromosome 3. The four QTLs were limited to 214.3–637.3 kb. These results revealed that these two chromosomal regions were potential QTL clusters for SER. The fine mapping of the four QTLs for SER laid the foundation for gene cloning.
Materials and methods
The SSSLs used
We have constructed a SSSL library with 2360 SSSLs by using 43 accessions of 7 species with rice AA-genome as donors and Huajingxian74 (HJX74), an elite indica variety, as recipient. Each SSSL carries only one chromosomal segment from a donor in the HJX74 genetic background (Zhang et al. 2004; Xi et al. 2006; He et al. 2017; Zhao et al. 2019; Zhang 2019). A set of 41 SSSLs carrying substitution segments from the donor of IR66897B (IB), a CMS maintainer line from International Rice Research Institute (IRRI), was selected from the HJX74-SSSL library. After surveying of SER, 7 SSSLs with higher SER than HJX74 were selected for this study (Table S1).
Field experiments
All plant materials were planted in the farm of South China Agriculture University, Guangzhou, China (23° 07′ N, 113° 15′ E). The materials were planted in nine cropping seasons from 2014 to 2018, two cropping seasons per year. The first cropping season (FCS) was from late February to middle July, the second cropping season (SCS) was from late July to middle November. Field cultivation and controlling of diseases and insect pests followed by conventional methods in South China.
Molecular markers and PCR protocol
SSR markers labeled “RM” were selected from online resources (https://archive.gramene.org/markers/). The “PSM” and “InDel” markers were designed using the software of Primer Premier 5.0 (Lalitha 2000) (Table S2). The GS3 gene was genotyped by a functional marker, SF28-PstI (Zhou et al. 2017a). Genomic DNA was extracted from freshly frozen leaves of plants using a reported method (Murray and Thompson 1980). Target DNA segments were amplified with the following program, 94 °C for 5 min, followed by 35 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 45 s, and a final extension of 72 °C for 5 min. The PCR products were separated on 6% denatured PAGE, and bands detected using the silver staining methods described by Fang et al. (2019).
Phenotyping of traits and statistical analysis
Stigma exsertion (SE) was classified into single stigma exsertion (SSE) and dual stigma exsertion (DSE). SER refers to the total stigma exsertion rate, including single stigma exsertion rate and dual stigma exsertion rate. For investigating SER, the 8–10 main panicles were sampled from plants of flourishing florescence. The SER per panicle was calculated based on the visual aspect of exserted stigmas (Liu et al. 2019). Grain traits were measured by the yield traits scorer (YTS), a rice phenotypic facility (Yang et al. 2014). The arcsine square root transformation of SER value was used for statistical analysis, one-way ANOVA. Dunnett t test treated one group as a control, and compared all other groups against it. Least significance range (LSR) was used for multiple rang test among multiple groups (Duncan 1955). The data analysis and figure making were done by SPSS statistics 23.0 and OriginPro 9.0 (https://www.originlab.com).
Substitution mapping of QTLs
The minimum length (Lmin) of a substitution segment refers to the length between two markers of donor genotype at the end of the substitution segment. The maximum length (Lmax) refers to the length between two markers flanking the two end of substitution segment with background genotype. The estimated length (Lest) = (Lmin + Lmax)/2 (Tan et al. 2020). Secondary SSSLs were developed by backcrossing primary SSSLs with HJX74. The QTLs for SER were mapped by the substitution mapping method (Eshed and Zamir 1995; Tan et al. 2020). When the phenotype of SER had significant difference between a SSSL and the recipient, a QTL for SER was located in the region of substitution segment of SSSL. Additive effect of a single gene was defined as half of the phenotypic difference between SSSL and HJX74. Epistatic effects among QTLs were estimated by the formula, \(i=\left({P}_{n}-{P}_{0}\right)-2\sum_{i=1}^{n}\left({a}_{i}\right)\), where i is an epistasis among the pyramided QTLs, Pn is a phenotype of a pyramiding line harboring n of QTLs, P0 is a phenotype of HJX74, ai (1 ≤ i ≤ n) is an additive effect of a single gene at the ith QTL. Epistatic effects among QTLs were tested in Student's t test under null hypothesis (H0) i = 0. QTLs were named by the method of McCouch et al. (1997). Linkage maps of markers were drawn by using MapChart2.2 (https://www.wur.nl/en/show/Mapchart.htm).
Gene annotation in the regions of QTLs
Two rice gene annotation databases, the Rice Annotation Project Database (RAP-DB) and the Rice Genome Annotation Project of Michigan State University (MSU-RAP), were used to identify the open reading frames (ORFs) within the target QTL regions of Nipponbare reference genome (IRGSP-1.0) from the Gramene database (http://www.gramene.org/).
Results
SER in the SSSLs derived from the CMS maintainer line
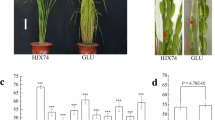
After investigating of SER from a set of SSSLs derived from the CMS maintainer line IB, seven SSSLs with higher SER than the HJX74 recipient were selected. Then, the SER of seven SSSLs was tested in nine cropping seasons from 2014 FCS to 2018 FCS. Compared with HJX74, the seven SSSLs were showed higher SER at the P ≤ 0.001 level in every cropping season. Average SER of seven SSSLs in the nine cropping seasons was from 44.8 to 55.3% with 17.0–27.5% higher than that of the control HJX74 (Fig. 1 and Table S1).

SER of seven SSSLs and HJX74. a SER of the SSSLs and control HJX74. Data were presented as mean ± S.E. in nine cropping seasons. Dunnett t test, *** P ≤ 0.001. b Exserted stigmas in the panicles of HJX74 and A88. Red arrows point the exserted stigmas. Scale bar, 1 cm. SSSL single-segment substitution line. SER stigma exsertion rate
The substitution segments of the seven SSSLs were detected by increasing the density of molecular markers (Table S2). The substitution segments were detected on chromosomes 2 and 3 with the estimated lengths from 1843.4 to 5162.6 kb, respectively (Table S3).
Eight agronomic traits in the seven SSSLs were investigated in two cropping seasons of 2018. Compared with HJX74, the SSSLs had no significant difference at P = 0.001 level in all traits, but had significant differences at P ≤ 0.01 level in some traits (Table S4). The results showed that the genetic background of the SSSLs was similar to HJX74 except for SER.
Two closely linked QTLs for SER were dissected on the substitution segment of chromosome 2
Six SSSLs, A7, A13, A35, A37, A41 and A42 with significantly higher SER than HJX74, carried the substitution segments in the region of RM12521-RM12865 on chromosome 2 (Fig. 2a, c). A42, the SSSL with the longest of substitution segment, was then selected to develop secondary SSSLs. Six secondary SSSLs were developed from a F2 population derived from the cross of HJX74/A42 (Fig. 2b–d). The substitution segments of six secondary SSSLs were divided into two sub-set. In the left sub-set, the secondary SSSL A42-45 carrying the substitution segment from markers RM12521 to ID02M23 had low SER as HJX74, while secondary SSSLs A42-69 and A42-34 carrying longer substitution segments had significantly higher SER than HJX74. These results indicated that there was a QTL for SER, qSER-2a, locating in the region from markers ID02M23 to RM3732 with the estimated length of 234.9 kb. In the right sub-set, the secondary SSSL A42-12 carrying the substitution segment from markers ID02MQ21 to RM12865 had low SER as HJX74, while the SSSL A41 and the secondary SSSL A42-17 carrying longer substitution segments had significantly higher SER than HJX74. These results indicated that there was another QTL for SER, qSER-2b, locating in the region from markers ID02MQ51 to ID02MQ21 with the estimated length of 214.3 kb. The two QTLs for SER, qSER-2a and qSER-2b, were closely linked in the region of 1288.0 kb (Fig. 2d).

Substitution mapping of QTLs for SER on chromosome 2. a The substitution segment of SSSL-A42 on chromosome 2. Physical distance (Mb) was shown under the chromosome. b Plant type of SSSL-A42 and HJX74, scale bar, 15 cm. c Substitution mapping based on the substitution segments of six SSSLs. d Secondary substitution mapping of QTLs based on the substitution segment of SSSL-A42. SER stigma exsertion rate. SER (%) was a mean ± S.E. in nine cropping seasons (c) and two cropping seasons (d). Different alphabet letters denote differences at 0.01 level of significance in Duncan test. White and black blocks on chromosomes represent genotypes of HJX74 and donor, respectively
Two closely linked QTLs for SER were dissected on the substitution segment of chromosome 3
The SSSL, A88, with significantly higher SER than HJX74, carried the substitution segment from markers PSM16 to RM15196 on chromosome 3 with the estimated length of 3874.7 kb (Fig. 3a–c and Table S3). A88 was used to develop secondary SSSLs. Three secondary SSSLs, A88-59, A88-107 and A88-141, were developed from the cross of A88/HJX74. The three secondary SSSLs carried different substitution segments showed significantly higher SER than HJX74 (Fig. 3c).

Substitution mapping of QTLs for SER on chromosome 3. a The substitution segment of SSSL-A88 on chromosome 3. Physical distance (Mb) was shown under the chromosome. b Plant type of HJX74 and SSSL-A88. Scale bar, 15 cm. c Secondary substitution mapping based on the substitution segment of SSSL-A88. d Secondary substitution mapping based on the substitution segment of A88-59 and A88-141, respectively. e Secondary substitution mapping of qSER-3a based on the substitution segment of A88-59-15. f Secondary substitution mapping of qSER-3b based on the substitution segment of A88-141-2. SER stigma exsertion rate. SER (%) was mean ± S.E. in two cropping seasons. Different alphabet letters denote differences at 0.01 level of significance in Duncan test. White and black blocks on chromosomes represent genotypes of HJX74 and donor, respectively. Δ, Location of GS3
A88-59 and A88-141 were then used to develop secondary SSSLs. Three secondary SSSLs, A88-59-11, A88-59-15 and A88-59-22, were developed from the A88-59 heterozygote. Only A88-59-15 showed high SER as A88-59, while A88-59-11 and A88-59-22 had low SER as HJX74. Other three secondary SSSLs, A88-141-2, A88-141-8 and A88-141-11, were developed from the A88-141 heterozygote. Only A88-141-2 showed high SER as A88-141, while A88-141-8 and A88-141-11 had low SER as HJX74. Based on the substitution segments of the two sets of secondary SSSLs, the substitution segment of A88 should have two QTLs for SER. One was located in the interval from markers ID79 to ID03M04-9 on the left, and another was located in the interval from markers ID03M04-7 to ID03Ma31 on the right (Fig. 3d).
To narrow the QTL interval from markers ID79 to ID03M04-9 on the left, A88-59-15 was used to develop secondary SSSLs. Five secondary SSSLs, A88-59-15-3, A88-59-15-10, A88-59-15-16, A88-59-15-19 and A88-59-15-20, were developed from the A88-59-15 heterozygote. Three secondary SSSLs, A88-59-15-3, A88-59-15-10 and A88-59-15-19, showed high SER as A88-59-15, while two secondary SSSLs, A88-59-15-16 and A88-59-15-20, had low SER as HJX74. Therefore, the QTL, qSER-3a, was located in the interval from markers ID61 to ID03M81 with estimated length of 319.1 kb (Fig. 3e).
To narrow the QTL interval from markers ID03M04-7 to ID03Ma31 on the right, A88-141-2 were used to develop secondary SSSLs. Four secondary SSSLs, A88-141-2-11, A88-141-2-12, A88-141-2-21 and A88-141-2-24, were developed from the A88-141-2 heterozygote. A88-141-2-11 and A88-141-2-21 showed high SER as A88-141-2, while A88-141-2-12 and A88-141-2-24 had low SER as HJX74. Because A88-141-2-21 carried the substitution segment from markers ID03M141-16 to ID03Ma31, the QTL, qSER-3b, was located in the interval with estimated length of 637.3 kb (Fig. 3f).
The two QTLs for SER, qSER-3a and qSER-3b, were closely linked in the region of 3575.5 kb (Fig. 3e-f). We noted that the GS3 locus controlling grain length was located at the right end of the qSER-3b interval between markers ID03Ma27 and RM15196. Genotyping of the GS3 gene by a functional marker showed that A88 carried the GS3 allele as same as HJX74 (Fig. S1). Phenotyping of A88 showed that the grain length was no significant difference with HJX74 (Table S4). These results indicated that the SER controlled by qSER-3b was not affected by the GS3 gene.
Additive and epistatic effects of the QTLs for SER
The two closely linked QTLs, qSER-2a and qSER-2b, were separated from the segment of chromosome 2. The secondary SSSLs only carrying qSER-2a or qSER-2b were used to evaluate the additive effects of the qSER-2a and qSER-2b, respectively (Table S5). The additive effects were 9.0% in qSER-2a and 8.8% in qSER-2b (Table 1). In the same way, the two closely linked QTLs, qSER-3a and qSER-3b, were dissected from the segment of chromosome 3. The sets of secondary SSSLs only carrying qSER-3a or qSER-3b were used to estimate the additive effects of the qSER-3a and qSER-3b, respectively (Table S6). The additive effects were 8.7% in qSER-3a and 7.9% in qSER-3b (Table 1).
In the same experiments, the four lines carrying the substitution segments with both qSER-2a and qSER-2b, A35, A37, A42 and A42-24, showed significantly higher SER than the lines only with qSER-2a or qSER-2b (Table S5 and Fig. 2d). Similarly, the SSSL A88 carrying both qSER-3a and qSER-3b had significantly higher SER than those only carrying qSER-3a or qSER-3b (Table S6 and Fig. 3d-f). These results indicated that the two pairs of QTLs had significantly additive effects.
To evaluate epistatic effects produced by the interaction of QTLs for SER, the SSSL A42 having qSER-2a and qSER-2b and the secondary SSSL A88-59-15 with qSER-3a were used to develop the pyramiding line P223 carrying qSER-2a, qSER-2b and qSER-3a (Fig. 4a, b). In single-QTL groups of SSSLs, S2a having qSER-2a, S2b having qSER-2b, S3a having qSER-3a and S3b having qSER-3b, showed SER of 46.3%, 46.0%, 43.5% and 43.7%, respectively, while two-QTL groups showed 51.3% SER in S22 carrying qSER-2a and qSER-2b, and 56.3% SER in S33 of qSER-3a and qSER-3b. SER of the two-QTL groups was significantly higher than those of the single-QTL groups (Table S7). Based on the SER data, the additive effect of the pair of qSER-3a and qSER-3b in S33 group was 14.6%, which was much greater than 12.2% of the qSER-2a and qSER-2b pair in S22 group. This was because the epistatic effect produced by the interaction of qSER-2a and qSER-2b was much greater than that of qSER-3a and qSER-3b, with the former being -13.9% and the latter being -3.9% (Fig. 4c). As a comparison, SER of the pyramiding line P223 with three QTLs was 67.2%, greatly higher than those of single-QTL groups and of two-QTL groups (Table S7). The epistatic effect in P223 was -14.6%, which seems mainly from the interaction of qSER-2a and qSER-2b (Fig. 4c).

Epistatic effects estimated in the lines with two or three QTLs for SER. a Plant type of HJX74 and pyramiding line P223. Scale bar, 15 cm. b Position of the three QTLs for SER in P223. Black part represents substitution segments from IR66897B (IB) with target QTLs, and white part is the genetic background of HJX74. c Epistatic effects estimated in S22, S33 and P223. S22 is a group of the SSSLs carrying qSER-2a and qSER-2b. S33 is a group of the SSSLs carrying qSER-3a and qSER-3b. P223 is a pyramiding line with qSER-2a, qSER-2b and qSER-3a. Student’s t test. SER stigma exsertion rate, SSSL single-segment substitution line
Gene annotation in the regions of qSER-2a, qSER-2b and qSER-3a
The ORFs within the maximum intervals of three fine mapped QTLs for SER were identified. From the RAP-DB and the MSU-RAP, 64 and 56 of ORFs were, respectively, identified within 370.7 kb of the qSER-2a interval. Among them, 45 ORFs were identified from the two databases. In the 291.5 kb of qSER-2b region, 51 and 45 of ORFs were, respectively, identified from the RAP-DB and the MSU-RAP and 36 of them were identified from the two databases. The qSER-3a region with 361.4 kb included 19 common ORFs, which from 22 and 51 ones identified from the RAP-DB and the MSU-RAP (Fig. S2).
In the regions of three QTLs, most identifiers only from the RAP-DB were hypothetical genes or non-protein coding transcripts, and most identifiers only from the MSU-RAP were hypothetical genes, retrotransposon genes or transposon genes (Tables S8-S10). Therefore, the common identifiers from both databases are more likely to be candidate genes of the QTLs.
Discussion
Many QTLs for SER and related traits have been identified and are distributed across all the 12 chromosomes. However, few of the QTLs were located within a 500 kb interval (Marathi and Jena 2015; Liu et al. 2019; Tan et al. 2020). In the present study, we detected two QTLs for SER in the region of 1288.0 kb on chromosome 2. qSER-2a was mapped in the estimated interval of 234.9 kb, and qSER-2b was located in 214.3 kb estimated interval. In previous studies, five QTLs for SER were detected around the region. Li et al. (2003) detected a QTL, qPES-2, in the interval of 1044 kb using a DH population. Deng et al. (2010) located qPES-2 in the region of about 770 kb using an F2 population. Li et al. (2017) mapped qPES2.2 in 4993 kb region using an F2: 3 population. Zou et al. (2020) identified qSERr2-1 in about 3221 kb region from O. rufipogon and qSERm2-1 in the region of about 6739 kb from O. meridionalis. On chromosome 3, we detected two QTLs for SER in the region of 3575.5 kb. qSER-3a was mapped in the estimated interval of 319.1 kb, and qSER-3b was located in 637.3 kb estimated interval. In previous studies, seven QTLs for SER were detected around the region. Miyata et al. (2007) detected a QTL, qES3, in the interval of 10554 kb using a CSSL population. Yue et al. (2009) located PES-3 in 7895 kb region using a F2 population. Feng et al. (2010) mapped qSPE3 in the region of about 600 kb using a F2 population. Li et al. (2014) identified qSSE3 in 14426 kb interval using a RIL population. Tan et al. (2020) detected qSER-3a in the region of 4632 kb from O. glumaepatula. Zou et al. (2020) identified qSERb3-1 in the region of 6629 kb from O. barthii and qSERm3-1 in the 2983 kb region from O. meridionalis. These results revealed that there are potential QTL clusters for SER in the two regions of chromosome 2 and chromosome 3. From the RAP-DB and the MSU-RAP, the open reading frames (ORFs) were identified within the maximum intervals of qSER-2a, qSER-2b and qSER-3a, respectively. The dissection of two pairs of closely linked QTLs for SER and the fine mapping of the QTLs laid a foundation for the cloning of genes for SER.
During the process of domestication, cultivated rice has already lost some traits of natural outcrossing (Parmar et al. 1979). Wild Oryza species have a strong outcrossing ability due to their larger stigma, longer style, greater exsertion of the stigma, and longer periods of spikelet opening (Marathi et al. 2015; Marathi and Jena 2015). It was found that cultivated rice tends to have a shorter stigma than the annual wild species, while annual wild species have shorter stigma than their perennial progenitors (Oka and Morishima 1967; Virmani and Athwal 1973; Parmar et al. 1979; Marathi et al. 2015). Previous studies revealed dominant differences for SER and floral traits between cultivated rice and wild rice (Virmani and Athwal 1973; Uga et al. 2003b). Recently, seven QTLs for SER from O. glumaepatula, a wild Oryza species, were located on five chromosomes by substitution mapping. The additive effects of seven QTLs ranged from 10.6 to 14.8% (Tan et al. 2020). In the present study, four QTLs for SER identified in cultivated rice had their additive effects from 7.9 to 9.0% (Table 1). Obviously, the additive effect of the QTLs for SER in cultivated rice was usually lower than that in wild rice. This may be one of the reasons for the decrease of outcrossing ability of cultivated rice during domestication.
In order to detect QTLs of rice by substitution mapping, we constructed a library containing 2360 SSSLs using 43 accessions from seven species of Oryza AA-genome as donors and Huajingxian74 (HJX74), an elite indica variety, as the recipient (Zhang et al. 2004; Xi et al. 2006; He et al. 2017; Zhao et al. 2019; Zhang 2019). Similar to NILs, each SSSL carries only one chromosomal segment from a donor under the genetic background of recipient (Zhang et al. 2004; Xi et al. 2006). Since the SSSLs have homozygous genotypes, the SSSLs can be used to test the phenotype in different cropping seasons. This increases the accuracy of phenotyping of complex traits (Tan et al. 2020). Because the QTLs for SER had minor effects and were closely linked, the QTLs were not easy to be dissected using traditional mapping methods. As mentioned above, we dissected two closely linked QTLs in the QTL cluster region of chromosomes 2 and 3, whereas only one QTL was detected in each previous study. These facts indicated that substitution mapping was a powerful tool for dissection of closely linked QTLs of complex traits.
Based on the HJX74-SSSL library, the platform of breeding by design in rice has been developed (Dai et al. 2015, 2016; Luan et al. 2019; Zhang 2019). In this study, we found that the qSER-2a and qSER-2b on chromosome 2 and the qSER-3a and qSER-3b on chromosome 3 had significant additive effects for SER. In comparison, the epistatic effect produced by the interaction of qSER-2a and qSER-2b was much greater than that of qSER-3a and qSER-3b. Therefore, the SSSLs carrying two closely linked QTLs, qSER-2a and qSER-2b on chromosome 2 or qSER-3a and qSER-3b on chromosome 3, will be favorable genetic resources for developing MSLs with high SER. Understanding the epistatic effect of QTL interaction is important for breeding by design.
References
Bakti C, Tanaka J (2019) Detection of dominant QTLs for stigma exsertion ratio in rice derived from Oryza rufipogon accession “W0120.” Breed Sci 69:143–150
Dai Z, Lu Q, Luan X, Cai J, Zhu H, Liu Z, Zeng R, Zhang Z, Wang S, Zheng L, Li J, Zhang G (2015) Development of a platform for breeding by design of CMS lines based on an SSSL library in rice (Oryza sativa L.). Euphytica 205:63–72
Dai Z, Lu Q, Luan X, Ouyang L, Guo J, Liang J, Zhu H, Wang W, Wang S, Zeng R, Liu Z, Zhang Z, Zhu X, Zhang G (2016) Development of a platform for breeding by design of CMS restorer lines based on an SSSL library in rice (Oryza sativa L.). Breed Sci 66:768–775
Deng Y, Ying J, Shi Y, Xiao C, Zhang H (2010) Mapping of QTLs for percentage of exserted stigma in rice. J. Hunan Agric Univers (Nat Sci) 36:373–376 (in Chinese with English abstract)
Duncan DB (1955) Multiple range and multiple F tests. Biometrics 11:1–42
Eshed Y, Zamir D (1995) An introgression line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated QTL. Genetics 141:1147–1162
Fan C, Xing Y, Mao H, Lu T, Han B, Xu C, Li X, Zhang Q (2006) GS3, a major QTL for grain length and weight and minor QTL for grain with and thickness in rice, encodes a putative transmembrane protein. Theor Appl Genet 112:1164–1171
Fang C, Li L, He R, Wang D, Wang M, Hu Q, Ma Q, Qin K, Feng X, Zhang G, Fu X, Liu Z (2019) Identification of S23 causing both interspecific hybrid male sterility and environment-conditioned male sterility in rice. Rice 12:10
Feng L, Jing Y, Huang C, Xu Z, Chen W (2010) QTL analysis of percentage of exserted stigma in rice (Oryza sativa L.). North Rice 40:20–22 (in Chinese with English abstract)
He N, Wu R, Pan X, Peng L, Sun K, Zou T, Zhu H, Zeng R, Liu Z, Liu G, Wang S, Zhang G, Fu X (2017) Development and trait evaluation of chromosome single-segment substitution lines of O. meridionalis in the background of O. sativa. Euphytica 213:281
Huang X, Kurata N, Wei X, Wang Z, Wang A, Zhao Q, Zhao Y, Liu K, Lu H, Li W, Guo Y, Lu Y, Zhou C, Fan D, Weng Q, Zhu C, Huang T, Zhang L, Wang Y, Feng L, Furuumi H, Kubo T, Miyabayashi T, Yuan X, Xu Q, Dong G, Zhan Q, Li C, Fujiyama A, Toyoda A, Lu T, Feng Q, Qian Q, Li J, Han B (2012) A map of rice genome variation reveals the origin of cultivated rice. Nature 490:497–501
Lalitha S (2000) Primer premier 5. Biotech Software Internet Rep 1:270–272
Li C, Sun C, Mu P, Chen L, Wang X (2001) QTL analysis of anther length and ratio of stigma exsertion, two key traits of classification for cultivated rice (Oryza sativa L.) and common wild rice (Oryza rufipogon Griff.). Acta Genet Sin 28:746–751
Li W, Dong G, Hu X, Teng S, Guo L, Zeng D, Qian Q (2003) QTL analysis for percentage of exserted stigma in rice (Oryza sativa L.). Acta Genet Sin 30:637–640
Li H, Gao F, Zeng L, Li Q, Lu X, Li Z, Ren J, Su X, Ren G (2010) QTL analysis of rice stigma morphology using an introgression line from Oryza longistaminata L. Mol Plant Breed 8:1082–1089 (in Chinese with English abstract)
Li P, Feng F, Zhang Q, Chao Y, Gao G, He Y (2014) Genetic mapping and validation of quantitative trait loci for stigma exsertion rate in rice. Mol Breed 34:2131–2138
Li W, Sheng Z, Zhu Z, Wei X, Shi L, Wu Y, Tang S, Wang J, Hu P (2017) QTL mapping of japonica rice stigma exsertion rate. Chin J Rice Sci 31:23–30 (in Chinese with English abstract)
Liu Y, Zhang A, Wang F, Kong D, Li M, Bi J, Zhang F, Wang J, Luo X, Pan Z, Yu X, Liu G, Luo L (2019) Fine mapping a quantitative trait locus, qSER-7, that controls stigma exsertion rate in rice (Oryza sativa L.). Rice 12:46
Luan X, Dai Z, Yang W, Tan Q, Lu Q, Guo J, Zhu H, Liu G, Wang S, Zhang G (2019) Breeding by design of CMS lines on the platform of SSSL library in rice. Mol Breed 39:126
Marathi B, Jena KK (2015) Floral traits to enhance outcrossing for higher hybrid seed production in rice: present status and future prospects. Euphytica 201:1–14
Marathi B, Ramos J, Hechanova SL, Oane RH, Jena KK (2015) SNP genotyping and characterization of pistil traits revealing a distinct phylogenetic relationship among the species of Oryza. Euphytica 201:131–148
McCouch SR, Cho YG, Yano M, Paul E, Blinstrub M, Mor-ishima H, Kinosita T (1997) II. Report from coordinators. (1) Report on QTL nomenclature. Rice Genet Newsl 14:11–12
Miyata M, Yamamoto TT, Nitta N (2007) Marker-assisted selection and evaluation of the QTL for stigma exsertion under japonica rice genetic background. Theor Appl Genet 114:539–548
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucl Acids Res 8:4321–4325
Oka HI, Morishima H (1967) Variations in the breeding systems of wild rice, Oryza perennis. Evolution 21:249–258
Parmar KS, Siddiq EA, Swaminathan MS (1979) Variation in components of flowering behavior of rice. Indian J Genet Plant Breed 39:542–550
Rahman MH, Zhang Y, Zhang K, Rahman MS, Barman HN, Riaz A, Chen Y, Wu W, Zhan X, Cao L, Cheng S, (2017) Genetic dissection of the major quantitative trait locus (qSE11), and its validation as the major influence on the rate of stigma exsertion in rice (Oryza sativa L.). Front Plant Sci 8:1818
Sui F, Zhao D, Zhu H, Gong Y, Tang Z, Huang X, Zhang G, Zhao F (2019) Map-based cloning of a new total loss-of-function allele of OsHMA3 causes high cadmium accumulation in rice grain. J Exp Bot 70:2857–2871
Takano-Kai N, Doi K, Yoshimura A (2011) GS3 participates in stigma exsertion as well as seed length in rice. Breed Sci 61:244–250
Tan Q, Zou T, Zheng M, Ni Y, Luan X, Li X, Yang W, Yang Z, Zhu H, Zeng R, Liu G, Wang S, Fu X, Zhang G (2020) Substitution mapping of the major quantitative trait loci controlling stigma exsertion rate from Oryza glumaepatula. Rice 13:37
Uga Y, Fukuta Y, Cai H, Iwata H, Ohsawa R, Morishima H, Fujimura T (2003a) Mapping QTLs influencing rice floral morphology using recombinant inbred lines derived from a cross between Oryza sativa L. and Oryza rufipogon Griff. Theor Appl Genet 107:218–226
Uga Y, Fukuta Y, Ohsawa R, Fujimura T (2003b) Variations of floral traits in Asian cultivated rice (Oryza sativa L.) and its wild relatives (O rufipogon Griff.). Breed Sci 53:345–352
Virmani SS (1994) Heterosis and hybrid rice breeding. Theor Appl Genet 115:301–304
Virmani SS, Athwal DS (1973) Genetic variability in floral characteristics influencing outcrossing in Oryza sativa L. Crop Sci 13:66–67
Wang S, Wu K, Yuan Q, Liu X, Liu Z, Lin X, Zeng R, Zhu H, Dong G, Qian Q, Zhang G, Fu X (2012) Control of grain size, shape and quality by OsSPL16 in rice. Nat Genet 44:950–954
Xi Z, He F, Zeng R, Zhang Z, Ding X, Li W, Zhang G (2006) Development of a wide population of chromosome single-segment substitution lines in the genetic background of an elite cultivar of rice (Oryza sativa L.). Genome 49:476–484
Xiong L, Liu K, Dai X, Xu C, Zhang Q (1999) Identification of genetic factors controlling domestication-related traits of rice using an F2 population of a cross between Oryza sativa and O. rufipogon. Theor Appl Genet 98:243–251
Yamamtot TT, Takemori N, Sue N (2003) QTL analysis of stigma exsertion in rice. Rice Genet Newsl 10:33–34
Yang W, Guo Z, Huang C, Duan L, Chen G, Jiang N, Fang W, Feng H, Xie W, Lian X, Wang G, Luo Q, Zhang Q, Liu Q, Xiong L (2014) Combining high-throughput phenotyping and genome-wide association studies to reveal natural genetic variation in rice. Nat Commun 5:5087
Yuan L (2017) Progress in super-hybrid rice breeding. The Crop J 5:100–102
Yue G, Mei H, Pan B, Lou J, Li M, Luo L (2009) Mapping of QTLs affecting stigma exsertion rate of Huhan 1B as a CMS maintainer of upland hybrid rice. Acta Agric Zhejiangensis 21:241–245 (in Chinese with English abstract)
Zhang G (2019) The platform of breeding by design based on the SSSL library in rice. Hereditas (Beijing) 41:754–760 (in Chinese with English abstract)
Zhang G, Zeng R, Zhang Z, Ding X, Li W, Liu G, He F, Tulukdar A, Huang C, Xi Z, Qin L, Shi J, Zhao F, Feng M, Shan Z, Chen L, Guo X, Zhu H, Lu Y (2004) The construction of a library of single segment substitution lines in rice (Oryza sativa L.). Rice Genet Newsl 21:85–87
Zhang Y, Yang J, Shan Z, Chen S, Qiao W, Zhu X, Xie Q, Zhu H, Zhang Z, Zeng R, Ding X, Zhang G (2012) Substitution mapping of QTLs for blast resistance with SSSLs in rice (Oryza sativa L.). Euphytica 184:141–150
Zhang K, Zhang Y, Wu W, Zhan X, Anis GB, Rahman MH, Hong Y, Riaz A, Zhu A, Cao Y, Sun L, Yang Z, Yang Q, Cao L, Cheng S (2018) qSE7 is a major quantitative trait locus (QTL) influencing stigma exsertion rate in rice (Oryza sativa L.). Sci Rep 8:14523
Zhao F, Zhu H, Zeng R, Zhang G, Xu S (2016) Detection of additive and additive x environment interaction effects of QTLs for yield-component traits of rice using single-segment substitution lines (SSSLs). Plant Breed 135:452–458
Zhao H, Sun L, Xiong T, Wang Z, Liao Y, Zou T, Zheng M, Zhang Z, Pan X, He N, Zhang G, Zhu H, Liu Z, He P, Fu X (2019) Genetic characterization of the chromosome single-segment substitution lines of O. glumaepatula and O. barthii and identification of QTLs for yield-related traits. Mol Breed 39:51
Zhou H, Li P, Xie W, Hussain S, Li Y, Xia D, Zhao H, Sun S, Chen J, Ye H, Hou J, Zhao D, Gao G, Zhang Q, Wang G, Lian X, Xiao J, Yu S, Li X, He Y (2017a) Genome-wide association analyses reveal the genetic basis of stigma exsertion in rice. Mol Plant 10:634–644
Zhou Y, Xie Y, Cai J, Liu C, Zhu H, Jiang R, Zhong Y, Zhang G, Tan B, Liu G, Fu X, Liu Z, Wang S, Zhang G, Zeng R (2017b) Substitution mapping of QTLs controlling seed dormancy using single segment substitution lines derived from multiple cultivated rice donors in seven cropping seasons. Theor Appl Genet 130:1191–1205
Zou T, Zhao H, Li X, Zheng M, Zhang S, Sun L, He N, Pan X, Liu Z, Fu X (2020) QTLs detection and pyramiding for stigma exsertion rate in wild rice species by using the single-segment substitution lines. Mol Breed 40:74
Funding
This work was supported by Grants from the National Natural Science Foundation of China (91735304 and 91435207).
Author information
Authors and Affiliations
Contributions
GZ and SW designed and supervised works. QT performed most of the experiments, analyzed experimental data and prepared the draft of manuscript. CW, XL, LZ, YN, WY and ZY conducted a part of experiments. HZ, RZ and GL developed the materials. GZ wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Takuji Sasaki.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tan, Q., Wang, C., Luan, X. et al. Dissection of closely linked QTLs controlling stigma exsertion rate in rice by substitution mapping. Theor Appl Genet 134, 1253–1262 (2021). https://doi.org/10.1007/s00122-021-03771-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-021-03771-9