
Abstract
Solitary fibrous tumors (SFTs) are NAB2-STAT6 fusion-associated neoplasms. There are several subtypes of NAB2-STAT6 fusions, but their clinical significances are still unclear. Moreover, the mechanisms of malignant progression are also poorly understood. In this study, using 91 SFT cases, we examined whether fusion variants are associated with clinicopathological parameters and also investigated the molecular mechanism of malignant transformation using whole-exome sequencing. We detected variant 1b (NAB2ex4-STAT6ex2) in 51/91 (56%) cases and variants 2a/2b (NAB2ex6-STAT6ex16/17) in 17/91 (19%) cases. The NAB2-STAT6 fusion variant types were significantly associated with their primary site (P < 0.001). In addition, a TERT promoter mutation was detected in 7/73 (10%) cases, and it showed a significant association with malignant SFTs (P = 0.003). To identify molecular changes during malignant progression, we selected an index patient to obtain parallel tissue samples from the primary and metastatic tumors. In the metastatic tissue, 10 unique molecular alterations, including those in TP53 and APAF1, were detected. In vitro functional experiments showed that APAF1 depletion increased the tumor potency of cells expressing NAB2-STAT6 fusion protein under treatment with staurosporine. We found that TP53 immunopositivity (P = 0.006) and loss of APAF1 immunoreactivity (P < 0.001) were significantly associated with malignant SFTs. Our study suggests that dysfunction of TP53 and APAF1 leads to impaired apoptotic function, and eventually contributes toward malignant SFT transformation.
Key messages
-
We firstly found that the TERT promoter mutation was strongly associated with malignant SFTs (P = 0.003) and the representative 1b (NAB2ex4-STAT6ex2) or 2a (NAB2ex6-STAT6ex16) fusion variants similarly contribute to tumorigenicity.
-
We also found that TP53 immunopositivity (P = 0.006) and loss of APAF1 immunoreactivity (P < 0.001) were significantly associated with malignant SFTs.
-
Our study suggests that dysfunction of TP53 and APAF1 leads to impaired apoptotic function, and eventually contributes toward malignant SFT transformation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Solitary fibrous tumors (SFTs) are uncommon mesenchymal tumors belonging to a group of fibroblastic/myofibroblastic tumors [1, 2]. In the past, they were considered as two distinct entities, namely SFT and hemangiopericytoma (HPC). However, because of the histological similarity and detection of the same NAB2-STAT6 fusions, they are now considered as the same entity as per the current WHO classifications [3,4,5]. According to the current WHO classifications, SFTs are classified into two types, borderline and malignant, except for the meningeal tumors, which are classified as benign, borderline, and malignant [3,4,5]. However, histological parameters for classification, such as mitotic counts, pleomorphism, tumor necrosis, and cellularity, are incomplete for precisely determining the malignant potential [6, 7]. Most SFTs are considered non-malignant neoplasms and are usually treated by surgical resection [8]. A total of 15–20% of SFTs are generally aggressive, showing metastasis or recurrence and are difficult to treat [9, 10]. Chemotherapy plus bevacizumab, pazopanib, sunitinib, figitumumab, or bevacizumab has recently emerged as a promising potential therapeutic strategy for the treatment of advanced SFT [11, 12]. However, the management of SFTs in patients who had developed locally recurrent or metastatic cancer has been challenging.
The NAB2-STAT6 fusion was first identified in two SFT cohorts by Robinson et al. and Chmielecki et al. through whole-exome sequencing (WES) studies [13, 14]. Subsequently, many groups have detected the NAB2-STAT6 fusion in SFT/HPC by WES, transcriptome sequencing, and reverse transcription polymerase chain reaction (RT-PCR). A recent meta-analysis found that more than 40 NAB2-STAT6 fusion variant types were present in up to 83% (452/546) of SFTs/HPCs, with NAB2ex6-STAT6ex16/17/18 and NAB2ex4-STAT6ex2/3 being the most frequent variants [15]. Interestingly, Barthelmess et al. suggested that NAB2ex6-STAT6ex16/17 was significantly associated with malignancy in SFTs/HPCs [16]. However, Tai et al., Chuang et al., and Yuzawa et al. reported that no fusion variants were associated with malignancy [17,18,19]. Therefore, the association between the fusion variants and malignant potentials is still unclear. In addition, there are few studies evaluating whether malignant SFTs have additional alterations as compared to that of non-malignant SFTs. Notably, recent reports have suggested that TERT promoter hot spot mutations are strongly associated with shorter disease-free survival, event-free survival, and high-risk clinicopathological characteristics in SFTs [6, 20, 21].
In this study, we investigated the incidence of NAB2-STAT6 fusion transcript variants and the associations between NAB2-STAT6 fusion variants or TERT promoter mutations and clinicopathological parameters in an SFT cohort. We performed a parallel WES to identify additional molecular changes in an index SFT case, which included a primary buttock sample and matching metastatic brain sample. We also explored whether the additional oncogenic mutations affected the malignancy of SFTs using stable cell lines expressing two representative fusion variants of NAB2-STAT6 by in vitro functional validation.
Methods
Patients
A total of 117 patients who had undergone surgical resection at Samsung Medical Center and diagnosed as SFT or HPC between 1995 and 2014 were included in our SFT cohort. Two pathologists reviewed all retrieved slides and confirmed the diagnosis with the morphology and the results of STAT6 and CD34 immunohistochemical staining. The 26 cases, with no available formalin-fixed paraffin-embedded (FFPE) tissue blocks or no STAT6 immunoreactivity, were excluded (Supplementary Fig. 1A). Then, we selected one case as the index case for WES. As a result, this study included 91 patients (men 53, women 38; median age 56 years, range 29–81 years) and one index case patient. Next, all cases were classified as non-malignant (including benign and borderline SFTs) or malignant SFT according to their mitotic activity. The cases with mitoses > 4/10 high-power fields (HPFs) were classified as malignant SFTs. Although meningeal SFT have different WHO criteria of the mitotic activity (> 5/10 HPFs), none of meningeal cases showed mitotic activity between > 4 and > 5 mitoses per 10 HPFs [5]. We were not able to use a recently proposed risk assessment model because we included meningeal SFTs in this study and this model was only validated in non-meningeal SFTs [6]. A 74-year-old female patient was selected as the index case of our study. This patient was diagnosed as SFT with a focal malignant change on the left buttock in August 2014, and showed a metastatic lesion in the brain in December 2014. The index patient with parallel tissue samples obtained from primary FFPE tissue and matched metastatic frozen tissue. This study was approved by the Institutional Review Board of Samsung Medical Center in Seoul, Korea (IRB file No. 2015–06-114).
Cell lines and reagents
NIH3T3 cells were obtained from the Korean Cell Line Bank. The cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (FBS) (Gibco, Grand Island, NY, USA) and 1% antibiotic–antimycotic (Gibco) at 37 °C and in 5% CO2.
DNA and RNA extraction and cDNA synthesis
Genomic DNA from FFPE tissue was extracted using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Total RNA from the FFPE tissues was isolated using the RNeasy FFPE Kit (Qiagen) according to the manufacturer’s instructions. cDNA from the total RNA was synthesized using the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA, USA).
RT-PCR and PCR
NAB2-STAT6 fusion variant-specific RT-PCR was performed using primer sets obtained from Barthelmess et al. (Supplementary Table 1A) [16]. RT-PCR was conducted using the Maxime RT-PCR premix kit (iNtRON Biotechnology, Seoul, Korea) with a temperature condition consisting of a pre-denaturation step at 95 °C for 5 min, followed by 50 cycles at 95 °C for 30 s, 57 °C for 45 s, and 72 °C for 1 min, and final extension step at 72 °C for 5 min. Amplified PCR products were confirmed by 1.5% agarose gel electrophoresis and purified for Sanger sequencing. To validate the mutations found in the index-case from the WES data, the genomic DNA of primary and metastatic tumors was used for PCR. The primer sets were designed using Primer3 and Primer-Blast (NCBI) (Supplementary Table 1B). PCR was performed at 95 °C for 10 min, followed by 40 cycles at 95 °C for 30 s, 55 °C for 45 s, and 72 °C for 30 s, and final extension step at 72 °C for 5 min. All PCR products were confirmed by Sanger sequencing.
To confirm TERT promoter mutations, PCR was conducted using PCR conditions and a primer set targeting regions surrounding two common TERT promoter hotspot mutations, -124C > T and -146C > T, for genomic DNA of an SFT cohort as previously described (Supplementary Table 1C) [22].
Bisulfite sequencing
Genomic DNA was isolated from FFPE blocks using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). Bisulfite conversion of DNA was performed using EZ DNA Methylation Kit (ZYMO Research, Irvine, CA, USA). The converted DNA samples were subjected to PCR amplification using a primer pair in a Furukawa et al.’ report (Supplementary Table 1D) [23].
RNA-Seq and WES
RNA was isolated from fresh-frozen metastatic brain tissue (malignant tumor tissue) and its CDNA libraries were synthesized using the Nextera XT DNA Sample Prep Kit (Illumina, San Diego, CA, USA) and sequenced on a HiSeq 2500 using the 100-bp paired-end mode of the TruSeq Rapid PE Cluster kit and TruSeq Rapid SBS kit. For chimeric splicing junction analysis, GSNAP was used to perform paired-end-mode mapping of the reads on the pair of gene sequences involved in the fusion, without allowing any mismatch, indel, or splicing. DNA was prepared from a matched primary buttock tumor FFPE sample and fresh-frozen metastatic brain tissue of the index case patient. WES was performed using the TruSeq Exome Enrichment Kit (Illumina) and SureSelect Human All Exon Kit (Agilent Technologies, Santa Clara, CA, USA). Paired-end libraries were sequenced on an Illumina HiSeq 2000. Raw reads in FASTQ format from WES were aligned to the reference genome hg19 using the Burrows-Wheeler Aligner and duplicates were removed with Picard. WES data were analyzed using two INDEL calling algorithms, (1) GATK and, (2) SnpEff, following the guidelines provided in the user manuals. INDELs were called with each algorithm and variants were annotated using the ANNOVAR program.
Immunohistochemistry (IHC)
Four-micrometer-thick sections from FFPE tissue blocks were cut with a microtome and routinely deparaffinized. The sections were incubated with 0.3% hydrogen peroxide. The antigen retrieval procedure was performed in 0.01 M of citrate buffer (pH 6.0) or Tris-EDTA Buffer (10 mM Tris, 1 mM EDTA, 0.03% Tween 20 (pH 9.0)) at 95 °C, and counterstaining was conducted with hematoxylin. The STAT6 antibody (Santa Cruz Biotechnology, Dallas, TX, USA, sc-621, 1:400 dilution) was used for STAT6 immunohistochemical staining. The CD34 antibody (Thermo Fisher Scientific, Inc., MA1-22646, 1:100 dilution) and Ki-67 antibody (Novocastra, Buffalo Grove, IL, USA, NCL-Li-Ki-67-MM1, 1:50 dilution) were used. The immunohistochemistry (IHC) for APAF1 was performed using an anti-APAF1 antibody (Sigma, St. Louis, MO, USA, PRS2015, 1:400 dilution) and IHC for TP53 was performed using the TP53 antibody (Vector Laboratories, Burlingame, CA, USA, VP-P958, 1:50 dilution). Loss of immunohistochemical reactivity for APAF1 was defined as no (0) or weak (1+) staining intensity of the tumor cells. Positive immunohistochemical reactivity for TP53 was defined as near complete absence of the immunoreactivity or positive immunoreactivity in more than 50% of the tumor cells.
Construction of expression plasmids of fusion variants of NAB2–STAT6
For the generation of NAB2ex4-STAT6ex2 and NAB2ex6–STAT6ex16, NAB2 cDNA was amplified using the pCDH1-NAB2 lentiviral vector provided by Monika C. Wolkers. STAT6 cDNA was generated from pCMV-STAT6-IRES-Neo (Addgene, Cambridge, MA, USA, Plasmid #35482). The PCR products were cloned into the N-terminal p3XFLAG-CMV-10 vector (Sigma-Aldrich, E7658). We confirmed the full sequence of wild-type NAB2 and wild-type STAT6 by the Sanger sequencing method. First-strand PCR was performed using the p3XFLAG-CMV vector containing two different wild-type fragments. The NAB2 exon 4 and NAB2 exon 6 fragments were synthesized with the following primer sets: forward, 3xFlag universal-F (ATGGACTACAAAGACCATGA) and reverse, NAB2 e4_bpR (GGACTTGGAGGTTGCCTCTTGTTTCAGCTTCTTCA) and NAB2 e6_bpR (CTATCTGTGGAGAGCCTGCGAGA GGTGGCTTCG). The STAT6 exon 2 and STAT6 exon 16 fragments were amplified with the following primer sets: forward, STAT6 e2-bpF (AAGCTGAAACAAGAGGCAACCTCCAAGTCCCAGAT) and STAT6 e16-bpF (AGCCACCTCTCGCAGGCTCTCCACAGATAGAGAACA), reverse, STAT6-R (TCACCAACTGGGGTTGGC). The second overlapping PCR was conducted using a mixture of two products as a template with the 3xFlag universal-F and STAT6-R primers. Full-length NAB2ex4–STAT6ex2 and NAB2ex6–STAT6 ex16 variants were cloned into a gateway entry vector pCR8/GW/Topo (Invitrogen, K250020) and then subcloned into pLenti6.3/V5-DEST (Invitrogen, V53306). Full-length sequences of NAB2-STAT6 fusion variants were validated by Sanger sequencing.
Transient transfection and generation of stable cell lines expressing NAB2-STAT6 fusion variants
NIH3T3 cells were plated in a 60-mm dish (5 × 105 cells) and then incubated in DMEM growth medium at 37 °C in 5% CO2. After 24 h, NIH3T3 cells were transfected with plasmids (10 μg) encoding the NAB2-STAT6 fusion and pLenti empty vector using Lipofectamine LTX (Invitrogen) according to the manufacturer’s instructions. The pLenti6.3/NAB2-STAT6 expression vector was transfected into 293FT cells using the ViraPower Packaging Mix (Invitrogen, K497500) to produce lentivirus. After 48 h, lentivirus was harvested and transduced into NIH3T3 cells in the presence of 8 μg/mL of polybrene. DMEM complete medium was transferred after 48 h, and medium containing blasticidin (5 μg/mL) was replaced after 24 h. Cells were selected for 2 weeks using selective medium. Stable expression of NAB2-STAT6 was confirmed by qRT-PCR and Western blotting. Total RNA from the cells expressing NAB2-STAT6 was isolated for qRT-PCR. qRT-PCR was conducted with SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA, 4367659) using NAB2-STAT6-specific primer sets. qRT-PCR data was normalized to mouse GAPDH or mouse HPRT as reference genes using the ΔCt method. For Western blotting, protein lysates from cells expressing NAB2-STAT6 were separated by 10% SDS-PAGE and transferred to polyvinylidene fluoride membranes (Bio-Rad, Hercules, CA, USA). Western blotting was performed with the following antibodies: monoclonal anti-FLAG M2 antibody (Sigma-Aldrich, F3165, 1:2000) and GAPDH antibody (Santa Cruz, sc-25778, 1:2000).
Apoptosis assay
Cell apoptosis was determined using the FITC Annexin V Apoptosis Detection Kit I (BD Biosciences, San Jose, CA, USA, 556547) following treatment with 1 μM of staurosporine (STS) (Sigma-Aldrich, S6942). Cell apoptosis was analyzed by an FACS Aria (BD Biosciences).
Cell proliferation and migration assays
The cell proliferation assay was performed with the EZ-CYTOX cell proliferation kit (Daeil Lab Service, Seoul, Korea, EZ-1000) according to the manufacturer instructions. NIH3T3 cells expressing an empty vector or NAB2ex4–STAT6ex2 or NAB2ex6–STAT6ex16 were plated in 96-well plates (5 × 102 cells/well). The 96-well plates were incubated with EZ-CYTOX reagent for 3 h at 37 °C after 1, 2, 3, 4, and 5 days. Absorbance was measured at 450 nm using a spectrophotometer. Twenty-four-well Transwell chambers (Corning Costar, Corning, NY, USA, #3422) with 8-μm polycarbonate membrane filters were used to determine cell migration ability. Next, 5 × 104 cells of NIH3T3-empty and NIH3T3-encoding NAB2-STAT6 fusion variants were seeded into the upper chamber in DMEM without FBS. The lower chamber contained 700 μL of DMEM containing 10% FBS. The Transwell chamber was incubated at 37 °C in 5% CO2. After 24 h of incubation, non-migrating cells on the upper filter surface were removed with a cotton swab and migrated cells were stained with 0.5% crystal violet.
Statistical analysis
Categorical variables were analyzed using the chi-square test or Fisher’s exact test. P values ≤ 0.05 (two-tailed) were used to establish statistical significance. All statistical analyses were conducted using SPSS version 21.0 (IBM SPSS, Armonk, NY, USA).
Results
Incidence of NAB2-STAT6 fusion variants in SFT cohorts
A total of 117 cases were diagnosed as SFTs or HPC, and of them, 91 cases were evaluated (Supplementary Fig. 1A). We detected three variants of the NAB2-STAT6 fusion transcript in 68 cases from the SFT cohort. The prevalence of the variant 1b (NAB2ex4-STAT6ex2) was 56% (n = 51), 2a (NAB2ex6-STAT6ex16) was 13% (n = 12), and 2b (NAB2ex6-STAT6ex17) was 6% (n = 5) (Supplementary Fig. 1A). The sequences in the other 23 cases could not be identified by RT-PCR using the five primer sets and were retained for the analysis of other NAB2-STAT6 fusion variants. The three different fusion variants were reconfirmed in 68 cases (75%) by Sanger sequencing. The corresponding fusion junctions of the NAB2-STAT6 variants are shown in Supplementary Fig. 1B.
Associations of clinicopathological characteristics with NAB2-STAT6 variants and TERT mutation status in SFTs
To investigate the associations between NAB2-STAT6 variants and clinicopathological data, we analyzed the clinicopathological characteristics of 91 histologically confirmed SFT patients displaying different NAB2-STAT6 variants (Table 1 and Supplementary Table 2). Variant 1b was significantly associated with pleural location (P < 0.001). Variants 2a/2b were significantly associated with meningeal location (P < 0.001). We found no significant association between any NAB2-STAT6 variant and any other clinicopathological parameter.
We wanted to evaluate whether SFTs progress to malignant transformation under TERT promoter mutations. The results were not available in 18 cases. In the remaining 73 SFT cases, only the -124C > T TERT promoter mutation was detected in seven cases (10%) and was strongly associated with malignant SFTs (P = 0.003) and the presence of necrosis (P = 0.036) (Supplementary Table 3, and Supplementary Fig. 2). However, the association between TERT promoter mutation and clinically aggressive behavior, which includes recurrence and/or metastasis, was not strong enough to show statistical significance (P = 0.205). All seven instances of TERT promoter mutations were discovered in older patients (≥ 56 years old) (Supplementary Table 3).
Characterization of genomic alterations between non-malignant and malignant SFTs
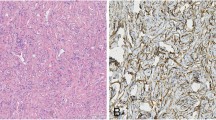
To identify additional oncogenic mutations in the malignant SFT, we selected an index patient to obtain parallel tissue samples from the primary and metastatic tumors. The index patient had a primary SFT in the buttock, and the primary tumor metastasized to the brain after 3 months (Fig. 1a). The primary tumor showed typical non-malignant SFT features in most areas, with a very focal area showing increased cellularity and mitotic activity. We selected a typical non-malignant SFT area from the primary case for molecular evaluation (Fig. 1d). The metastatic tumor presented as a totally histologic malignant SFT (Fig. 1d). We identified the same fusion variant type 2a in both the primary and metastatic tumors by RNA-sequencing, and RT-PCR, and Sanger sequencing (Fig. 1b, c). We evaluated the immunohistochemical expression of CD34 as a mesenchymal cell marker, STAT6 as an SFT diagnostic marker, and Ki-67 as a proliferation marker in the index case (Fig. 1d). The metastatic tumor cells expressed elevated levels of Ki-67, and STAT6, but decreased levels of CD34 relative to those in primary tumor cells (Fig. 1d). The TERT promoter hot spot mutations (-124C > T and -146C > T) were not detected in the index case (data not shown).

Characterization of the molecular and histopathological features of the index SFT case. a Diagram of the primary and metastatic sites of the index case. b A schematic pre-mRNAs of the fusion variant 2a from the RNA-sequencing experiment. Bottom sequences in black are the reads that map onto the chimeric exon-exon splicing junction. c The fusion variant 2a was confirmed by RT-PCR and Sanger sequencing. d Comparison of CD34, Ki-67, and STAT6 immunohistochemical staining and counterpart H&E staining in the primary and metastatic tissues
Next, we performed parallel WES on the primary tissue and matched metastatic tissue from this index case patient. These samples were sequenced to meet a minimum coverage of all targeted regions, with a minimum of > 81.1% exhibiting > 20× coverage. The primary samples contained 21,727 single-nucleotide variations and 14,453 small insertions and deletions (INDELs), while the metastatic samples contained 21,088 single-nucleotide variations and 14,375 INDELs (data not shown). Among all somatic variations, mutations were found only in the primary tumor. The synonymous mutations and unknown mutations with variant allele fractions < 20% were excluded from further analysis. Following this filtering, 12 genes were selected for re-sequencing (Sanger sequencing, primers shown in Supplementary Table 1B) (Fig. 2a–c). The corresponding mutations were confirmed in 10 genes, excluding DNAH8 and SYNJ2 (Supplementary Fig. 3). We then evaluated whether these 10 mutations had previously been reported in The Cancer Genome Atlas (TCGA) database. Mutations in APAF1 (c. 1669C > T), KLHL22 (c.1655G > A), and TP53 (c.313G > T) were found in several cancer subtypes, and the TP53 mutation was predicted to be pathogenic based on its FATHMM algorithm score (0.99) (Fig. 2a). We then focused on APAF1, which had a variant allele fraction (VAF) of 43.82% (expected for a heterozygous gain of a stop codon mutation), and TP53, which had a VAF of 95.45% (expected for a homozygous mutation) (Fig. 2a). The molecular changes and corresponding immunohistochemical expression for APAF1 and TP53 were evaluated using archived formalin-fixed paraffin-embedded (FFPE) samples. We confirmed the presence of the APAF1 (c.1669C > T) and TP53 (c.313G > T) mutations in FFPE samples of metastatic tumors by Sanger sequencing (Fig. 2c, right panel). Interestingly, APAF1 immunoreactivity was detected in the primary tumor but was reduced in the metastatic tumor. Conversely, TP53 protein expression was faintly detected in the sole focal areas of the primary tumors, while the metastatic tumors showed strong and diffuse immunoreactivity of TP53 (Fig. 2c, left panel).

Characterization of genomic alterations between primary and metastatic tissues in the index SFT case. a Genes mutated only in the metastatic tissue and incidences of each mutation from TCGA cancer subtype. b The mutated genes were confirmed by Sanger sequencing in the primary and metastatic tissues of the index case. c Molecular changes and protein expression of APAF1 and TP53 were validated by Sanger sequencing and immunohistochemical assay, respectively. dAPAF1 promoter DNA methylation on the SP1 binding motif was detected only in the metastatic tissues by bisulfite sequencing. e Reduction in APAF1 mRNA was quantified in primary and metastatic tissues of the index case using qRT-PCR (n = 3; bar represents the SE, Student’s t test. f APAF1 and TP53 protein expression were monitored through IHC of tissues obtained from patients with SFTs. Comparisons of TP53 immunopositivity and loss of APAF1 immunoreactivity in non-malignant and malignant solitary fibrous tumors are shown
We further screened for APAF1 (c. 1669C > T in exon 12) and TP53 (c.313G > T in exon 4) mutations in the 19 malignant SFT tissues. In addition, we tested for the previously reported mutations in APAF1, particularly focusing on exons 9–14 corresponding to the hinge region including exon 12, and in TP53, focusing on exons 5–8 corresponding to the reported hot spot areas responsible for the DNA-binding domain. TP53 mutation analysis failed in 3 cases, and revealed TP53 mutation in 6 (6/16, 37.5%) cases (including one c.313G > T, six c.742C > T, one c.818G > A, and one c.832C > T mutation) (Supplementary Tables 4 and 5).
APAF1 is inactivated by DNA methylation in several cancers and leukemia [24,25,26]. The putative binding sites of known transcription factors, TP53, SP1, and E2F, have been identified in the APAF1 promoter region [27, 28]. As APAF1 was altered by a heterozygous mutation resulting in the gain of a stop codon in the index case, we assessed whether these DNA-binding motif sequences were methylated in the remaining allele. DNA methylation at the SP1 binding motif of the APAF1 promoter was detected only in the metastatic tissues using bisulfite sequencing (Fig. 2d and Supplementary Table 1D). APAF1 mRNA expression was also decreased in the metastatic tissues as compared to that in the primary tissue (Fig. 2e).
Next, we evaluated TP53 and APAF1 protein expression status in our cohort (19 malignant SFTs and 23 non-malignant SFTs). TP53 immunopositivity (P = 0.006) and loss of APAF1 immunoreactivity (P < 0.001) were significantly associated with the malignant SFTs (Fig. 2f). Clinically aggressive behavior, which includes known history of recurrence and/or metastasis, showed statistically significant association with TP53 immunopositivity (P = 0.006), but not with the loss of APAF1 immunoreactivity (P = 0.256) (Fig. 2f). However, the cases with loss of APAF1 expression showed a higher rate of recurrence and/or metastasis (27.8%, 5/18) as compared to those with intact APAF1 expression (12.5%, 3/24).
In vitro functional validation of APAF1 as a driver of malignant transformation in SFT
Based on the inactivation of APAF1 in the metastatic tissue of the index SFT case, we used in vitro functional studies to evaluate whether this inactivation acts as an additional oncogenic hit inducing malignant SFT. First, we generated stable NIH-3T3 cell lines expressing representative variant 1b or 2a NAB2-STAT6 fusions and empty control cells (Fig. 3a and b).

Comparison of representative 1b and 2a NAB2-STAT6 fusion variants’ roles on tumor progression. a Diagram of representative 1b or 2a NAB2-STAT6 fusion variants. b The ectopic-expression of representative variant 1b or 2a fusion proteins was validated by immunoblotting. c The cell proliferation was evaluated using WST-1 assay in stable NIH-3T3 cell lines expressing representative variant 1b or 2a forms (gray; empty, orange; long, greenish blue; short) (n = 3; bar represents the SE, Student’s t test was performed, P values are presented in comparison with empty and variant 1b or 2a forms). d Migration assay was performed in the Transwell chamber. The images of crystal violet-stained cells expressing the indicated protein on the membranes are depicted
Next, we compared the tumorigenic functional effects of the 1b or 2a fusion variants. We conducted proliferation and migration assays using these stable NIH-3T3 cells. Expression of either variant 1b or 2a fusion protein increased cell proliferation (P < 0.001) and migration as compared to that in the control cells; however, there was no significant difference in tumorigenesis between cells expressing variant 1b and those expressing 2a (Fig. 3c and d).
APAF1 forms one of the central apoptosomes in the apoptotic regulatory pathway [29]. To investigate the role of APAF1 inactivation in malignant SFT progression, we evaluated whether APAF1 depletion affects the viability, growth, and motility of stable cells expressing the 1b or 2a fusion variants under apoptotic conditions induced by treatment with staurosporine. APAF1 depletion accelerated the inhibition of apoptosis in cells expressing variant 1b or 2a and in empty control cells as compared to that in the non-depleted cells (Fig. 4a). Moreover, as compared to the empty cells, the cells expressing the variant 1b or 2a fusion protein similarly exhibited reduced apoptosis (Fig. 4a). Notably, APAF1 depletion increased the growth and motility of cells expressing variant 1b or 2a fusion protein relative to that in empty cells (Fig. 4b–c). Our findings suggest that APAF1 inactivation accelerates cell survival by inhibiting apoptosis and enhancing cell proliferation and migration. APAF1 inactivation may trigger an increase in SFT malignancy with similar potency in the common NAB2-STAT6 fusion variant subtypes.

Evaluation of the potential role of APAF1 depletion on malignant progression. a Upper panel: Effects of APAF1 knockdown on apoptotic signaling was monitored via Annexin V assay using FACS, after treatment with 1 μM STS for 24 h. The numbers indicate the percentage of apoptotic cells in each quadrant. Lower panel: The values indicate the levels of apoptotic cells from the upper panel. b Increased cell viability of the indicated cells after STS treatment was evaluated by WST-1 assay (n = 3; bar represents the SE; *P < 0.05, **P < 0.05, Student’s t test. P values represent the indicated comparisons). c Migration assay was performed using Transwell chamber
Discussion
The goals of our study were to determine the incidence and clinical significance of NAB2-STAT6 fusion transcript variants and TERT promoter mutations in the SFT cohort and explore the possibility of additional molecular alterations associated with malignant SFTs using an index patient.
We identified three variants of the NAB2-STAT6 fusion transcript in 68 cases among 91 samples using RT-PCR and Sanger sequencing. The common fusion variants were variants 1b (NAB2ex4-STAT6ex2) in 51 (56%) SFTs, 2a (NAB2ex6-STAT6ex16) in 12 (13%) SFTs, and 2b (NAB2ex6-STAT6ex17) in 5 (6%) SFTs. In a review of the previous reports, our incidence rates were similar to those reported by Barthelmess et al. and Akaike et al. [16, 20]. In our cohort, variants 1b and 2a/2b were significantly associated with primary site tumors, which was also consistent with the previous studies (Supplementary Table 6) [13, 17, 20, 30, 31].
Most of the previous studies have reported that NAB2-STAT6 fusion variants were not associated with malignant potentials [13, 17,18,19,20, 30, 32,33,34,35,36,37] (Supplementary Table 7). We also found no significant association between variants 1b or 2a/2b and malignant potentials. By performing in vitro functional validation, we first demonstrated that representative variant 1b or 2a fusion proteins demonstrated similar tumorigenicities. These results indicate that the representative 1b or 2a fusion variants contributed similarly to tumor cell proliferation and migration. Taken together, these results strongly suggest that the type of NAB2-STAT6 fusion variant is not associated with the malignant behavior of SFTs; rather, malignant SFT may have additional oncogenic alterations in comparison with non-malignant SFT.
Recent reports have suggested that TERT promoter mutations are associated with old age, large tumor size, high-risk classifications, and short event-free survival [20, 21]. According to previous reports, the frequencies of TERT promoter mutations in SFT patients were 13% (5/40), 28% (26/94), 13% (4/31), and 20% (2/10) [21, 22, 38, 39]. Although we failed to find a significant association between TERT promoter mutation and recurrence, we found that the TERT promoter mutation (-124C > T) was strongly associated with malignant SFTs (P = 0.003) and the presence of necrosis (P = 0.036). These findings suggest that TERT promoter mutations promote aggressive tumor progression in SFTs. In addition, we found TERT promoter mutations only in elderly patients (≥ 56 years), which was also consistent with the findings of previous studies.
While TERT promoter mutations might correlate with the aggressiveness of SFT, a TERT promoter mutation was only found in a part of the malignant SFTs. To identify unspecified additional molecular changes in the malignant SFTs, we selected an index patient with a typical primary SFT in the buttock and metastatic brain tumor with an apparent malignant histological appearance. Through WES analysis followed by validation using Sanger sequencing, we identified 10 mutations in APAF1, AKR1C2, DAPK3, KLC1, KLHL22, PMPCB, RABEP2, TP53, TUBA3D, and ZNF717 as unique molecular alterations in the metastatic SFT brain tissue. Among these 10 mutations, we especially focused on APAF1 and TP53 mutation.
TP53 mutation has been already suggested to be associated with the malignant transformation of the SFTs in several previous reports [32, 40]. We also found statistically significant association between TP53 immunohistochemical positivity and malignant SFTs (P = 0.006) and clinical history of recurrence and/or metastasis (P = 0.006). In addition, we found TP53 mutations in 41% (7/17) of the malignant SFTs in our cohort by molecular analysis. Interestingly, all found mutations (six c.742C > T, c.818G > A and c.832C > T of TP53) resulted in increased migration and proliferation [41]. Our findings suggest that TP53 mutations are associated with malignant SFTs.
TP53 is one of the most intensively investigated tumor suppressor genes, with hotspot mutations leading to loss of function. However, since the first report that the transduction of mutant TP53 protein into TP53-deficient cells enhanced tumorigenicity, numerous studies have demonstrated that a variety of TP53 mutations can cause a neomorphic gain of function [41, 42]. Oncogenic TP53 mutations are associated with inhibition of apoptosis by upregulating the transcription of various genes such as early growth response 1 (EGR1), a well-known upstream regulator of NAB2, and BCL2-associated athanogene (BAG1), a well-established blocker of apoptosis [43, 44]. It has been well established that wild-type TP53 protein is maintained at very low levels in most of the normal or cancer cells through strict regulation by MDM2, an E3 ubiquitin ligase targeting TP53, which forms a negative feedback loop [45, 46]. However, mutant TP53 proteins frequently accumulate at high levels in the tumor cells, and many reports have suggested that multiple events inhibit the degradation of mutant TP53 proteins during tumorigenesis, although mutant TP53 is mainly regulated by mechanisms consistent with those of the wild-type TP53 protein [47,48,49]. Interestingly, in the index case, the primary non-malignant tissue expressed minimal wild-type TP53 protein, while the metastatic brain tissue showed higher expression of mutant TP53 protein. Taken together, our data suggest that the mutant TP53 protein has an oncogenic function that may contribute to malignant progression.
In the metastatic brain SFT tissue of our index case, we detected APAF1 (c.1669C > T) with a heterozygous mutation resulting in the gain of a stop codon. We found that APAF1 protein was expressed in the primary tumor, but was only minimally expressed in the mutant metastatic tumor. We also discovered that promoter DNA methylation inhibited APAF1 mRNA expression, which might have resulted in the low expression of APAF1 protein observed in the metastatic brain tissue. Interestingly, we confirmed that the low expression of APAF1 protein is not a unique phenomenon in our index case by immunohistochemical staining of malignant SFTs in our cohort (14/21, 66.7%). We also revealed the statistically significant association between the low expression of APAF1 protein and the malignant SFTs (P = 0.002). The cases with low APAF1 expression also showed a higher rate of recurrence and/or metastasis (27.8%, 5/18) as compared to those with intact APAF1 expression (12.5%, 3/24). Unfortunately, the association was not strong enough to show statistical significance (P = 0.256) in this study. In addition, through functional validation using stable cells expressing variant 1b or 2a fusion forms, we demonstrated that APAF1 inactivation promotes cell survival through inhibition of apoptosis signaling and enhances tumorigenesis, which may trigger an increase in the tumor malignancy of SFT.
Our study has several limitations in finding a general mechanism of how SFTs turn malignant. Only one index case was included to identify additional molecular alterations in malignant SFTs and epigenetic changes in the whole genome were not determined. Each case may be unique in the process of evolving into malignant tumors. Our study confirmed events in one case, and although these results were only immunohistochemically elucidated in other cases, we think these findings will contribute to understanding the tumor evolution and malignant transformation of SFTs.
Notwithstanding these limitations, based on our findings from the index case and those of Robinson et al., we suggest a model in which NAB2-STAT6 constitutively activates tumor proliferation and migration via EGR1 pathway under control of intact apoptosis signaling in non-malignant SFT. In contrast, the stabilized mutant TP53 and inactivated APAF1 with impaired apoptotic function trigger additional malignant features in malignant SFTs (Fig. 5) [13].

Model of TP53 and APAF1 roles in SFT with the NAB2-STAT6 fusion variant onto malignant transformation
Abbreviations
- SFT :
-
Solitary fibrous tumors
- HPC :
-
Hemangiopericytoma
- WES :
-
Whole-exome sequencing
- FFPE :
-
Formalin-fixed paraffin-embedded
- HPF :
-
High-power field
- IHC :
-
Immunohistochemistry
- INDEL :
-
Insertions and deletions
References
Koelsche C, Schweizer L, Renner M, Warth A, Jones DT, Sahm F, Reuss DE, Capper D, Knosel T, Schulz B et al (2014) Nuclear relocation of STAT6 reliably predicts NAB2-STAT6 fusion for the diagnosis of solitary fibrous tumour. Histopathology 65:613–622
Kang S, Kim HS, Choi ES, Han I (2015) Incidence and treatment pattern of extremity soft tissue sarcoma in Korea, 2009-2011: a nationwide study based on the Health Insurance Review and Assessment Service database. Cancer Res Treat 47:575–582
Fletcher CDM, Bridge JA, Lee J-C (2013) Extrapleural solitary fibrous tumour. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F (eds) . World Health Organization classification of tumours IARC Press, Lyon, pp 80–84
Fletcher CDM, Gibbs A (2015) Solitary fibrous tumour. In: Travis WD, Brambilla E, BA P, Marx A, Nicholson AG (eds) WHO classification of tumours of lung, pleura, thymus and heart. IARC Press, Lyon, pp 178–179
Giannini C, Rushing EJ, Hainfellner JA, Bouvier C, Figarella-Branger D, von Deimling A, Wesseling P, Antonescu CR (2016) Solitary fibrous tumour / Haemangiopericytoma. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Ellison DW, Figarella-Branger D, Perry A, Reifenberger G, von Deimling A (eds) WHO classification of tumours of the central nervous system. IARC press, Lyon, pp 249–254
Demicco EG, Park MS, Araujo DM, Fox PS, Bassett RL, Pollock RE, Lazar AJ, Wang WL (2012) Solitary fibrous tumor: a clinicopathological study of 110 cases and proposed risk assessment model. Mod Pathol 25:1298–1306
Yang JW, Song DH, Jang IS, Ko GH (2014) Dedifferentiated solitary fibrous tumor of thoracic cavity. Korean J Pathol 48:250–253
Thway K, Ng W, Noujaim J, Jones RL, Fisher C (2016) The current status of solitary fibrous tumor: diagnostic features, variants, and genetics. Int J Surg Pathol 24:281–292
Lorigan JG, David CL, Evans HL, Wallace S (1989) The clinical and radiologic manifestations of hemangiopericytoma. AJR Am J Roentgenol 153:345–349
Hajdu M, Singer S, Maki RG, Schwartz GK, Keohan ML, Antonescu CR (2010) IGF2 over-expression in solitary fibrous tumours is independent of anatomical location and is related to loss of imprinting. J Pathol 221:300–307
Stacchiotti S, Tortoreto M, Baldi GG, Grignani G, Toss A, Badalamenti G, Cominetti D, Morosi C, Dei Tos AP, Festinese F, Fumagalli E, Provenzano S, Gronchi A, Pennacchioli E, Negri T, Dagrada GP, Spagnuolo RD, Pilotti S, Casali PG, Zaffaroni N (2014) Preclinical and clinical evidence of activity of pazopanib in solitary fibrous tumour. Eur J Cancer 50:3021–3028
Park MS, Patel SR, Ludwig JA, Trent JC, Conrad CA, Lazar AJ, Wang WL, Boonsirikamchai P, Choi H, Wang X, Benjamin RS, Araujo DM (2011) Activity of temozolomide and bevacizumab in the treatment of locally advanced, recurrent, and metastatic hemangiopericytoma and malignant solitary fibrous tumor. Cancer 117:4939–4947
Robinson DR, Wu YM, Kalyana-Sundaram S, Cao X, Lonigro RJ, Sung YS, Chen CL, Zhang L, Wang R, Su F, Iyer MK, Roychowdhury S, Siddiqui J, Pienta KJ, Kunju LP, Talpaz M, Mosquera JM, Singer S, Schuetze SM, Antonescu CR, Chinnaiyan AM (2013) Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat Genet 45:180–185
Chmielecki J, Crago AM, Rosenberg M, O'Connor R, Walker SR, Ambrogio L, Auclair D, McKenna A, Heinrich MC, Frank DA, Meyerson M (2013) Whole-exome sequencing identifies a recurrent NAB2-STAT6 fusion in solitary fibrous tumors. Nat Genet 45:131–132
Nakada S, Minato H, Nojima T (2016) Clinicopathological differences between variants of the NAB2-STAT6 fusion gene in solitary fibrous tumors of the meninges and extra-central nervous system. Brain Tumor Pathol 33:169–174
Barthelmess S, Geddert H, Boltze C, Moskalev EA, Bieg M, Sirbu H, Brors B, Wiemann S, Hartmann A, Agaimy A et al (2014) Solitary fibrous tumors/hemangiopericytomas with different variants of the NAB2-STAT6 gene fusion are characterized by specific histomorphology and distinct clinicopathological features. Am J Pathol 184:1209–1218
Tai HC, Chuang IC, Chen TC, Li CF, Huang SC, Kao YC, Lin PC, Tsai JW, Lan J, Yu SC, Yen SL, Jung SM, Liao KC, Fang FM, Huang HY (2015) NAB2-STAT6 fusion types account for clinicopathological variations in solitary fibrous tumors. Mod Pathol 28:1324–1335
Chuang IC, Liao KC, Huang HY, Kao YC, Li CF, Huang SC, Tsai JW, Chen KC, Lan J, Lin PC (2016) NAB2-STAT6 gene fusion and STAT6 immunoexpression in extrathoracic solitary fibrous tumors: the association between fusion variants and locations. Pathol Int 66:288–296
Yuzawa S, Nishihara H, Wang L, Tsuda M, Kimura T, Tanino M, Tanaka S (2016) Analysis of NAB2-STAT6 gene fusion in 17 cases of meningeal solitary fibrous tumor/hemangiopericytoma: review of the literature. Am J Surg Pathol 40:1031–1040
Akaike K, Kurisaki-Arakawa A, Hara K, Suehara Y, Takagi T, Mitani K, Kaneko K, Yao T, Saito T (2015) Distinct clinicopathological features of NAB2-STAT6 fusion gene variants in solitary fibrous tumor with emphasis on the acquisition of highly malignant potential. Hum Pathol 46:347–356
Bahrami A, Lee S, Schaefer IM, Boland JM, Patton KT, Pounds S, Fletcher CD (2016) TERT promoter mutations and prognosis in solitary fibrous tumor. Mod Pathol 29:1511–1522
Liu X, Bishop J, Shan Y, Pai S, Liu D, Murugan AK, Sun H, El-Naggar AK, Xing M (2013) Highly prevalent TERT promoter mutations in aggressive thyroid cancers. Endocr Relat Cancer 20:603–610
Furukawa Y, Sutheesophon K, Wada T, Nishimura M, Saito Y, Ishii H, Furukawa Y (2005) Methylation silencing of the Apaf-1 gene in acute leukemia. Mol Cancer Res 3:325–334
Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M, Opitz-Araya X, McCombie R, Herman JG, Gerald WL, Lazebnik YA, Cordón-Cardó C, Lowe SW (2001) Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature 409:207–211
Fu WN, Bertoni F, Kelsey SM, McElwaine SM, Cotter FE, Newland AC, Jia L (2003) Role of DNA methylation in the suppression of Apaf-1 protein in human leukaemia. Oncogene 22:451–455
Loginov VI, Pronina IV, Burdennyi AM, Pereyaslova EA, Braga EA, Kazubskaya TP, Kushlinskii NE (2017) Role of methylation in the regulation of apoptosis genes APAF1, DAPK1, and BCL2 in breast cancer. Bull Exp Biol Med 162:797–800
Furukawa Y, Nishimura N, Furukawa Y, Satoh M, Endo H, Iwase S, Yamada H, Matsuda M, Kano Y, Nakamura M (2002) Apaf-1 is a mediator of E2F-1-induced apoptosis. J Biol Chem 277:39760–39768
Moroni MC, Hickman ES, Lazzerini Denchi E, Caprara G, Colli E, Cecconi F, Muller H, Helin K (2001) Apaf-1 is a transcriptional target for E2F and p53. Nat Cell Biol 3:552–558
Riedl SJ, Li W, Chao Y, Schwarzenbacher R, Shi Y (2005) Structure of the apoptotic protease-activating factor 1 bound to ADP. Nature 434:926–933
Vogels RJ, Vlenterie M, Versleijen-Jonkers YM, Ruijter E, Bekers EM, Verdijk MA, Link MM, Bonenkamp JJ, van der Graaf WT, Slootweg PJ et al (2014) Solitary fibrous tumor—clinicopathologic, immunohistochemical and molecular analysis of 28 cases. Diagn Pathol 9:224
Ronchi A, Cozzolino I, Zito Marino F, Accardo M, Montella M, Panarese I, Roccuzzo G, Toni G, Franco R, De Chiara A (2018) Extrapleural solitary fibrous tumor: a distinct entity from pleural solitary fibrous tumor. An update on clinical, molecular and diagnostic features. Ann Diagn Pathol 34:142–150
Dagrada GP, Spagnuolo RD, Mauro V, Tamborini E, Cesana L, Gronchi A, Stacchiotti S, Pierotti MA, Negri T, Pilotti S (2015) Solitary fibrous tumors: loss of chimeric protein expression and genomic instability mark dedifferentiation. Mod Pathol 28:1074–1083
Fritchie KJ, Jin L, Rubin BP, Burger PC, Jenkins SM, Barthelmess S, Moskalev EA, Haller F, Oliveira AM, Giannini C (2016) NAB2-STAT6 gene fusion in meningeal hemangiopericytoma and solitary fibrous tumor. J Neuropathol Exp Neurol 75:263–271
Mohajeri A, Tayebwa J, Collin A, Nilsson J, Magnusson L, von Steyern FV, Brosjo O, Domanski HA, Larsson O, Sciot R et al (2013) Comprehensive genetic analysis identifies a pathognomonic NAB2/STAT6 fusion gene, nonrandom secondary genomic imbalances, and a characteristic gene expression profile in solitary fibrous tumor. Genes Chromosom Cancer 52:873–886
Huang SC, Li CF, Kao YC, Chuang IC, Tai HC, Tsai JW, Yu SC, Huang HY, Lan J, Yen SL, Lin PC, Chen TC (2016) The clinicopathological significance of NAB2-STAT6 gene fusions in 52 cases of intrathoracic solitary fibrous tumors. Cancer Med 5:159–168
Kao YC, Lin PC, Yen SL, Huang SC, Tsai JW, Li CF, Tai HC, Lan J, Chuang IC, Yu SC, Huang HY (2016) Clinicopathological and genetic heterogeneity of the head and neck solitary fibrous tumours: a comparative histological, immunohistochemical and molecular study of 36 cases. Histopathology 68:492–501
Guseva NV, Tanas MR, Stence AA, Sompallae R, Schade JC, Bossler AD, Bellizzi AM, Ma D (2016) The NAB2-STAT6 gene fusion in solitary fibrous tumor can be reliably detected by anchored multiplexed PCR for targeted next-generation sequencing. Cancer Genet 209:303–312
Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr, Friedman AH, Friedman H, Gallia GL, Giovanella BC et al (2013) TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A 110:6021–6026
Koelsche C, Renner M, Hartmann W, Brandt R, Lehner B, Waldburger N, Alldinger I, Schmitt T, Egerer G, Penzel R, Wardelmann E, Schirmacher P, von Deimling A, Mechtersheimer G (2014) TERT promoter hotspot mutations are recurrent in myxoid liposarcomas but rare in other soft tissue sarcoma entities. J Exp Clin Cancer Res 33:33
Yokoi T, Tsuzuki T, Yatabe Y, Suzuki M, Kurumaya H, Koshikawa T, Kuhara H, Kuroda M, Nakamura N, Nakatani Y et al (1998) Solitary fibrous tumour: significance of p53 and CD34 immunoreactivity in its malignant transformation. Histopathology 32:423–432
Muller PA, Vousden KH (2014) Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 25:304–317
Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, Finlay C, Levine AJ (1993) Gain of function mutations in p53. Nat Genet 4:42–46
Weisz L, Zalcenstein A, Stambolsky P, Cohen Y, Goldfinger N, Oren M, Rotter V (2004) Transactivation of the EGR1 gene contributes to mutant p53 gain of function. Cancer Res 64:8318–8327
Yang X, Pater A, Tang SC (1999) Cloning and characterization of the human BAG-1 gene promoter: upregulation by tumor-derived p53 mutants. Oncogene 18:4546–4553
Poyurovsky MV, Prives C (2006) Unleashing the power of p53: lessons from mice and men. Genes Dev 20:125–131
Oren M, Rotter V (2010) Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol 2:a001107
Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, Van Pelt CS, Lozano G (2008) The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev 22:1337–1344
Bartek J, Bartkova J, Vojtesek B, Staskova Z, Lukas J, Rejthar A, Kovarik J, Midgley CA, Gannon JV, Lane DP (1991) Aberrant expression of the p53 oncoprotein is a common feature of a wide spectrum of human malignancies. Oncogene 6:1699–1703
Suh YA, Post SM, Elizondo-Fraire AC, Maccio DR, Jackson JG, El-Naggar AK, Van Pelt C, Terzian T, Lozano G (2011) Multiple stress signals activate mutant p53 in vivo. Cancer Res 71:7168–7175
Acknowledgements
We would like to thank Tae Won Yun of LOGONE Bio Convergence Research Foundation.
Funding
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean Government (grant number 2013R1A1A2011536, 2016R1A2B2012975, and 2016R1A5A2945889).
Author information
Authors and Affiliations
Contributions
Conception and design: YJK and YLC.
Analysis and interpretation: HKP, DBY, ESO, YJK, and YLC.
Data collection: HKP, DBY, MJS, MGK, JYS, MSL, KS, KSJ, KWN, SBA, DHN, and YJK.
Writing the article: HKP, DBY, YJK and YLC.
Final approval of the article: HKP, YJK and YLC.
Corresponding authors
Ethics declarations
This study was approved by the Institutional Review Board of Samsung Medical Center in Seoul, Korea (IRB file No. 2015–06-114).
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Figure 1
(A) Flow chart of the incidences of NAB2-STAT6 fusion variants in our cohort. The variant types were confirmed by RT-PCR and Sanger sequencing. (B) The three representative NAB2-STAT6 fusion variants were confirmed by RT-PCR and Sanger sequencing. (PPTX 71 kb)
Supplementary Figure 2
The TERT promoter mutation (-124C>T) was validated by Sanger sequencing in the malignant tissues. (PPTX 44 kb)
Supplementary Figure 3
The mutations of DNAH8 and SYNJ2 were not confirmed in the metastatic tissues. (PPTX 58 kb)
ESM 1
(XLSX 21 kb)
ESM 2
(DOCX 62 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Park, H.K., Yu, D.B., Sung, M. et al. Molecular changes in solitary fibrous tumor progression. J Mol Med 97, 1413–1425 (2019). https://doi.org/10.1007/s00109-019-01815-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-019-01815-8