
Abstract
Aluminum (Al) toxicity in acid soils is a major limitation to the production of alfalfa (Medicago sativa subsp. sativa L.) in the USA. Developing Al-tolerant alfalfa cultivars is one approach to overcome this constraint. Accessions of wild diploid alfalfa (M. sativa subsp. coerulea) have been found to be a source of useful genes for Al tolerance. Previously, two genomic regions associated with Al tolerance were identified in this diploid species using restriction fragment length polymorphism (RFLP) markers and single marker analysis. This study was conducted to identify additional Al-tolerance quantitative trait loci (QTLs); to identify simple sequence repeat (SSR) markers that flank the previously identified QTLs; to map candidate genes associated with Al tolerance from other plant species; and to test for co-localization with mapped QTLs. A genetic linkage map was constructed using EST-SSR markers in a population of 130 BC1F1 plants derived from the cross between Al-sensitive and Al-tolerant genotypes. Three putative QTLs on linkage groups LG I, LG II and LG III, explaining 38, 16 and 27% of the phenotypic variation, respectively, were identified. Six candidate gene markers designed from Medicago truncatula ESTs that showed homology to known Al-tolerance genes identified in other plant species were placed on the QTL map. A marker designed from a candidate gene involved in malic acid release mapped near a marginally significant QTL (LOD 2.83) on LG I. The SSR markers flanking these QTLs will be useful for transferring them to cultivated alfalfa via marker-assisted selection and for pyramiding Al tolerance QTLs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alfalfa (Medicago sativa subsp. sativa) is one of the most important forage crops in the world. Often called the “Queen of the Forages,” it is the fourth most widely grown crop in the United States behind corn (Zea mays L.), wheat (Triticum aestivum L.) and soybean (Glycine max L.). Alfalfa not only has a high nutritional quality as animal feed, but has potential to be used in soil-bioremediation, bio-fuel for the production of electricity, and for the production of industrial enzymes. An estimated 30–40% of the world’s arable soils have a pH below 5.5 and therefore possess aluminum (Al) toxicity hazards (von Uexkull and Mutert 1995). Soil acidity is common to soils where rainfall is high enough to leach appreciable amounts of exchangeable bases from the soil surface layers which lower the soil pH (Brady 1974). In low pH, Al, the third most abundant element, becomes soluble in its toxic forms and is absorbed by plants. The root tips of plants show the most sensitivity to Al toxicity, inhibiting cell division and cell elongation, and thereby also inhibiting the root and shoot growth (Ryan et al. 1993; Sivaguru and Horst 1998). Alfalfa productivity is highly limited by Al toxicity related to acid soils across the world and in the United States (Rechigl et al. 1988). Even in the areas where liming is used to raise the soil pH, the subsoil may still remain acidic, reducing root growth, thus resulting in plants that are stunted and drought susceptible (Sumner et al. 1986). A cost effective alternative is growing Al-tolerant cultivars in problem soils combined with soil amendments (Foy 1988).
In several crop species, the genetic variability for Al tolerance has been exploited to develop Al-tolerant varieties and to explore the function of the genes involved in Al tolerance. The tolerance strategies involve either a mechanism of exclusion of Al from the root apex by excretion of organic acids that chelate Al, or a mechanism that allows the plants to tolerate Al within the cells. In wheat, barley (Hordeum vulgare L) and sorghum (Sorghum bicolor L. Moench), Al tolerance was attributed to the action of a single dominant gene, involved in Al-activated malate transportation (Delhaize et al. 1993; Minella and Sorrells 1997; Magalhaes et al. 2004). However, in the wheat cv. Chinese Spring, three quantitative trait loci (QTLs) that enhanced root growth under Al stress were identified, suggesting that inheritance of Al resistance is polygenic (Ma et al. 2006). Approximately nine Al tolerance QTLs in rice (Oryza sativa L) and five QTLs in soybean have been identified (Nguyen et al. 2001; Bianchi-Hall et al. 2000). Two QTLs for Al tolerance were identified in Arabidopsis thaliana, both of which co-segregated with Al-activated malate release from roots (Kobayashi and Koyama 2002; Hoekenga et al. 2003).
Extensive effort has been directed to screening and selecting alfalfa for resistance to acidic, Al-containing soil (Baligar et al. 1989; Bouton 1996; Dall’Agnol et al. 1996). Nevertheless, no M. sativa subsp. sativa cultivar or plant introduction is currently available that shows Al tolerance and does not suffer a decline in biomass under acid conditions. This lack of Al tolerance in primary alfalfa germplasm dictates the need for identifying genes or QTLs for Al tolerance in relatives of alfalfa that could be transferred to cultivated alfalfa. Genotypes with Al tolerance have been identified among wild diploid M. sativa subspecies (Bouton 1996) and in M. truncatula germplasm (Sledge et al. 2005a). Recent QTL mapping using a diploid alfalfa, M. sativa subsp. coreulea, a relative of cultivated alfalfa, has provided insights into the inheritance of Al tolerance. Three F2 populations were developed from a cross between tolerant and sensitive diploid alfalfa accessions (Sledge et al. 2002). Four restriction fragment length polymorphism (RFLP) markers, in conjunction with callus growth bioassay, were found to be associated with Al tolerance using a single marker analysis. Two of the RFLP markers were confirmed to be present in genotypes of high Al tolerance in a backcross population derived from the same tolerant accession, using both a callus growth bioassay and a soil-based assay. RFLP markers, however, are problematic for high throughput genotyping efforts for effectively introgressing the QTL. Polymerase chain reaction (PCR) based markers linked to Al tolerance QTL that are useful for marker assisted selection (MAS) have not yet been reported, nor have markers flanking the QTL been identified in the diploid M. sativa subsp. coreulea population. In addition, since single marker analysis does not provide precise QTL locations, it is possible that the QTL can be lost through recombination between the marker and the QTL. Thus it is critical to do a whole genome coverage to identify additional Al-tolerance QTLs and to flank the previously identified QTL using simple sequence repeat (SSR) markers to be used for efficient marker-assisted introgression of the QTLs into commercially useful populations.
The ultimate goal underlying QTL mapping is often to identify the specific genes responsible for phenotypic variation. One method of doing this is the placement of candidate genes that are associated with desirable phenotypes from other species on to genetic maps to look for coincidence of map position. Several studies to identify genes associated with Al tolerance have been carried out in wheat, maize, Arabidopsis, sugarcane and other grass species. However, in Medicago, no specific candidate gene(s) conditioning Al tolerance have been reported. Genes involved in Al-tolerance mechanisms from other species may also be involved in tolerance mechanisms in Medicago species. Drummond et al. (2001) used this reasoning and identified candidate genes by prospecting the sugarcane expressed sequence tag (SUCEST) data bank for sugarcane genes with homology to known Al tolerance genes from other plant species and yeast. In this study we used the same approach to search the M. truncatula database to identify genes and DNA sequences with high homology to Al tolerance genes identified in other plant species, to be used as candidate genes for genetic mapping in alfalfa.
The objectives of this study were to (a) develop a genetic linkage map of a diploid alfalfa backcross population segregating for Al tolerance using PCR based SSR markers; (b) identify additional Al-tolerance QTLs and flank the previously identified QTL using SSR markers to be used for MAS in alfalfa breeding; (c) to map candidate genes associated with Al tolerance from other plant species; and (d) to test for co-localization of candidate genes with mapped QTLs.
Materials and methods
Population development and screening
The backcross population (BC1F1) developed by Sledge et al. (2002) was used in this study. It was derived from a cross between diploid alfalfa (M. sativa subsp. coreulea) genotype PI 440501-2 (Al-sensitive) designated as AL1 and PI 464724-25 (Al-tolerant) designated as AL2 from the USDA collections. The sensitive parent was crossed as the recurrent female parent to F1 (designated as AL4) to make the first backcross. The parents and the 130 backcross progenies were screened for Al tolerance by means of a callus growth bioassay by Sledge et al. (2002). Briefly the calli from the leaf tissues of the parents and population were established in modified Blaydes medium with and without Al. A ratio of growth in Al relative to growth without Al was used as an Al tolerance score. The higher values for the ratios indicate increased tolerance to Al toxicity. Analysis of variance (ANOVA) was performed in this study to demonstrate the statistical differences between the genotypes.
Molecular marker analysis
Genomic DNA from the backcross progenies was extracted using a CTAB extraction method (Saghai-Maroof et al. 1984). A polymorphism survey of the parents was carried out using SSR markers from different sources including EST-SSRs from M. truncatula (Eujayl et al. 2004; Julier et al. 2003; Sledge et al. 2005b), alfalfa genomic SSRs (Diwan et al. 2000), and BAC-SSRs, identified in the Mt2Genome using the U.C. Davis Index of MTGS SSRs (http://www.mtgenome.ucdavis.edu).
A two-step approach was devised to select primers from the above sources for the polymorphism survey on the parents. In the first step, bulks of the population were prepared based on the presence or absence of the RFLP marker associated with Al tolerance identified in Sledge et al. (2002). Three bulks were prepared for each of two RFLP markers by pooling equal amounts of DNA from five to eight genotypes. One bulk consisted of individuals homozygous for the positive allele of the RFLP marker, one was heterozygous, and the last was homozygous for the negative or neutral allele. The DNA bulks and the two parental lines were initially screened with a random set of SSR markers. Markers that were polymorphic among the bulks and parents were screened on the backcross population to establish linkage with the RFLP markers previously identified as being associated with Al tolerance. In the second approach, all of the markers that were mapped in the genetic linkage map of tetraploid alfalfa (Sledge et al. 2005b) were screened for polymorphism. A total of 314 EST-SSR primer pairs were chosen for screening the parents for polymorphism. The DNA probes for the four RFLP markers that were associated with increased Al tolerance in Sledge et al. (2002) were obtained from the UGA alfalfa genetic map (Brummer et al. (1991).
Candidate gene primers
The list of candidate genes published by Drummond et al. (2001) for Al tolerance in sugarcane was used as a guide in selecting potential candidate genes. Selected genes included those coding for proteins involved in organic acid and signal transduction pathways, and those involved in alleviating oxidative stress. For these groups of genes, the M. truncatula database was prospected to identify orthologous regions for the chosen candidate genes. The TIGR database was searched using two algorithms from the BLAST (basic local alignment search tool) family of programs, TBLASTN and TBLASTX, for amino acid or nucleotide sequence queries, respectively. Clusters were accepted as related to a particular gene when e-values fell below 10−5. Fifteen genes known to be involved in organic acid pathways, oxidative stress and pathogen defense mechanisms in response to Al stress were selected for candidate gene mapping (Table 1).
Two groups of EST primer pairs were designed from the identified orthologous regions of M. truncatula. Group one consisted of one primer pair for each of two genes, ‘malate dehydrogenase’ (Genbank accession: AF020272) and ‘citrate synthase’ (Table 1). The full length genomic sequence of the M. truncatula ortholog for malate dehydrogenase and partial sequence of M. truncatula ortholog for citrate synthase were obtained from M. truncatula sequence data (http://www.mtgenome.ucdavis.edu/index.html). The primers in this group were designed based on the ‘exon/intron’ junction site information, inferred from the pairwise alignments (http://www.ncbi.nlm.nih.gov/BLAST/) between the alfalfa (M. sativa) ESTs, and their M. truncatula ortholog full length genomic sequences. They were designed using the Primer3 software program (http://www.frodo.wi.mit.edu/cgi-bin/primer3.html) to anneal in exon sequences and amplify across intron regions. This strategy of focusing on non-coding regions could maximize the opportunities for detecting single-nucleotide polymorphism (SNP) variation. In the second group of primers, SSRs within each of the identified ESTs were found using the SSR identification tool (http://www.bioinfo.noble.org/phpssrminer/). Primers were designed using the same software to amplify fragments that contained the SSR region. Primers were designed to have an average length of 20 nucleotides, melting temperatures of 58–64°C, and theoretical PCR amplicons of 150–500 bp. A total of 68 candidate gene primer pairs were developed.
PCR reaction, genotyping and sequencing
The forward primers were synthesized with an additional 18 nucleotides from the M13 universal primer appended to the 5′ end of the forward primer (Schuelke 2000). The M13 universal primer was labeled either with blue (6-FAM), green (VIC), yellow (NED), or red (PET) fluorescent tags. PCR products (3 μl) with different fragment size and different fluorescent labels were pooled and combined with 10 μl deionized formamide and 0.5 μl GeneScan-250LIZ internal size standard and analyzed on an ABI PRISM® 3730 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The reactions were visualized and scored with GeneMapper 3.7 software. The PCR were performed as explained in Sledge et al. (2005b). For sequencing, PCR products were prepared in a reaction volume of 25 μl which contained 50 ng template DNA, 2.5 mM MgCl2, 1 × PCR buffer II (Applied Biosystems, Foster City, CA, USA), 0.15 mM dNTPs, 1.0 pmol of each forward and reverse primer, and 0.5 U AmpliTaq Gold® DNA polymerase (Applied Biosystems, Foster City, CA, USA). Reaction products were sequenced on the ABI PRISM® 3730 Genetic Analyzer.
Detection and evaluation of polymorphism pattern
Primer pairs that amplified polymorphic fragments were visualized and scored using the GeneMapper software. The alleles generated by the candidate gene primers that were monomorphic in size were sequenced to search for SNPs. The primers that produced multiple bands were separated on PAGE gels and each band was reamplified by PCR. All PCR products were gel-purified using QIAquick spin columns (QIAGEN, Valencia, CA, USA) and sequenced from both ends. The resulting genomic sequences were aligned with the corresponding EST sequences via BLAST (http://www.ncbi.nlm.nih.gov/blast/bl2seq/wblast2.cgi) to confirm the amplification of the required region. DNA sequences from the two parental alleles were aligned using the software program DNASTAR to identify SNPs.
Single-nucleotide polymorphisms were converted to cleaved amplified polymorphic sequences (CAPS) by identifying SNPs that showed differential restriction enzyme sites between the two parental lines. In cases in which a suitable restriction enzyme site was not identified, a single nucleotide mismatch was designed adjacent to the polymorphic position, such that a restriction site was created in the PCR product of one parent, but not in the other (dCAPS; see Neff et al. 1998). The PCR product was digested at 37°C for 2–4 h and the cleaved DNA fragments were analyzed on 2.3% SFR agarose gels (Genemate, ISC Bioexpress) stained with ethidium bromide.
Linkage analysis
The BC1F1 population and the parents were genotyped using 177 primers, of which 165 were fluorescently labeled primers and four were RFLP markers. For the seven CAPS and dCAPS primers, the population was genotyped using 2% agarose gel electrophoresis, and was manually scored. JoinMap 3.0 (van Ooijen and Voorips 2001) was used for constructing a linkage map. Linkage groups (LOD = 5.0) were identified on the basis of the assignment of known markers mapped in a previously published tetraploid alfalfa map (Sledge et al. 2005b). The data were analyzed as population type CP and map distances were calculated using the Kosambi function. Since the parents were non-inbred, four single-locus segregation ratios were tested: (1:1 3:1, 1:2:1, and 1:1♀ and 1:1♂) (JoinMap 3.0) using a chi-square goodness of fit. The charts of genetic linkage and the overall chromosomal maps were drawn using the MapChart (Voorrips 2002).
QTL analysis
All the components of the QTL analysis were performed with the MapQTL 5.0 software program (van Oojien 2004). First, the non-parametric Kruskal–Wallis (KW) test was employed to detect association between markers and traits individually. In a second step, interval mapping (IM) analysis was performed to select markers significantly associated with the trait to constitute an initial set of cofactors. A backward elimination procedure was applied to the initial set of cofactors. Only significant markers at P < 0.02 were used as cofactors in the multiple QTL method (MQM) (Jansen and Stam 1994) analysis for QTL detection. A mapping step size of 2 cM was used for both the IM and MQM analyses. Log of odds (LOD) thresholds for genome-wide (P < 0.05) were empirically determined for the trait using the PERMUTATION test of MAPQTL with 5,000 iterations. Based on the permutation tests, a threshold LOD value of 3.1 was used to declare the presence of a QTL. Regions with a LOD score above 2.5 were considered as suggestive of a QTL.
Results
Phenotypic data
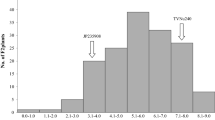
The sensitive parent (AL1) had an Al tolerance score of 0.48 and the tolerant parent (AL2) had a score of 0.72. Among all genotypes of the BC1F1 population, mean Al tolerance score values ranged between 0.32 and 1.15, with an average of 0.69 (Sledge et al. 2002). The histogram exhibited a continuous and normal distribution consistent with polygenic inheritance, characteristic of a quantitative trait (Fig. 1). The ANOVA showed significant variation (P < 0.001) among genotypes for Al tolerance score. High heritability (0.93) for Al tolerance using the callus bioassay was previously reported (Sledge et al. 2002).

Distribution of the Al tolerance score evaluated using a tissue culture method across the backcross population of diploid alfalfa including the parental lines
Marker analysis and polymorphism
From among the 314 EST-SSR selected primers and the four RFLP markers, 162 (51.3%) detected polymorphism between the two parents. In addition, there were 68 candidate gene primers designed from 15 candidate genes selected for this study. In the first group of primers (13 primer pairs) designed from intron sequences, ten primer pairs amplified products that ranged from 140 to 520 bp with GT-AG sequences at the intron junctions. Out of the ten intron-based primers, six of them (60%) were polymorphic. In the second group of primers (55 primer pairs) designed to amplify SSR repeat regions, 32 resulted in PCR amplification with a product size that ranged from 125 to 346 bp. In this group, 12 primer pairs gave a product that was different by sequence and size than what was expected. In the second group of primers 14 out of 32 pairs (44%) were polymorphic. Of the 42 primers that gave a product from groups 1 and 2, nine primer pairs were mapped in the population by virtue of their inherent fragment size or presence/absence criteria, between parental alleles (Table 2). Sequence information obtained from the amplification of the remaining 33 primer products from both groups showed the presence of six SNPs and four indels (Table 2). The primers that showed an insertion/deletion pattern of polymorphism were not genotyped. Five of the SNP alleles were converted to CAPS markers by using the appropriate restriction enzyme and one SNP was converted to a dCAPS primer to obtain polymorphism between the parents. Thus, a total of 15 out of the original 68 candidate gene primer pairs, representing ten different genes, were informative and were used for genotyping (Table 2). In summary, a total of 177 primers including the candidate gene primers were used for genotyping the entire BC1F1 population. There were 15 markers that did not produce distinct segregation patterns in the population, and these were eliminated from the analysis.
Linkage map
Five segregation patterns, 1:1 ♀, 1:1 ♂, 3:1, 1:2:1 and 1:1:1:1 were tested for fit in the backcross population developed from non-inbred parents. A genetic linkage map of the BC1F1 population (Fig. 2) was constructed using 162 segregating loci. A total of 132 loci were placed into ten linkage groups and matched to the eight linkage groups of the previously published EST-SSR tetraploid alfalfa map (Sledge et al. 2005b). Of these 132 loci, 121 loci were placed at LOD ≥ 5.0, and eight were placed at LOD ≥ 3.0. There were three loci that were placed on the map in LG I at a LOD ≥ 2.5 and 30 loci that remained unlinked. The linkage groups LG II and LG V were each represented by two distinct linkage groups. The marker assignments to the eight linkage groups were mostly similar to the tetraploid alfalfa map, except for LG III, VI and VIII. This genetic linkage map spanned a distance of 764 cM covering 84% of the estimated genome length (Chakravarti et al. 1991) with coverage ranging from 25 markers in LG I to 5 markers in LG V (Fig. 2). Coverage of the individual linkage groups was 92% (LG I), 90% (LG III), 86% (LG IIA, IV, VI, VII and VIII) and 66% for LG IIB, LG VA and VB (data not shown). Significantly skewed segregation ratios (P < 0.01) were observed in 43.2% of the loci of which 48% of the distorted markers were due to the presence of an excess of Al-sensitive parent alleles and 52% of the distorted alleles were due to the presence of heterozygotes. The majority of the distorted loci were spread across LG I, LG III and LG VII. There were only three skewed markers in LG IV and LG VIII, and five markers in LG VI. LG II and LG VI each had two non-contiguous distorted markers. Out of the four RFLP markers, UGAc502 mapped to LG I, UGAc471 and UGAc191 to LG II and UGAc141 to LG VIII.

Quantitative trait loci (QTL) and linkage map of the BC1F1 diploid M. sativa subsp. coreulea population. QTL analysis is based on the interval mapping method and MQM method of analysis. The marker name and map distances in centiMorgans are shown on the right and left sides of the linkage group, respectively. The candidate gene markers are in boldface. †Indicates loci mapped at LOD ≤ 3.0 and ≤ 2.5. QTL likelihood plots are shown on the right side of the linkage group. The horizontal dotted line indicates the threshold LOD score = 3.0. Areas along the linkage groups marked in gray are genomic regions encompassed by distorted markers
QTL associations
The non-parametric (Kruskal–Wallis) test suggested the existence of QTLs for Al tolerance in three linkage groups (LG I, LG II and LG III) using P ≤ 0.05 as a threshold criterion for QTL detection (Table 3). Two regions were associated with the Al tolerance phenotype in LG I. One region was spread across several markers in the middle of the linkage group, extending approximately 46 cM. In addition, there were two markers at the proximal region of this linkage group that were significantly associated (P ≤ 0.05) with the trait. Four markers in the proximal region of LG II were significantly associated with Al tolerance (P ≤ 0.05). In the case of linkage group LG III, three markers in the proximal region of the group showed a significant association (P ≤ 0.05). The two markers in linkage group IV were less statistically supported (P ≤ 0.05) for their association with Al tolerance. Four additional markers, one in LG V and three from the unlinked set of markers, also showed a significant association with the trait (P < 0.01) using the Kruskal–Wallis test.
The permutation test to obtain empirical significant threshold levels resulted in a LOD interval of 3.1 or above (genome-wide, P < 0.05) which was used to declare the presence of a segregating QTL in the interval and MQM analyses. The genetic location, corresponding LOD scores, and the variation for each QTL identified using the interval and MQM analysis, is shown in Table 4. Interval mapping identified at least four genomic regions linked to Al tolerance in linkage groups I, II and III (Table 4). LG I had two genomic regions separated by 60 cM. The LOD score for the QTL on LG II (2.86) using interval mapping analysis was slightly below the significance threshold.
The Multiple QTL model included background markers in the QTL analysis as cofactors. The first QTL spanned a region of 50–71 cM in LG I between BE93-2 and UGAc502 and explained 37.9% of the variation using marker BE103 as a co-factor in the MQM model (Table 4; Fig. 2). This major locus on LG I increased the Al tolerance score over the sensitive parent by 17%. For the two linked markers in this region, the increased Al tolerance score was due to the absence of the allele from the sensitive parent (Table 4). A second locus affecting the trait at the proximal region of the linkage group was suggested from the Kruskal–Wallis and interval analysis. However inclusion of the marker in this region as a co-factor in the MQM analysis revealed that this second QTL had a slightly lower LOD score (2.83) than the significance threshold level (Table 4; Fig. 2). The absence of the allele from the sensitive parent for marker MTR58 and the presence of the allele in a heterozygous state for marker 122161_4 contributed to increased Al tolerance (Table 4). The Al tolerance due to this minor QTL was increased by 12%.
In LG II, the LOD score of marker AL99 did not reach the LOD threshold value of 3.1 (genome-wide, P ≤ 0.05), but the inclusion of this marker as a co-factor in the MQM model raised the LOD value to 3.47. The QTL associated with marker AL99 at map position 70 cM explained 16% of the phenotypic variation. The absence of the allele from the sensitive parent at this locus increased the Al tolerance score (Table 4). This QTL in LG II increased the Al tolerance by 14%. There were two potential markers associated with Al tolerance in the proximal region of LG III. Inclusion of marker AW369 alone as a co-factor in the MQM analysis revealed a significant QTL in this region which explained 27% of the phenotypic variation. The QTL region, between BF220 and AW369 at 43–50 cM, explained 26% of the phenotypic variation. The absence of the allele from the sensitive parent for marker BF220 and the presence of allele in the heterozygous state for marker AW369 contributed to higher Al tolerance in the population (Table 4). The effect of this QTL in terms of increase in Al tolerance score was by 9%. Further analysis with marker AA05 in LG V as a cofactor in the MQM analysis did not reveal any more QTLs.
Discussion
Phenotypic data collection
In this study, the Al tolerance score for the population was obtained by the callus growth assay method. The advantage of this approach is that it minimizes environmental variation and G X E interactions, which increases the efficiency of the study. Such in vitro selection must correspond to practical selection procedures, such as the soil based assay or solution culture methods. In a study to compare the cell culture and soil based selection procedures, the cell culture technique, was as efficient as the soil assays in selecting the tolerant germplasms (Dall’Agnol et al. 1996). Sledge et al. (2002) identified an RFLP marker associated with Al tolerance using the tissue culture method and confirmed the presence of the same marker while phenotyping using the soil assay method.
The parents chosen for this study varied greatly for Al tolerance using the tissue culture method, and the population developed from these parents segregated for Al tolerance (Fig. 1). Sledge et al. (2002) reported a high heritability value (93%) for this trait using a tissue culture method, which suggests that the QTLs identified in this study could be efficiently transferred to another population by marker-assisted backcrossing procedures.
Linkage map construction
The linkage map developed in this study showed good agreement with a tetraploid alfalfa genetic map (Sledge et al. 2005b). Despite the overall similarity, there were differences in a few loci in LG III, VI and VIII. There were two distinct groups for linkage groups II and V. Recently we established a genetic linkage map of Medicago truncatula from an F2 population obtained from a cross between Jemalong A17 and PI 566890 (unpublished). In our study the distinct grouping in LG II and V and the marker content in these two groups followed a pattern similar to that observed in the unpublished M. truncatula genetic linkage map. The marker density and genome coverage of this map is high for all groups except LG V. In particular, the 87–92% genome coverage of linkage groups I, IIA and III which carry the QTL, was high and therefore adequate for QTL identification. The polymorphism rate in this study (51%) using the EST-SSRs was comparable to that reported for an alfalfa genetic linkage map (Sledge et al. 2005b), and a M. truncatula genetic linkage map (unpublished). In all three genetic maps, the genetic distance between the flanking markers for a marker linked to the QTL was less than 20 cM. In a simulation study to determine the effect of marker spacing, number of markers and population size, Darvasi et al. (1993), reported that the power for detecting a QTL is virtually the same for a marker spacing of 10 cM as for an infinite number of markers, and was only slightly decreased for marker spacing of 20 cM.
Outcrossing species often exhibit a high level of segregation distortion, as observed in pine (Kubisiak et al. 1995), peach (Dettori et al. 2001), apple (Liebhard et al. 2002), willow (Hanley et al. 2002) and rose (Yan et al. 2005). Diploid alfalfa, an out-crossing species, carries a high proportion of heterozygous loci, and when inbred, shows evidence of inbreeding depression. Previous genetic mapping studies in diploid alfalfa reported distorted segregation (15–63%) due to the excess of heterozygotes that survived in F2 segregating populations (Brummer et al. 1993; Kiss et al. 1993; Kalo et al. 2000) or a backcross population (Echt et al. 1994). Thus, the high level of segregation distortion observed in this study (43%) may be related to the association between heterozygosity and plant vigor in alfalfa. The proximal portion of LG I, LG VII and a major portion of LG III had markers showing significant segregation distortion.
QTL detection and comparison
This study identified at least three putative QTLs in linkage groups I, II and III. The most important QTL with the largest effect (R 2 = 38%) was observed in LG I. In this linkage group, the QTL was centered on marker BE103 and spanned 20 cM. Using interval mapping, multiple markers in this region (BE90, BE103 and BE93-2) were significantly associated with Al tolerance, with LOD scores between 3.12 and 5.47, well above the threshold level of LOD 3.1. Using MQM mapping, however, a single major QTL influencing the trait was revealed in this region (Fig. 2). In the proximal region of this linkage group, a second QTL associated with two markers (MTR58 and 122161_4) was identified using interval mapping analysis. With MQM mapping, however, the QTL exhibited a LOD of 2.83, slightly below the threshold level. It could be that the gap of approximately 30 cM and the segregation distortion in this region may have caused underestimation of the effects and that the mapping of more markers in this region may better resolve this QTL. The presence of multiple loci in the same linkage group does not strictly imply the presence of multiple QTLs; however, the possibility of the presence of multiple QTLs cannot be ruled out in this study. Sledge et al. (2002) had earlier identified the RFLP marker (UGAc502) in a segregating F2 population associated with Al tolerance trait using a single marker analysis which was confirmed in the backcross population. The BC1 population used in this study was derived from the same parents of that used to develop the F2 population. It is noteworthy that the same RFLP marker UGAc502 lies proximal at a distance of 13.4 cM to the most significant marker BE103 in this QTL region and slightly outside the QTL region and significant using single marker analysis. Another marker, BE90, maps at a distance of 6.8 cM proximal to BE103, and is significantly linked to this QTL.
A second significant QTL located on the proximal portion of LG II accounted for 16% of the phenotypic variation. The likelihood of the presence of this QTL (LOD = 3.47) using interval-mapping analysis was slightly below the threshold level. However, the sparse coverage around the QTL region may have underestimated the effects of this QTL. Better marker coverage in this region could help to resolve the QTL and its effect. The RFLP marker, UGAc471, previously reported to be loosely linked with Al tolerance and UGAc191 (Sledge et al. 2002) maps 33 cM away from this QTL and is not linked to Al tolerance trait. The third most significant QTL was identified on linkage group III, between markers BF220 and AW369, and explained 27% of phenotypic variation. There were no prior reports of the existence of an Al tolerance QTL on this linkage group. On linkage group V, one marker was highly significant using the Kruskal–Wallis test, but the interval mapping analysis did not support it. However linkage group V had consisted of two linkage groups, and marker coverage was less than for the other groups.
Efforts to saturate this linkage group using markers from other genetic linkage maps, such as that of tetraploid alfalfa (Sledge et al. 2005b) have thus far been unsuccessful. Linkage group IV had two markers that showed a significant effect using the Kruskal–Wallis test. Marker-trait associations were not detected with other analyses, even though the genome coverage in the linkage group was high (86%), with 14 markers. Thus, the results from the Kruskal–Wallis test could be a false positive in LG IV. A significant association was observed between Al tolerance and three unlinked markers using the Kruskal-Wallis test of significance (Table 3). These, three markers, MTIC 447, AL108 and BE28, map to LG I in the tetraploid alfalfa linkage map (Sledge et al. 2005b). It is possible that these three markers could be linked to LG I if additional markers are added to this group.
Candidate gene mapping
The co-localization of candidate genes with a QTL can support the hypothesis that the gene is responsible for variation in the phenotype. However, this approach is limited by the understanding of the physiology and biochemistry of the trait of interest and by the previous identification of genes potentially involved in trait expression. In our study we used the extensive sequence data available for M. truncatula to choose three groups of candidate genes that have been associated with Al tolerance in other plant species. The three groups included the set of genes that coded for proteins responsible for organic acid synthesis, genes involved in signal transduction, and genes that code for enzymes that alleviate oxidative stress.
Choi et al. (2004) developed intron-targeted primers for EST-based markers by aligning the Medicago truncatula ESTs with the genomic sequences of Arabidopsis orthologs to take advantage of the DNA polymorphisms in introns. We adopted a similar strategy for two M. sativa genes, cytosolic malate dehydrogenase and citrate synthase, using their EST alignments with genomic sequences of M. truncatula. Of the few organic acids produced by plants in response to Al stress, citric acid has the highest binding capacity, followed by malate, oxalate and succinate (Hue et al. 1986). Al tolerant lines of wheat and corn release citric and malic acid in response to Al stress (Ryan et al. 2001; Zhang et al. 2001). Sasaki et al. (2004) cloned the wheat gene Al-activated malate transporter (ALMT1) using the near-isogenic wheat lines that differ in Al tolerance. Kochian et al. (2005) summarized several reports showing high levels of Al-activated release of carboxylates correlated with Al resistance in a large number of plant species. When all the evidence in support of Al-activated root carboxylate release as a major resistance mechanism is examined, a very strong case in support of this concept is seen. For this reason, the closest M. truncatula ortholog was used to infer intron position for malate dehydrogenase and citrate synthase, and thereby aid primer design.
The full-length genomic sequence for the malate dehydrogenase gene and a partial sequence for the citrate synthase gene were available. For primers designed from intron/exon junction site information, it was anticipated that PCR primers designed to anneal in conserved exon regions and to amplify across the more highly diverged intron regions would result in a higher rate of polymorphism. Choi et al. (2004), while analyzing 47 EST markers, found that the intron targeted mapping strategy was an efficient method to map transcribed genes because of a higher polymorphism rate. Wei et al. (2005) reported 63% polymorphism rate using an intron based marker strategy to develop a Rhododendron genetic linkage map. We developed a small set of intron based primers which exhibited a high (60%) rate of polymorphism. Two of the intron based primers designed from the sequences of the M. truncatula ortholog for malate dehydrogenase mapped to LG I. One of them, 122161_4, co-localized with a putative QTL that was identified using the interval mapping analysis, but which fell slightly short of the threshold values when using the MQM analysis (Fig. 2). Although the candidate gene marker mapped to a second locus on LG I, proximal to the major QTL, it is worth examining this region thoroughly in order to confirm that the QTL resides in this region. Similar results were found in fine-scale mapping of a QTL for Al tolerance in A. thaliana in which a gene named AtALMT1 was found located proximal to the major QTL on chromosome 1 (Hoekenga et al. 2006). The physical and genetic location for AtALMT1 while not consistent with the location for the principal Al tolerance locus, indicated that AtALMT1 is an essential factor for Al tolerance in Arabidopsis but does not represent the major Al tolerance QTL.
Another candidate gene, 149493_2, also an intron based marker designed from the sequences of M. truncatula ortholog encoding for malate dehydrogenase, mapped at the distal end of the linkage group and showed a significant association (P ≤ 0.01) with the Kruskal–Wallis analysis (Table 3). However, this marker did not reach significance in the interval mapping analysis. The other candidate genes mapped to other linkage groups (LG IV, V and VI) but were not associated with Al-tolerance in our population. In addition, six of the candidate gene primers remained unlinked and were not associated with Al tolerance in this population. Although the most compelling evidence has focused on a resistance mechanism based on chelation and exclusion of extracellular Al via Al-activated root organic acid release, a number of other potential Al-resistance mechanisms have begun to receive attention. Mechanisms such as Al-chelating ligands and internal detoxification of Al (Ma et al. 1997; Ma and Hiradate 2000), Al-induced alkalinization of rhizosphere pH (Degenhardt et al. 1998), and root exudation of phenolic compounds (Kidd et al. 2001) need to be investigated.
Conclusion
This study revealed three genomic regions in LG I, LG II and LG III in wild diploid alfalfa that increased the Al tolerance score using a tissue culture assay. The putative QTLs identified in this study are flanked by PCR-based SSR markers, which can aid in marker assisted transfer of these QTLs to cultivated alfalfa. These QTLs are being isolated in near isogenic lines by crossing and backcrossing to tetraploid alfalfa, and selecting for the lines carrying the QTL using flanking markers. The co-localization of a candidate gene marker designed from malate dehyrogenase with a putative QTL suggests that this gene may be responsible for part of the variation in Al tolerance observed in this study. The association between candidate genes and Al tolerance in this mapping population are only suggestive, however, and require more rigorous validation in a cultivated alfalfa background to evaluate their utility.
References
Baligar VC, Elgin JH, Foy CD (1989) Variability in alfalfa for growth and mineral uptake and efficiency ratios under aluminum stress. Agron J 81:223–229
Bianchi-Hall CM, Carter TE Jr, Bailey MA, Mian MAR, Rufty TW, Ashley DA, Boerma HR, Arellano C, Hussey RS, Parrot WA (2000) Aluminum tolerance associated with quantitative trait loci derived from soybean PI 416937 in hydroponics. Crop Sci 40:538–545
Bouton JH (1996) Screening the alfalfa core collection for acid soil tolerance. Crop Sci 36:198–200
Brady NC (1974) The nature and property of soils. Macmillian Publishing Co. Inc., New York
Brummer EC, Kochert G, Bouton JH (1991) RFLP variation in diploid and tetraploid alfalfa. Theor Appl Genet 83:89–96
Brummer EC, Bouton JH, Kochert G (1993) Development of an RFLP map in diploid alfalfa. Theor Appl Genet 86:329–332
Chakravarti A, Lasher LK, Reefer JE (1991) A maximum likelihood method for estimating genome length using genetic linkage data. Genetics 128:175–182
Choi HK, Kim DJ, Uhm T, Limpens E, Lim H, Mun JH, Kalo P, Penmetsa RV, Seres A, Kulikova O, Roe BA, Bisseling T, Kiss GB, Cook DR (2004) A sequence-based genetic map of Medicago truncatula and comparison of marker colinearity with M. sativa. Genetics 166:1463–1502
Dall’Agnol M, Bouton JH, Parrott WA (1996) Screening methods to develop alfalfa germplasms tolerant of acid, aluminum toxic soils. Crop Sci 36:64–70
Darvasi A, Weinreb A, Minke V, Weller JI, Soller M (1993) Detecting marker-QTL linkage and estimating QTL gene effect and map location using a saturated genetic map. Genetics 134:943–951
Degenhardt J, Larsen PB, Howell SH, Kochian LV (1998) Aluminum resistance in the Arabidopsis mutant alr-104 is caused by aluminum induced increase in rhizosphere pH. Plant Physiol 117: 19–27
Delhaize E, Craig S, Beaton CD, Bennet RJ, Jagadish VC, Randall PJ (1993) Aluminum tolerance in wheat (Triticum aestivum L.). Uptake and distribution of aluminum in root apices. Plant Physiol 125:2059–2067
Dettori MT, Quarta R, Verde I (2001) A peach linkage map integrating RFLPs, SSRs, RAPDs, and morphological markers. Genome 44:783–790
Diwan N, Bouton J, Kochert G, Cregan P (2000) Mapping of simple sequence repeat (SSR) DNA markers in diploid and tetraploid alfalfa. Theor Appl Genet 101:165–172
Drummond RD, Guimaraes CT, Felix J, Ninamango-Cárdenas FE, Carneiro NP, Paiva E, Menossi M. (2001). Prospecting sugarcane genes involved in aluminum tolerance. Genet Mol Biol 24:221–230
Echt CS, Kidwell KK, Knapp SJ, Osborn TC, McCoy TJ (1994) Linkage mapping in diploid alfalfa (Medicago sativa). Genome 37:61–71
Eujayl I, Sledge MK, Wang L, May G, Chekhovskiy K, Zwonitzer J, Mian R (2004) Medicago truncatula EST-SSRs reveal cross-species genetic markers for Medicago spp. Theor Appl Genet 108:414–422
Foy CD (1988) Plant adaptation to acid, aluminum toxic soils. Comm Soil Sci Plant Anal 19:958–987
Hanley S, Barker JHA, Van Ooijen JW, Aldam C, Harris SL, Ahman I, Larsson S, Karp A (2002) A genetic linkage map of willow (Salix viminalis) based on AFLP and microsatellite markers. Theor Appl Genet 105:1087–1096
Hoekenga OA, Vision TJ, Shaff JE, Monforte AJ, Lee GP, Howell SH, Kochian LV (2003) Identification and characterization of Aluminum tolerance loci in Arabidopsis (Landsberg erecta x Columbia) by quantitative trait locus mapping. A physiologically simple but genetically complex trait. Plant Physiol 132:1–13
Hoekenga OA, Maron LG, Pineros MA, Cancado GMA, Shaff Jon, Kobayashi Y, Ryan PR, Dong B, Delhaize E, Sasaki T, Matsumoto H, Yamamoto Y, Koyama H, Kochian LV (2006) AtALMT1, which encodes a malate transporter is identified as one of several genes critical for aluminum tolerance in Arabidopsis. Proc Natl Acad Sci USA 103:9738–9743
Hue NV, Craddock GR, Adams F (1986) Effect of organic acids on aluminum toxicity in subsoil. Soil Sci Soc Am J 50:28–34
Jansen RC, Stam P (1994) High resolution of quantitative traits into multiple loci via interval mapping. Genetics 136:1447–1455
Julier B, Flajoulot S, Barre P, Cardinet G, Santoni S, Huguet T, Huyghe C (2003) Construction of two genetic linkage maps in cultivated tetraploid alfalfa (Medicago sativa) using microsatellite and AFLP markers. BMC Plant Biol 3:19
Kalo P, Endre L, Zimanyi G, Csanadi G, Kiss GB (2000) Construction of an improved linkage map of diploid alfalfa (Medicago sativa). Theor Appl Genet 100:641–657
Kidd PS, Llugany M, Poschenrieder C, Gunse B, Barcelo J (2001) The role of root exudates in aluminum resistance and silicon-induced amelioration of aluminum toxicity in three varieties of maize (Zea mays L.). J Exp Bot 52:1339–1352
Kiss BG, Csanadi G, Kalman K, Kalo P, Okresz L (1993) Construction of a basic genetic map for alfalfa using RFLP, RAPD, isozyme and morphological markers. Mol Genet 238:129–137
Kobayashi Y, Koyama H (2002) QTL Analysis of Al tolerance in recombinant inbred lines of Arabidopsis thaliana. Plant Cell Physiol 43:1526–1533
Kochian LV, Pineros MA, Hoekenga OA (2005) The physiology, genetics and molecular biology of plant aluminum resistance and toxicity Plant Soil 274:175–195
Kubisiak TL, Nelson CD, Nance WL, Stine M (1995) RAPD linkage mapping in a longleaf pine × slash pine F1 family. Theor Appl Genet 90:1119–1127
Liebhard R, Gianfranceschi L, Koller B, Ryder CD, Tarchini R, Van de Weg E, Gessler C (2002) Development and characterisation of 140 new microsatellites in apple (Malus × domestica Borkh). Mol Breed 10:217–241
Ma JF, Hiradate S (2000) Form of aluminum for uptake and translocation in buckwheat (Fagopyrum esculentum Moench). Planta 211:355–360
Ma JF, Hiradate S, Nomoto K, Iwashita T, Matsumoto H (1997) Internal detoxification mechanism in Hydrangea. Identification of the Al form in leaves. Plant Physiol 113:1033–1039
Ma H-X, Bai GH, LuW-Z (2006) Quantitative trait loci for Aluminum resistance in wheat cultivar Chinese Spring. Plant Soil 283:239–249
Magalhaes JV, Garvin DF, Wang Y, Sorrells ME, Klein PE, Schaffert RE, Li L, Kochian LV (2004) Comparative mapping of a major aluminum tolerance gene in sorghum and other species in the Poaceae. Genetics 167:1905–1914
Minnella E, Sorrells ME (1997) Inheritance and chromosome location of Alp, a gene controlling aluminum tolerance in ‘Dayton’ barley. Plant Breed 116:465–469
Nguyen VT, Burow MD, Nguyen HT, Le BT, Le TD, Paterson AH (2001) Molecular mapping of genes conferring aluminum tolerance in rice (Oryza sativa L.). Theor Appl Genet 102:1002–1010
Neff MM, Neff JD, Chory J, Pepper AE (1998) dCAPS, a simple technique for the genetic analysis of single nucleotide polymorphisms: experimental applications in Arabidopsis thaliana. Plant J 14:387–392
Rechcigl JE, Reneau Jr RB, Zelazny LW (1988) Soil solution Al as a measure of Al toxicity to alfalfa in acid soil. Commun Soil Sci Plant Anal 19:989–1001
Ryan PR, DiTomaso JM, Kochian LV (1993) Aluminium toxicity in roots: an investigation of spatial sensitivity and the role of the root cap. J Exp Bot 44:437–446
Ryan PR, Delhaize E, Jones DL (2001) Function and mechanism of organic anion exudation from plant roots. Annu Rev Plant Physiol Plant Mol Biol 52:527–560
Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW (1984) Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci USA 81:8014–8018
Sasaki T, Yamamoto Y, Ezaki B, Katsuhara M, Ahn SJ, Ryan PR, Delhaize E, Matsumoto H (2004) A wheat gene encoding an aluminum-activated malate transporter. Plant J 37:645–653
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234
Sledge MK, Bouton JH, Dall’Agnoll M, Parrott WA, Kochert G (2002) Identification and confirmation of aluminum tolerance QTL in diploid Medicago sativa subsp. coerulea. Crop Sci 42:1121–1128
Sledge MK, Pechter P, Payton ME (2005a) Aluminum tolerance in Medicago truncatula germplasm. Crop Sci 45:2001–2004
Sledge MK, Ray IM, Jiang G (2005b) An expressed sequence tag SSR map of tetraploid alfalfa ( Medicago sativa L). Theor Appl Genet 111:980–992
Sivaguru M, Horst WJ (1998) The distal part of the transition zone is the most aluminum-sensitive apical root zone of maize. Plant Physiol 116:155–163
Sumner ME, Shahandeh H, Bouton J, Hammel J (1986) Amelioration of an acid soil profile through deep liming and surface. Soil Sci Soc Am J 50:1254–1258
van Ooijen JW (2004) Software for the calculation of QTL positions on genetic maps, MAPQTL 5.0 edn, Wageningen, Netherlands
van Ooijen JW, Voorrips RE (2001) JoinMap® 3.0, Software for the calculation of genetic linkage maps. Plant Research International, Wageningen, The Netherlands
von Uexküll HR, Mutert E (1995) Global extent, development and economic impact of acid soils. Plant Soil 171:1–15
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Wei H, Fu Y, Arora R (2005) Intron flanking EST-PCR markers: from genetic marker development to gene structure analysis in Rhododendron. Theor Appl Genet 111:1347–1356
Yan Z, Denneboom C, Hattendorf A, Dolstra O, Debener T, Stam P, Visser PB (2005) Construction of an integrated map of rose with AFLP, SSR, PK, RGA, RFLP, SCAR and morphological markers Theor Appl Genet 110:766–777
Zhang WH, Ryan PR, Tyerman SD (2001) Malate-permeable channels and cation channels activated by aluminum in the apical cells of wheat roots. Plant Physiol 125: 1459–1472
Acknowledgments
We thank Drs. Mark Sorrells and Malay C. Saha for critical reading of the manuscript. This research was supported by The Samuel Roberts Noble Foundation, Inc.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by H. T. Nguyen.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Narasimhamoorthy, B., Bouton, J.H., Olsen, K.M. et al. Quantitative trait loci and candidate gene mapping of aluminum tolerance in diploid alfalfa. Theor Appl Genet 114, 901–913 (2007). https://doi.org/10.1007/s00122-006-0488-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-006-0488-7