
Abstract
Only approximately 10 % of genetically unselected patients with chemorefractory metastatic colorectal cancer experience tumor regression when treated with the anti-epidermal growth factor receptor (EGFR) antibodies cetuximab or panitumumab (“primary” or “de novo” resistance). Moreover, nearly all patients whose tumors initially respond inevitably become refractory (“secondary” or “acquired” resistance). An ever-increasing number of predictors of both primary and acquired resistance to anti-EGFR antibodies have been described, and it is now evident that most of the underlying mechanisms significantly overlap. By trying to extrapolate a unifying perspective out of many idiosyncratic details, here, we discuss the molecular underpinnings of therapeutic resistance, summarize research efforts aimed to improve patient selection, and present alternative therapeutic strategies that are now under development to increase response and combat relapse.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer is the second commonest cancer worldwide, and the metastatic disease accounts for up to 20 % of newly diagnosed patients or further develops in 50 % of cases, with a median overall survival (OS) of approximately 20 months [1-5].
The clinical outcome of patients with metastatic colorectal cancer (mCRC) has been improved by the introduction of cetuximab and panitumumab, two monoclonal antibodies (moAbs) targeting the epidermal growth factor receptor (EGFR/ErbB1/HER1), given in combination with chemotherapy or, when other options are exhausted, as monotherapy [6-8].
EGFR is a member of the ErbB family of receptor tyrosine kinases (RTKs), which also includes HER2/neu (ERBB2), HER3 (ErbB3), and HER4 (ErbB4) [9]. EGF or other EGF-like ligands trigger homo- and hetero-dimerization of EGFR with other ErbB members, which activates a mitogenic and antiapoptotic signaling cascade via several pathways, including not only the RAS-RAF-MEK-ERK and the PI3K-AKT-mTOR axes but also SRC family kinases, PLCγ-PKC, and STATs [9, 10]. Such activation stimulates key processes involved in tumor growth and progression, including proliferation, angiogenesis, invasion, and metastasis [11] (Fig. 1).

EGFR signaling pathways. a Upon ligand binding and consequent homo- and hetero-dimerization, ErbB family members can activate a number of pathways, including the RAS-RAF-MEK-ERK and the PI3K-AKT-mTOR axes, the SRC family kinases (SFKs), PLCγ-PKC, and STATs, driving cell proliferation and/or influencing apoptosis. b By binding the extracellular domain of EGFR, both cetuximab and panitumumab prevent ligand-induced activation of downstream signaling
When used as monotherapy in genetically unselected patients with chemotherapy-refractory mCRC, cetuximab and panitumumab achieve clinically meaningful response rates (RRs) of approximately 10 % [7, 8, 12]. Unlike other tumor types such as non-small cell lung cancers (NSCLCs) or melanomas, in which target mutations are associated with massive regressions following treatment with specific inhibitors [13, 14], genetic alterations of EGFR are extremely infrequent in colorectal tumors.
The complex and thin boundary between primary and acquired resistance is determined by the evidence of an initial response to treatment. If refractoriness to therapy is present at baseline, this is defined as primary (also known as de novo) resistance and can be explained by resistance-conferring factors preexisting in the bulk of tumor cells. Acquired (or secondary) resistance refers to disease progression in the face of ongoing treatment that was initially effective and can be caused by mutations arising during treatment as well as through other various adaptive nongenetic responses [15, 16]. In the case of colorectal cancer, acquired resistance typically occurs within 3–18 months after treatment initiation [7, 8].
Starting with seminal observations in 2006–2007 [17, 18], a large body of evidence has described different biomarkers of primary resistance to anti-EGFR moAbs in mCRC patients, leading to exclusion from treatment of a number of molecularly defined nonresponders [19, 20]. The field of acquired resistance has received preclinical and clinical attention much more recently, with the emergence of new insights only in the last 2 years.
In this review, we will appraise the current knowledge on primary and acquired resistance to anti-EGFR moAbs in mCRC, from initial mechanistic exploration to clinical applications, and will highlight emerging lines of investigation aimed at improving response and delay relapse in this tumor setting.
Molecular mechanisms of resistance to anti-EGFR antibodies in patients with metastatic colorectal cancer
In general terms, the commonest mechanisms of resistance to inhibition of receptor tyrosine kinases (RTKs) involve genomic alterations affecting downstream effectors, such as KRAS and PIK3CA mutations, with consequent constitutive pathway hyperactivation. Notably, the KRAS and PI3K signaling cascades can also be activated by upstream RTKs other than EGFR [21], leading to an oncogenic shift [22]. In both cases, the primary drug target remains unaltered and continues to be inhibited while an alternative signal transducer becomes activated, bypassing the consequences of EGFR inhibition [16, 23] (Fig. 2a, b).

Mechanisms of resistance to anti-EGFR moAbs in mCRC. a Activating mutations of EGFR effectors, such as KRAS (by either point mutations or gene amplification), BRAF and PI3KCA, or PTEN loss of function, cause persistent activation of downstream signaling despite EGFR inhibition. b Aberrant activation (by either receptor gene amplification or high ligand levels) of alternative receptors, such as HER2 or MET (not shown), can bypass EGFR inhibition and mediate downstream pathway activation. c Additional genetic alterations within the target oncogene may abrogate drug binding. The EGFR S492R mutation inhibits cetuximab but not panitumumab binding, mediating acquired resistance to the former but not the latter in mCRC patients. d Other mechanisms of resistance may be “pathway independent,” such as altered angiogenesis (through increased secretion of VEGF or activation of VEGFR-1/2), dysregulation of EGFR recycling (with consequent increase of EGFR degradation), or tumor-stroma interactions (i.e., through increased release of antiapoptotic growth factors and cytokines, such as HGF)
Importantly, it is increasingly recognized that tumors can contain a high degree of genetic and molecular heterogeneity within the same lesion [24]. Thus, secondary resistance can arise not only through acquisition of de novo genetic lesions over the course of therapy but also through treatment-induced selection of resistant minor subpopulations of cells that are intrinsically insensitive and already present in the original tumor [25]. If secondary resistance may be nothing but the emergence, under drug pressure, of rare tumor subsets featuring primary resistance, then most of the molecular mechanisms of primary and acquired resistance should overlap. Accordingly, hereinafter, we provide a description of resistance predictors as a whole, specifying for each biomarker when it has been reported in both cases. We will also focus on current research efforts aimed at developing alternative strategies to circumvent such resistances in patients with no other therapeutic options. Table 1 summarizes the main biomarkers of primary and acquired resistance observed in mCRC patients and describes potential alternative strategies proposed by different approaches.
RAS
The RAS family includes three small GTPases (KRAS, NRAS, and HRAS) responsible for coupling EGFR to the RAF/MEK/ERK pathway [22]. Several retrospective analyses have described KRAS mutations in exon 2 (codons 12 and 13), which are found in approximately 40–45 % of CRCs [20, 26], as major determinants of primary resistance to cetuximab or panitumumab [17, 27-29]. The robust predictive power of such correlations, despite being obtained in retrospective studies, was sufficient to convince both the US Food and Drug Administration and the European Medicines Agency to approve the use of anti-EGFR moAbs only in the subset of KRAS wild-type colorectal cancers [26, 30-34].
Although exclusion of patients with KRAS (exon 2)-mutant tumors has arithmetically increased the percentage of responders up to 13–17 %, most KRAS wild-type tumors still do not respond to anti-EGFR moAbs [26, 32]. Additional rare mutations of KRAS, as well as mutations of NRAS, have been associated with primary resistance to treatment. The relatively high cumulative frequency of rare KRAS mutations and NRAS mutations, coupled with initial successful validation in prospective trials, strongly advocates prompt incorporation of such biomarkers into clinical practice as negative predictors [35]. A very low frequency of KRAS amplification (0.7 %) has also been reported and found to correlate with primary resistance [36].
KRAS point mutations and gene copy number gains are responsible not only for primary but also for acquired resistance in 38–60 % of patients who relapse on cetuximab or panitumumab [37-39]. Intriguingly, such mutations presumably are either present in a clonal subpopulation within the tumor before treatment initiation [37, 38] or raise as a consequence of continued mutagenesis over the course of therapy [38, 39]. KRAS alterations could be identified noninvasively 5–10 months before radiographic disease progression by analyzing cell-free circulating tumor DNA (ctDNA) [37, 38]. Using this approach, two recent studies have highlighted the emergence of several independent clones carrying heterogeneous patterns of KRAS and NRAS mutations concomitantly associated with acquired resistance to EGFR blockade [40, 41].
Currently, KRAS-mutant patients are treated with chemotherapy (with or without antiangiogenic therapy using the anti-VEGF moAb bevacizumab), but if intensive regimens are not tolerated or relapse occurs, the remaining treatment option is best supportive care [42]. To date, direct inhibitors of mutant KRAS protein are not yet available; therefore, multiple efforts have been made at the preclinical level by approaches as different as targeting downstream effectors such as MEK and PI3K [43], exploiting synthetic lethal interactions [44-49] or using high-throughput drug screens [50]. Of note, most of these studies showed that simultaneous targeting of two different pathways induced some responses in KRAS mutant CRC mouse models, albeit rarely with overt tumor regressions [51] (see Table 1); most of these approaches are currently under evaluation in phase I/II clinical trials (NCT01085331, http://clinicaltrials.gov/ct2/show/NCT01085331?term=NCT01085331&rank=1; NCT01390818, http://clinicaltrials.gov/ct2/results?term=NCT01390818&Search=Search; NCT02039336, http://clinicaltrials.gov/ct2/show/NCT02039336?term=NCT02039336&rank=1). In the case of secondary resistance due to RAS mutations, preclinical evidence suggests that early initiation of a combinational targeting of EGFR and MEK could delay or reverse the emergence of resistance [40].
BRAF
Mutations of BRAF, which encodes the cytoplasmic serine/threonine kinase immediately downstream of RAS, are found in 4–13 % of advanced CRCs and are usually mutually exclusive with KRAS mutations [20, 52].
The BRAF V600E mutation has been described as a predictor of tumor aggressiveness in metastatic disease [33, 52, 53] and also of low RRs to cetuximab and panitumumab [18, 20, 52, 53]. However, the predictive impact of BRAF mutations is tempered by their low prevalence and is further biased by the prominent role of mutant BRAF as a negative prognostic biomarker [54]. Overall, the predictive power of this alteration remains immature and requires further prospective endorsement before clinical applicability [20, 33, 52, 55].
Although, unlike RAS, BRAF can be efficiently blocked by clinically approved small-molecule inhibitors, no targeted therapeutic options are currently available for BRAF mutant CRC. In contrast to dramatic responses obtained in BRAF V600E-mutant melanomas (RR of 48 to 67 %) [13, 56], selective BRAF inhibitors such as vemurafenib have failed in BRAF-mutant CRCs (RR of 5 %) [57]; this lack of efficacy has been ascribed to the feedback activation of EGFR, which ensues as a consequence of BRAF inactivation and leads to EGFR-dependent compensatory signals [58, 59]. Accordingly, preclinical studies have provided a proof of principle that the combined inhibition of EGFR and BRAF can be synergistic in BRAF-mutant CRCs; however, it is worth noting that the best responses of CRC cell xenografts to such combinations were only disease stabilizations or mild tumor regressions [52, 58-60]. At the clinical level, a recent case report sustained the rationale of combined therapy with vemurafenib and cetuximab in BRAF V600E-mutant mCRC patients [61], and a pilot study of vemurafenib and panitumumab in this disease setting is currently recruiting participants (NCT01791309, http://clinicaltrials.gov/ct2/results?term=NCT01791309&Search=Search). From the diagnostic viewpoint, the feedback activation of EGFR upon BRAF inhibition likely implies that EGFR expression and phosphorylation levels may be potential predictors of response to vemurafenib monotherapy in BRAF-mutant mCRC patients [58, 59]. Other combinatorial approaches needing further testing or already under clinical evaluation [50, 62-64] are listed in Table 1.
BRAF mutations could be also detected noninvasively by ctDNA analysis, together with concomitant KRAS and NRAS mutations [40, 41], in patients who objectively responded to anti-EGFR therapy but subsequently relapsed. This indicates that the emergence of BRAF-mutant-resistant subclones also sustains secondary resistance.
PI3K-AKT-PTEN pathway
PI3Ks are a family of lipid kinases; in particular, class IA PI3Ks can be activated by different RTKs [65], but also through RAS association [66] or signaling from G protein-coupled receptors [9].
Class IA PI3Ks consist of heterodimeric proteins composed of a regulatory (p85) and a catalytic (p110) subunit [67]. Activating mutations of PIK3CA (encoding p110α) have been found in 10–20 % of CRCs [20, 68-70]; most of them occur in exons 9 and 20, respectively, in the helical and kinase domain [68, 71]. Sartore-Bianchi and colleagues performed a retrospective analysis of 110 mCRC patients treated with cetuximab or panitumumab, reporting a statistically significant association between PIK3CA mutations and primary resistance to treatment within KRAS wild-type tumors. In this study, 11 out of 15 mutations were found in exon 20 (73.3 %) and only 4 in exon 9 (26.7 %) [72]. Another study, in which a majority of exon 9 mutations was reported, did not confirm such correlation [70]. These conflicting reports were then reconciled by a large retrospective consortium analysis on 1,022 tumor samples which showed that, in the KRAS wild-type subpopulation, only PIK3CA exon 20 mutations may be predictive of lack response to cetuximab (RR of 0 % in mutant vs 36.8 % in wild-type cases) [20]. This study also described a strong association between PIK3CA exon 9 (but not exon 20) mutations and KRAS mutations, suggesting the lack of independent influence of PIK3CA exon 9 mutations on cetuximab efficacy.
Loss of function of PTEN, which antagonizes PI3K activity, occurs in 30 % of sporadic CRCs through a variety of mechanisms [73, 74]. PTEN inactivation (usually assessed as lack of protein expression) has been associated with nonresponsiveness to anti-EGFR moAbs in mCRC patients in several studies [19, 73, 75, 76], whereas others have only reported a prognostic role [53]. In summary, both PIK3CA exon 20 mutations and loss of PTEN expression are promising predictors of tumor suitability for anti-EGFR therapies. However, due to the low incidence of exon 20 mutations (2–5 %) [77] and lack of a consensus method for PTEN expression analysis [20, 73, 76, 78, 79], further prospective trials are required to challenge the clinical utility of PI3K pathway activation as a negative response predictor.
In principle, patients harboring PIK3CA mutations or PTEN loss of function, without concomitant KRAS/BRAF mutations, may benefit from targeted treatments against PI3K or PI3K-downstream effectors such as mTOR or AKT [80]; however, emerging clinical data have shown only minimal single-agent activity of such inhibitors at tolerated doses [81-83]. It is likely that mTOR kinase, AKT, pan-PI3K, or isoform-specific PI3K inhibitors will provide greater therapeutic index when combined with RTK inhibitors [84]. Phase I/II studies testing mTOR inhibitors, such as everolimus or temsirolimus, in combination with RTK inhibitors or anti-EGF moAbs plus chemotherapy in mCRC patients are underway (NCT01154335, http://clinicaltrials.gov/ct2/show/NCT01154335?term=colorectal+cancer&rank=33; NCT01139138, http://clinicaltrials.gov/ct2/show/NCT01139138?term=colorectal+cancer&rank=67; NCT01387880, http://clinicaltrials.gov/ct2/show/NCT01387880?term=everolimus+AND+colorectal+cancer&rank=2; NCT00827684, http://clinicaltrials.gov/ct2/show/NCT00827684?term=everolimus+AND+colorectal+cancer&rank=9).
Finally, recent observational studies have shown that adjuvant low-dose aspirin improved survival in patients with PIK3CA-mutant tumors [85-87]; this sensitivity requires further prospective evaluation and could be at least partially explained by the fact that PI3K-AKT seems to induce NF-ĸB-dependent transcriptional upregulation of COX2, which has been demonstrated to display pro-survival activity in CRC cells [87-89]. Therefore, a PIK3CA-mutant context may render CRC cells susceptible to apoptosis by aspirin-mediated COX2 inhibition.
To our knowledge, no alterations in the PI3K/AKT pathway have been associated with acquired resistance thus far.
HER2
It has been calculated that, among nonresponsive patients, 70 % bear tumors harboring at least one genetic alteration in the four abovementioned markers: KRAS, NRAS, BRAF, and PIK3CA [19]; therefore, the remaining 30 % of “quadruple negative” resistant cases display still-unidentified features that sustain lack of response.
HER2 is the only member of the ErbB family that does not bind ligands; it is activated via hetero-dimerization with the other ligand-bound receptors [10], with the strongest mitogenic signals created by HER2-HER3 heterodimers; HER2 overexpression, usually caused by gene amplification, allows HER2 activation even in the absence of ligand bound to the other partners [90].
Two independent studies have recently shown that HER2 amplification is a predictor of poor sensitivity to anti-EGFR antibodies [91, 92]. By performing genotype-response correlations in a preclinical platform of patient-derived metastatic CRC xenografts (xenopatients), Bertotti and colleagues identified HER2 amplification as a biomarker of resistance to cetuximab within a quadruple negative population. Concomitantly, using a combination of resistant clones from cetuximab-sensitive cell lines and plasma and tissue samples from cetuximab-treated mCRC patients, Yonesaka and colleagues reported aberrant HER2 signaling (by either HER2 amplification or through overproduction of the HER3-activating ligand heregulin) as a mediator of lack of response [92]. In retrospective studies, patients with tumors featuring HER2 amplification or heregulin overexpression and treated with cetuximab or panitumumab experienced disease progression and shorter progression-free and overall survival compared with HER2 wild-type cases [91-93].
Interestingly, in patients with acquired resistance, HER2 amplification was present in a small percentage of pretreatment tumor cells (14 %) that considerably increased in posttreatment samples (71 %). Similarly, heregulin levels, evaluated both in plasma and tumor specimens, were found to be significantly higher in patients that relapsed on anti-EGFR therapy [92]. This indicates that enhanced HER2 signaling confers both primary and acquired resistance.
Active HER2 also contributes to unleashing the oncogenic properties of HER3 mutations, which have been recently identified in about 11 % of colon cancers [94]. A “dosage effect” may be envisioned whereby low-grade HER2 amplification or low levels of heregulin, which alone would be insufficient to sustain therapeutic resistance, might in fact decrease responsiveness to EGFR inhibition by collaborating with coexisting HER3 mutations. Anti-HER3 antibodies and small molecules are now available and have been shown to effectively impair HER3-mediated signals and tumor progression in preclinical studies in vivo [94]. Therefore, HER3 mutations in CRC deserve further exploration as new potential biomarkers of resistance to EGFR targeted therapies as well as new predictors of response to alternative treatment options.
Therapeutically, only the dual targeting of HER2 and EGFR by combining a small-molecule inhibitor, such as the dual EGFR/HER2 inhibitor lapatinib, with a moAb, such as cetuximab or pertuzumab, induced overt and long-lasting tumor regressions in proof-of-concept trials in HER2-amplified xenopatients [91]. This finding led to the design and execution of a clinical trial that is currently assessing the activity and efficacy of a trastuzumab-lapatinib combination in mCRC patients with KRAS wild-type, HER2-amplified, cetuximab-resistant tumors (https://www.clinicaltrialsregister.eu/ctr-search/trial/2012-002128-33/IT). A similar study, in which a combination of trastuzumab and irinotecan was tested in patients with HER2-overexpressing advanced colorectal cancers, has been recently completed (NCT00003995, http://clinicaltrials.gov/ct2/results?term=NCT00003995&Search=Search). It is likely that also heregulin-driven tumors lacking HER2 amplification may benefit from HER2-directed therapies [92, 95, 96], although the definition of proper cutoff levels for ligand expression will be necessary before starting further clinical studies.
MET
The RTK MET and its ligand, hepatocyte growth factor (HGF), can activate a number of pathways, including the RAS-BRAF-ERK cascade, the PI3K-AKT axis, SRC, and STAT signaling [97]; these signaling networks collectively influence multiple key processes in cancer such as proliferation, apoptosis, invasion, and angiogenesis [98, 99]. Aberrant activation of MET may occur by several mechanisms, including MET amplification and/or increased HGF expression/activity [97], and has been widely described as a cause of both primary and acquired resistance to EGFR inhibitors in NSCLCs carrying EGFR mutations [100-102].
HGF-induced MET activation as a mechanism of attenuated sensitivity to cetuximab in CRC has been reported by preclinical studies using either CRC cell lines [103, 104] or, more recently, CRC spheroids enriched in cancer stem cells [105]. In these studies, only the simultaneous blockade of both MET and EGFR effectively impaired tumor growth in vivo. Based on publicly available gene expression data, cetuximab resistance mediated by HGF overexpression may be also relevant in mCRC patients [105]. However, similar to that discussed for heregulin, the investigation of such candidate biomarker requires the definition of methods and the categorization of expression cutoffs before further clinical evaluations.
The role of MET amplification as a mechanism of primary resistance to cetuximab and panitumumab in mCRC patients has been recently elucidated by Bardelli and colleagues [104]. MET amplification was retrospectively found in around 1 % of mCRC samples, in line with previous reports [106]. However, this frequency increased to 12.5 % in a subpopulation of cetuximab-resistant xenopatients bearing wild-type forms of KRAS, NRAS, BRAF, PIK3CA, and HER2. Notably, only focal, high-grade amplification of the MET locus associated with lack of response; conversely, cetuximab proved to be active in tumors with modest gene copy number gains or polysomy of chromosome 7, where the MET gene lies [106]. This suggests that resistance is driven by a dosage effect. Multi-arm preclinical trials in MET-positive CRC cell lines and patient-derived xenografts revealed that long-lasting abolition of tumor growth could be achieved through MET inhibition, with or without concurrent interception of EGFR [104, 107]. In coherence, a phase II clinical trial with the primary objective to assess the antitumor efficacy of the dual MET-ALK inhibitor crizotinib in patients with solid tumors (including CRCs) harboring MET alterations is currently recruiting participants (NCT02034981, http://clinicaltrials.gov/ct2/results?term=NCT02034981&Search=Search).
MET amplification was also found in three out of seven patients who developed acquired resistance, showing mutual exclusivity with secondary KRAS mutations. Of note, the MET amplicon was detected in circulating, cell-free DNA as early as 3 months after initiation of therapy, before relapse was clinically evident. Like HER2 amplification and KRAS mutations, rare MET-amplified cells were found in pretreatment tumor material from one out of three patients with MET-driven acquired resistance, suggesting that preexisting clones were selected under the pressure of anti-EGFR therapy.
EGFR
Additional genetic alterations within the target oncogene itself, which prevent drug binding and lead to kinase activation even in the presence of the inhibitor, are a common mechanism of both primary and acquired resistance in cancer; a paradigmatic example is provided by the T790M “gatekeeper” secondary mutation in the EGFR gene, which installs resistance to reversible EGFR inhibitors in EGFR-mutant NSCLC [108]. In colorectal cancer patients, a mutation in the extracellular domain of EGFR (S492R), which abrogates cetuximab binding but retains panitumumab sensitivity, has been recently described as a mechanism of acquired resistance [109, 110] (Fig. 2c). Two mutations in the EGFR kinase domain (codons 714 and 794), which were not detected before EGFR blockade, were identified as circulating mutations by cell-free DNA analysis. Although the functional relevance of these alterations in affecting sensitivity to anti-EGFR moAbs remains to be determined, it is conceivable that they contribute to the onset of secondary resistance [41].
Other potential biomarkers of drug sensitivity and resistance
The step forward into refining mCRC patient stratification presumably will be the validation of new candidate positive and negative predictors of response to EGFR moAbs. Increased EGFR gene copy number could predict response among KRAS wild-type patients [53, 91, 111-113], but EGFR FISH in mCRCs still needs interlaboratory standardization [75, 114, 115].
Different EGFR-specific ligands could differently influence the clinical activity of cetuximab: while high mRNA levels of either amphiregulin or epiregulin may predict a better response [21, 116-119], high levels of TGF-α as well as HB-EGF could confer lack of sensitivity [107, 118]. These findings, together with the role of other growth factors mentioned in this review, i.e., HGF, sustain the potential but understudied contribution of tumor-stromal interactions in influencing drug response in mCRCs [104, 120, 121].
Controversial data have been reported regarding the predictive role of a number of prognostic biomarkers: for example, IGF-1R overexpression seems to be a favorable prognostic factor [79, 106, 122] while high levels of EphA2, a pro-angiogenic RTK [123, 124], have been associated with poor outcome in mCRC patients treated with cetuximab-based therapy [125]. Biologically, altered tumor angiogenesis as a way to escape cetuximab antitumor activity has been previously reported in CRC cellular models and ascribed to either VEGF protein overexpression or increased VEGFR-1 and VEGFR-2 activation [126, 127]; taken together, these findings suggest that increased expression of pro-angiogenic ligands and cognate receptors (including VEGFs, VEGFRs, and Eph receptors) may dictate sensitivity to anti-EGFR therapy in colorectal tumors. Other “pathway-independent” mechanisms could also have a role in modifying drug response, for example, deregulation of EGFR ubiquitination which affects receptor recycling and expression at the cell membrane [128] (Fig. 2d).
Ongoing research and challenges
New therapeutic opportunities are currently being offered by genome-scale analyses of CRCs: recurrent mutations, rearrangements, and copy-number alterations have been proposed as therapeutically actionable drivers of colorectal tumorigenesis [129, 130] and will receive further biological validation by future integrated proteogenomics [131]. Promising hints are also emerging from treatments aimed at disrupting immune evasion strategies. As a means to instigate immune suppression, tumor cells often engage immune checkpoint molecules, such as CTLA-4 and PD1, which downregulate pathways of T cell activation. Antibodies against CTLA-4 (ipilimumab) or PD1 (nivolumab) have been shown to induce quick and intense tumor regression in melanoma and NSCLC patients and are currently under clinical investigation in other solid tumors, including CRC [132, 133] (NCT01975831, http://clinicaltrials.gov/ct2/show/NCT01975831).
Although an ever-increasing number of primary and acquired resistance mechanisms have been described until now, mutant KRAS is the only validated biomarker in routine practice for selection of mCRC patients to be treated with EGFR-targeted therapies. Thereby, there is a need to develop new models for clinical trials in order to facilitate and accelerate the introduction of other potentially useful biomarkers into clinical practice. Translational research in this context has an unquestionable role. Despite the lack of defined successful endpoints for preclinical models [134], arrest of cell growth and induction of apoptosis in vitro, and especially tumor regression in vivo (ideally in patient-derived xenografts), could have great impact to help design new cancer drug trials. Basket trials, in which patients are treated with different regimens based on their specific genetic profiles, may also optimize outcomes.
Further trial shaping could be provided by genomic analysis from serial biopsies to monitor response evolution and acquisition of genetic or adaptive resistance. However, tumor biopsy may not be representative of the intratumoral and intermetastatic heterogeneity and posttreatment tumor tissue is almost invariably unavailable. Such limitations could be overcome by less-invasive analysis of circulating tumor DNA, which can offer a high degree of sensitivity and specificity to monitor the emergence of drug resistance during the course of treatment [41, 135]. The mechanisms by which ctDNA is released into the circulation and whether multiple metastases shed ctDNA homogeneously are still unclear; however, the proof of principle that such an approach could complement tumor biopsy and provide an early warning of acquired resistance has been established [38, 41, 136].
One way for cancer to escape therapy is by continuous adaptation to the selective pressure of the drug, mainly through tumor genetic heterogeneity and biochemical or transcriptional activation of compensatory feedback loops [137]; exploiting these observations to create a “balance” between drug activity and graded responsiveness of different clones could be useful to delay the onset of resistance and, ideally, to turn cancer into a chronic disease. In this scenario, the lessons learned from metronomic treatment strategies for BRAF V600E melanomas as well as EGFR-mutant NSCLCs suggest that discontinuous dosing of the drug could be a strategy to prevent or retard acquired resistance [138, 139].
Early detection of disease progression calls for hypothesis-driven approaches to contrast outcompetition by subclones exhibiting resistance-conferring mutations. Nonetheless, resistance is pervasive, and observations so far let us conclude that, in most cases, progression to alternative strategies of drug elusion will inevitably occur [109, 140]. Thereby, there is a need to design adaptive drug combinations to achieve tumor response, reduce chances of relapse, and prolong patient survival. In line with this, bioinformatics and systems biology approaches interrogating the huge amount of patient datasets produced until now may provide models and signatures that are more comprehensive and predictive than the mutation status alone [21, 141, 142]. Rational combination therapies guided by real-time monitoring of tumor evolution along treatment, coupled with integrated omics approaches, will ultimately inform trial design to improve patients care in the coming years.
Final remarks
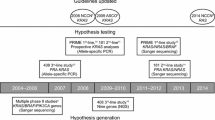
A decade after the introduction of cetuximab in the treatment of mCRC, much is known about the genetic determinants of primary resistance to anti-EGFR moAbs and initial insights are emerging about the mechanisms underlying acquisition of secondary resistance. The unifying concept is that the very same genetic alterations that account for intrinsic refractoriness also appear to foster progressive lack of response along treatment (Fig. 3), likely due to the presence of preexisting drug-insensitive subclones that are positively selected by continuous EGFR blockade. Future studies are needed to address cogent issues such as modeling tumor heterogeneity along cancer progression and under drug pressure, designing rational combination therapies to target concurrent mutations in the same cells or in different subpopulations, and improving early detection of disease progression.

Overlap between molecular biomarkers of primary and acquired resistance in mCRC. Most of the primary and acquired mechanisms of resistance to EGFR-targeted therapies in mCRC overlap. To date, no alterations of the PI3K pathway have been associated with acquired resistance; on the contrary, EGFR mutations have never been detected before exposure to EGFR monoclonal antibodies
References
Ferlay J, Autier P, Boniol M, Heanue M, Colombet M, Boyle P (2007) Estimates of the cancer incidence and mortality in Europe in 2006. Ann Oncol 18:581–592
Poston GJ, Figueras J, Giuliante F, Nuzzo G, Sobrero AF, Gigot JF, Nordlinger B, Adam R, Gruenberger T, Choti MA et al (2008) Urgent need for a new staging system in advanced colorectal cancer. J Clin Oncol 26:4828–4833
Segal NH, Saltz LB (2009) Evolving treatment of advanced colon cancer. Annu Rev Med 60:207–219
Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, Forman D, Bray F (2013) Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer 49:1374–1403
Garden OJ, Rees M, Poston GJ, Mirza D, Saunders M, Ledermann J, Primrose JN, Parks RW (2006) Guidelines for resection of colorectal cancer liver metastases. Gut 55(Suppl 3):iii1-8
Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, Berry SR, Krahn M, Price T, Simes RJ et al (2007) Cetuximab for the treatment of colorectal cancer. N Engl J Med 357:2040–2048
Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C et al (2004) Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 351:337–345
Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, Canon JL, Van Laethem JL, Maurel J, Richardson G et al (2007) Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol 25:1658–1664
Yarden Y, Sliwkowski MX (2001) Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2:127–137
Hynes NE, Lane HA (2005) ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 5:341–354
Ciardiello F, Tortora G (2001) A novel approach in the treatment of cancer: targeting the epidermal growth factor receptor. Clin Cancer Res 7:2958–2970
Siena S, Sartore-Bianchi A, Di Nicolantonio F, Balfour J, Bardelli A (2009) Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J Natl Cancer Inst 101:1308–1324
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M et al (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364:2507–2516
Sequist LV, Martins RG, Spigel D, Grunberg SM, Spira A, Jänne PA, Joshi VA, McCollum D, Evans TL, Muzikansky A et al (2008) First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol 26:2442–2449
Garraway LA, Jänne PA (2012) Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov 2:214–226
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG (2013) Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 13:714–726
Lièvre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M et al (2006) KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 66:3992–3995
Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, Zanon C, Moroni M, Veronese S, Siena S, Bardelli A (2007) Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 67:2643–2648
Sartore-Bianchi A, Di Nicolantonio F, Nichelatti M, Molinari F, De Dosso S, Saletti P, Martini M, Cipani T, Marrapese G, Mazzucchelli L et al (2009) Multi-determinants analysis of molecular alterations for predicting clinical benefit to EGFR-targeted monoclonal antibodies in colorectal cancer. PLoS One 4:e7287
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P et al (2010) Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 11:753–762
Tian S, Simon I, Moreno V, Roepman P, Tabernero J, Snel M, van’t Veer L, Salazar R, Bernards R, Capella G (2013) A combined oncogenic pathway signature of BRAF, KRAS and PI3KCA mutation improves colorectal cancer classification and cetuximab treatment prediction. Gut 62:540–549
Brand TM, Iida M, Wheeler DL (2011) Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol Ther 11:777–792
Chong CR, Jänne PA (2013) The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med 19:1389–1400
Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P et al (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366:883–892
Turner NC, Reis-Filho JS (2012) Genetic heterogeneity and cancer drug resistance. Lancet Oncol 13:e178–e185
Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S et al (2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 359:1757–1765
Lièvre A, Blons H, Laurent-Puig P (2010) Oncogenic mutations as predictive factors in colorectal cancer. Oncogene 29:3033–3043
Di Fiore F, Blanchard F, Charbonnier F, Le Pessot F, Lamy A, Galais MP, Bastit L, Killian A, Sesboüé R, Tuech JJ et al (2007) Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br J Cancer 96:1166–1169
Bardelli A, Siena S (2010) Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol 28:1254–1261
Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J et al (2010) Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol 28:4697–4705
Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, André T, Chan E, Lordick F, Punt CJ et al (2010) Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol 28:4706–4713
Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R et al (2008) Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 26:1626–1634
Van Cutsem E, Köhne CH, Láng I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S et al (2011) Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol 29:2011–2019
Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF, Schilsky RL (2009) American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol 27:2091–2096
Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J et al (2013) Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 369:1023–1034
Valtorta E, Misale S, Sartore-Bianchi A, Nagtegaal ID, Paraf F, Lauricella C, Dimartino V, Hobor S, Jacobs B, Ercolani C et al (2013) KRAS gene amplification in colorectal cancer and impact on response to EGFR-targeted therapy. Int J Cancer 133:1259–1265
Diaz LA, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA et al (2012) The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486:537–540
Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G et al (2012) Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 486:532–536
Osumi H, Matsusaka S, Shinozaki E, Suenaga M, Mingyon M, Saiura A, Ueno M, Mizunuma N, Yamaguchi T (2013) Acquired drug resistance conferred by a KRAS gene mutation following the administration of cetuximab: a case report. BMC Res Notes 6:508
Misale S, Arena S, Lamba S, Siravegna G, Lallo A, Hobor S, Russo M, Buscarino M, Lazzari L, Sartore-Bianchi A et al (2014) Blockade of EGFR and MEK intercepts heterogeneous mechanisms of acquired resistance to anti-EGFR therapies in colorectal cancer. Sci Transl Med 6:224ra226
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM et al (2014) Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 6:224ra224
Siena S, Van Cutsem E, Li M, Jungnelius U, Romano A, Beck R, Bencardino K, Elez ME, Prenen H, Sanchis M et al (2013) Phase II open-label study to assess efficacy and safety of lenalidomide in combination with cetuximab in KRAS-mutant metastatic colorectal cancer. PLoS One 8:e62264
Migliardi G, Sassi F, Torti D, Galimi F, Zanella ER, Buscarino M, Ribero D, Muratore A, Massucco P, Pisacane A et al (2012) Inhibition of MEK and PI3K/mTOR suppresses tumor growth but does not cause tumor regression in patient-derived xenografts of RAS-mutant colorectal carcinomas. Clin Cancer Res 18:2515–2525
Steckel M, Molina-Arcas M, Weigelt B, Marani M, Warne PH, Kuznetsov H, Kelly G, Saunders B, Howell M, Downward J et al (2012) Determination of synthetic lethal interactions in KRAS oncogene-dependent cancer cells reveals novel therapeutic targeting strategies. Cell Res 22:1227–1245
Scholl C, Fröhling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, Silver SJ, Tamayo P, Wadlow RC, Ramaswamy S et al (2009) Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 137:821–834
Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ (2009) A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 137:835–848
Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, Greninger P, Brown RD, Godfrey JT, Cohoon TJ et al (2013) Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell 23:121–128
Singh A, Sweeney MF, Yu M, Burger A, Greninger P, Benes C, Haber DA, Settleman J (2012) TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell 148:639–650
Ebi H, Corcoran RB, Singh A, Chen Z, Song Y, Lifshits E, Ryan DP, Meyerhardt JA, Benes C, Settleman J et al (2011) Receptor tyrosine kinases exert dominant control over PI3K signaling in human KRAS mutant colorectal cancers. J Clin Invest 121:4311–4321
Faber AC, Coffee EM, Costa C, Dastur A, Ebi H, Hata AN, Yeo AT, Edelman EJ, Song Y, Tam AT et al (2014) mTor inhibition specifically sensitizes colorectal cancers with KRAS or BRAF mutations to BCL-2/BCL-XL inhibition by suppressing MCL-1. Cancer Discov 4:42–52
Dunn EF, Iida M, Myers RA, Campbell DA, Hintz KA, Armstrong EA, Li C, Wheeler DL (2011) Dasatinib sensitizes KRAS mutant colorectal tumors to cetuximab. Oncogene 30:561–574
Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S et al (2008) Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol 26:5705–5712
Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet JB, Lecomte T, Rougier P, Lievre A, Landi B, Boige V et al (2009) Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol 27:5924–5930
Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M, Celik I, Köhne CH (2012) Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer 48:1466–1475
Karapetis CS, Jonker D, Daneshmand M, Hanson JE, O’Callaghan CJ, Marginean C, Zalcberg JR, Simes J, Moore MJ, Tebbutt NC et al (2014) PIK3CA, BRAF, and PTEN status and benefit from cetuximab in the treatment of advanced colorectal cancer—results from NCIC CTG/AGITG CO.17. Clin Cancer Res. doi:10.1158/1078-0432.CCR-13-0606
Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K et al (2010) Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 363:809–819
Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Lee RJ et al (2010) PLX4032 in metastatic colorectal cancer patients with mutant BRAFtumors. J Clin Oncol 28(suppl):5s, Abstract nr 3534
Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R (2012) Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483:100–103
Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D, Hung KE et al (2012) EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov 2:227–235
Yang H, Higgins B, Kolinsky K, Packman K, Bradley WD, Lee RJ, Schostack K, Simcox ME, Kopetz S, Heimbrook D et al (2012) Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res 72:779–789
Connolly K, Brungs D, Szeto E, Epstein RJ (2014) Anticancer activity of combination targeted therapy using cetuximab plus vemurafenib for refractory BRAF (V600E)-mutant metastatic colorectal carcinoma. Curr Oncol 21:e151–e154
Mao M, Tian F, Mariadason JM, Tsao CC, Lemos R, Dayyani F, Gopal YN, Jiang ZQ, Wistuba II, Tang XM et al (2013) Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res 19:657–667
Coffee EM, Faber AC, Roper J, Sinnamon MJ, Goel G, Keung L, Wang WV, Vecchione L, de Vriendt V, Weinstein BJ et al (2013) Concomitant BRAF and PI3K/mTOR blockade is required for effective treatment of BRAF (V600E) colorectal cancer. Clin Cancer Res 19:2688–2698
Zecchin D, Boscaro V, Medico E, Barault L, Martini M, Arena S, Cancelliere C, Bartolini A, Crowley EH, Bardelli A et al (2013) BRAF V600E is a determinant of sensitivity to proteasome inhibitors. Mol Cancer Ther 12:2950–2961
De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S (2011) KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol 12:594–603
Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J (1994) Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 370:527–532
Kraus S, Nabiochtchikov I, Shapira S, Arber N (2014) Recent advances in personalized colorectal cancer research. Cancer Lett. doi:10.1016/j.canlet.2014.01.025
Liao X, Morikawa T, Lochhead P, Imamura Y, Kuchiba A, Yamauchi M, Nosho K, Qian ZR, Nishihara R, Meyerhardt JA et al (2012) Prognostic role of PIK3CA mutation in colorectal cancer: cohort study and literature review. Clin Cancer Res 18:2257–2268
Karakas B, Bachman KE, Park BH (2006) Mutation of the PIK3CA oncogene in human cancers. Br J Cancer 94:455–459
Prenen H, De Schutter J, Jacobs B, De Roock W, Biesmans B, Claes B, Lambrechts D, Van Cutsem E, Tejpar S (2009) PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin Cancer Res 15:3184–3188
Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, Vogelstein B, Gabelli SB, Amzel LM (2007) The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science 318:1744–1748
Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P, De Dosso S, Mazzucchelli L et al (2009) PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res 69:1851–1857
Perrone F, Lampis A, Orsenigo M, Di Bartolomeo M, Gevorgyan A, Losa M, Frattini M, Riva C, Andreola S, Bajetta E et al (2009) PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann Oncol 20:84–90
Goel A, Arnold CN, Niedzwiecki D, Carethers JM, Dowell JM, Wasserman L, Compton C, Mayer RJ, Bertagnolli MM, Boland CR (2004) Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability-high sporadic colorectal cancers. Cancer Res 64:3014–3021
Frattini M, Saletti P, Romagnani E, Martin V, Molinari F, Ghisletta M, Camponovo A, Etienne LL, Cavalli F, Mazzucchelli L (2007) PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer 97:1139–1145
Loupakis F, Pollina L, Stasi I, Ruzzo A, Scartozzi M, Santini D, Masi G, Graziano F, Cremolini C, Rulli E et al (2009) PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J Clin Oncol 27:2622–2629
Okada Y, Miyamoto H, Goji T, Takayama T (2014) Biomarkers for predicting the efficacy of anti-epidermal growth factor receptor antibody in the treatment of colorectal cancer. Digestion 89:18–23
Negri FV, Bozzetti C, Lagrasta CA, Crafa P, Bonasoni MP, Camisa R, Pedrazzi G, Ardizzoni A (2010) PTEN status in advanced colorectal cancer treated with cetuximab. Br J Cancer 102:162–164
Inno A, Di Salvatore M, Cenci T, Martini M, Orlandi A, Strippoli A, Ferrara AM, Bagalà C, Cassano A, Larocca LM et al (2011) Is there a role for IGF1R and c-MET pathways in resistance to cetuximab in metastatic colorectal cancer? Clin Colorectal Cancer 10:325–332
Di Nicolantonio F, Arena S, Tabernero J, Grosso S, Molinari F, Macarulla T, Russo M, Cancelliere C, Zecchin D, Mazzucchelli L et al (2010) Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J Clin Invest 120:2858–2866
Ganesan P, Janku F, Naing A, Hong DS, Tsimberidou AM, Falchook GS, Wheler JJ, Piha-Paul SA, Fu S, Stepanek VM et al (2013) Target-based therapeutic matching in early-phase clinical trials in patients with advanced colorectal cancer and PIK3CA mutations. Mol Cancer Ther 12:2857–2863
Altomare I, Hurwitz H (2013) Everolimus in colorectal cancer. Expert Opin Pharmacother 14:505–513
Fruman DA, Rommel C (2014) PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 13:140–156
Courtney KD, Corcoran RB, Engelman JA (2010) The PI3K pathway as drug target in human cancer. J Clin Oncol 28:1075–1083
Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M, Imamura Y, Qian ZR, Baba Y, Shima K et al (2012) Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med 367:1596–1606
Domingo E, Church DN, Sieber O, Ramamoorthy R, Yanagisawa Y, Johnstone E, Davidson B, Kerr DJ, Tomlinson IP, Midgley R (2013) Evaluation of PIK3CA mutation as a predictor of benefit from nonsteroidal anti-inflammatory drug therapy in colorectal cancer. J Clin Oncol 31:4297–4305
Fuchs CS, Ogino S (2013) Aspirin therapy for colorectal cancer with PIK3CA mutation: simply complex! J Clin Oncol 31:4358–4361
Hutti JE, Pfefferle AD, Russell SC, Sircar M, Perou CM, Baldwin AS (2012) Oncogenic PI3K mutations lead to NF-κB-dependent cytokine expression following growth factor deprivation. Cancer Res 72:3260–3269
Kaur J, Sanyal SN (2010) PI3-kinase/Wnt association mediates COX-2/PGE(2) pathway to inhibit apoptosis in early stages of colon carcinogenesis: chemoprevention by diclofenac. Tumour Biol 31:623–631
Tzahar E, Waterman H, Chen X, Levkowitz G, Karunagaran D, Lavi S, Ratzkin BJ, Yarden Y (1996) A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol Cell Biol 16:5276–5287
Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, Corà D, Di Nicolantonio F, Buscarino M, Petti C et al (2011) A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov 1:508–523
Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, Ercan D, Rogers A, Roncalli M, Takeda M et al (2011) Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med 3:99ra86
Martin V, Landi L, Molinari F, Fountzilas G, Geva R, Riva A, Saletti P, De Dosso S, Spitale A, Tejpar S et al (2013) HER2 gene copy number status may influence clinical efficacy to anti-EGFR monoclonal antibodies in metastatic colorectal cancer patients. Br J Cancer 108:668–675
Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, Chaudhuri S, Pujara K, Guillory J, Edgar KA et al (2013) Oncogenic ERBB3 mutations in human cancers. Cancer Cell 23:603–617
Wilson TR, Lee DY, Berry L, Shames DS, Settleman J (2011) Neuregulin-1-mediated autocrine signaling underlies sensitivity to HER2 kinase inhibitors in a subset of human cancers. Cancer Cell 20:158–172
Agus DB, Akita RW, Fox WD, Lewis GD, Higgins B, Pisacane PI, Lofgren JA, Tindell C, Evans DP, Maiese K et al (2002) Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell 2:127–137
Comoglio PM, Giordano S, Trusolino L (2008) Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov 7:504–516
Benvenuti S, Comoglio PM (2007) The MET receptor tyrosine kinase in invasion and metastasis. J Cell Physiol 213:316–325
Bussolino F, Di Renzo MF, Ziche M, Bocchietto E, Olivero M, Naldini L, Gaudino G, Tamagnone L, Coffer A, Comoglio PM (1992) Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J Cell Biol 119:629–641
Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S et al (2007) MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 104:20932–20937
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J et al (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316:1039–1043
Cappuzzo F, Jänne PA, Skokan M, Finocchiaro G, Rossi E, Ligorio C, Zucali PA, Terracciano L, Toschi L, Roncalli M et al (2009) MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann Oncol 20:298–304
Liska D, Chen CT, Bachleitner-Hofmann T, Christensen JG, Weiser MR (2011) HGF rescues colorectal cancer cells from EGFR inhibition via MET activation. Clin Cancer Res 17:472–482
Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, Sartore-Bianchi A, Scala E, Cassingena A, Zecchin D et al (2013) Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov 3:658–673
Luraghi P, Reato G, Cipriano E, Sassi F, Orzan F, Bigatto V, De Bacco F, Menietti E, Han M, Rideout WM et al (2014) MET signaling in colon cancer stem-like cells blunts the therapeutic response to EGFR inhibitors. Cancer Res. doi:10.1158/0008-5472.CAN-13-2340-T
Cappuzzo F, Varella-Garcia M, Finocchiaro G, Skokan M, Gajapathy S, Carnaghi C, Rimassa L, Rossi E, Ligorio C, Di Tommaso L et al (2008) Primary resistance to cetuximab therapy in EGFR FISH-positive colorectal cancer patients. Br J Cancer 99:83–89
Troiani T, Martinelli E, Napolitano S, Vitagliano D, Ciuffreda LP, Costantino S, Morgillo F, Capasso A, Sforza V, Nappi A et al (2013) Increased TGF-α as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin Cancer Res 19:6751–6765
Pao W, Chmielecki J (2010) Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer 10:760–774
Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, Salido M, Gallen M, Marsters S, Tsai SP et al (2012) Identification of a mutation in the extracellular domain of the epidermal growth factor receptor conferring cetuximab resistance in colorectal cancer. Nat Med 18:221–223
Esposito C, Rachiglio AM, La Porta ML, Sacco A, Roma C, Iannaccone A, Tatangelo F, Forgione L, Pasquale R, Barbaro A, et al. (2013) The S492R EGFR ectodomain mutation is never detected in KRAS wild type colorectal carcinoma before exposure to EGFR monoclonal antibodies. Cancer Biol Ther 14
Scartozzi M, Bearzi I, Mandolesi A, Pierantoni C, Loupakis F, Zaniboni A, Negri F, Quadri A, Zorzi F, Galizia E et al (2009) Epidermal growth factor receptor (EGFR) gene copy number (GCN) correlates with clinical activity of irinotecan-cetuximab in K-RAS wild-type colorectal cancer: a fluorescence in situ (FISH) and chromogenic in situ hybridization (CISH) analysis. BMC Cancer 9:303
Moroni M, Veronese S, Benvenuti S, Marrapese G, Sartore-Bianchi A, Di Nicolantonio F, Gambacorta M, Siena S, Bardelli A (2005) Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol 6:279–286
Cappuzzo F, Finocchiaro G, Rossi E, Jänne PA, Carnaghi C, Calandri C, Bencardino K, Ligorio C, Ciardiello F, Pressiani T et al (2008) EGFR FISH assay predicts for response to cetuximab in chemotherapy refractory colorectal cancer patients. Ann Oncol 19:717–723
Italiano A, Follana P, Caroli FX, Badetti JL, Benchimol D, Garnier G, Gugenheim J, Haudebourg J, Keslair F, Lesbats G et al (2008) Cetuximab shows activity in colorectal cancer patients with tumors for which FISH analysis does not detect an increase in EGFR gene copy number. Ann Surg Oncol 15:649–654
Sartore-Bianchi A, Fieuws S, Veronese S, Moroni M, Personeni N, Frattini M, Torri V, Cappuzzo F, Vander Borght S, Martin V et al (2012) Standardisation of EGFR FISH in colorectal cancer: results of an international interlaboratory reproducibility ring study. J Clin Pathol 65:218–223
Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, Wong TW, Huang X, Takimoto CH, Godwin AK et al (2007) Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol 25:3230–3237
Jacobs B, De Roock W, Piessevaux H, Van Oirbeek R, Biesmans B, De Schutter J, Fieuws S, Vandesompele J, Peeters M, Van Laethem JL et al (2009) Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol 27:5068–5074
Tabernero J, Cervantes A, Rivera F, Martinelli E, Rojo F, von Heydebreck A, Macarulla T, Rodriguez-Braun E, Eugenia Vega-Villegas M, Senger S et al (2010) Pharmacogenomic and pharmacoproteomic studies of cetuximab in metastatic colorectal cancer: biomarker analysis of a phase I dose-escalation study. J Clin Oncol 28:1181–1189
Pentheroudakis G, Kotoula V, De Roock W, Kouvatseas G, Papakostas P, Makatsoris T, Papamichael D, Xanthakis I, Sgouros J, Televantou D et al (2013) Biomarkers of benefit from cetuximab-based therapy in metastatic colorectal cancer: interaction of EGFR ligand expression with RAS/RAF, PIK3CA genotypes. BMC Cancer 13:49
McMillin DW, Negri JM, Mitsiades CS (2013) The role of tumour-stromal interactions in modifying drug response: challenges and opportunities. Nat Rev Drug Discov 12:217–228
Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT et al (2012) Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 487:500–504
Scartozzi M, Mandolesi A, Giampieri R, Pierantoni C, Loupakis F, Zaniboni A, Galizia E, Giustini L, Silva RR, Bisonni R et al (2010) Insulin-like growth factor 1 expression correlates with clinical outcome in K-RAS wild type colorectal cancer patients treated with cetuximab and irinotecan. Int J Cancer 127:1941–1947
Brantley DM, Cheng N, Thompson EJ, Lin Q, Brekken RA, Thorpe PE, Muraoka RS, Cerretti DP, Pozzi A, Jackson D et al (2002) Soluble Eph A receptors inhibit tumor angiogenesis and progression in vivo. Oncogene 21:7011–7026
Sáinz-Jaspeado M, Huertas-Martinez J, Lagares-Tena L, Martin Liberal J, Mateo-Lozano S, de Alava E, de Torres C, Mora J, Del Muro XG, Tirado OM (2013) EphA2-induced angiogenesis in ewing sarcoma cells works through bFGF production and is dependent on caveolin-1. PLoS One 8:e71449
Strimpakos A, Pentheroudakis G, Kotoula V, De Roock W, Kouvatseas G, Papakostas P, Makatsoris T, Papamichael D, Andreadou A, Sgouros J et al (2013) The prognostic role of ephrin A2 and endothelial growth factor receptor pathway mediators in patients with advanced colorectal cancer treated with cetuximab. Clin Colorectal Cancer 12:267–274.e262
Ciardiello F, Bianco R, Caputo R, Damiano V, Troiani T, Melisi D, De Vita F, De Placido S, Bianco AR, Tortora G (2004) Antitumor activity of ZD6474, a vascular endothelial growth factor receptor tyrosine kinase inhibitor, in human cancer cells with acquired resistance to antiepidermal growth factor receptor therapy. Clin Cancer Res 10:784–793
Bianco R, Rosa R, Damiano V, Daniele G, Gelardi T, Garofalo S, Tarallo V, De Falco S, Melisi D, Benelli R et al (2008) Vascular endothelial growth factor receptor-1 contributes to resistance to anti-epidermal growth factor receptor drugs in human cancer cells. Clin Cancer Res 14:5069–5080
Lu Y, Li X, Liang K, Luwor R, Siddik ZH, Mills GB, Mendelsohn J, Fan Z (2007) Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab. Cancer Res 67:8240–8247
Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, Chaudhuri S, Guan Y, Janakiraman V, Jaiswal BS et al (2012) Recurrent R-spondin fusions in colon cancer. Nature 488:660–664
Network CGA (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487:330–337
Ellis MJ, Gillette M, Carr SA, Paulovich AG, Smith RD, Rodland KK, Townsend RR, Kinsinger C, Mesri M, Rodriguez H et al (2013) Connecting genomic alterations to cancer biology with proteomics: the NCI clinical proteomic tumor analysis consortium. Cancer Discov 3:1108–1112
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC et al (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363:711–723
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB et al (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366:2443–2454
Bertotti A, Trusolino L (2013) From bench to bedside: does preclinical practice in translational oncology need some rebuilding? J Natl Cancer Inst 105:1426–1427
Diaz LA, Bardelli A (2014) Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol 32:579–586
Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong AS et al (2013) Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 497:108–112
Trusolino L, Bertotti A (2012) Compensatory pathways in oncogenic kinase signaling and resistance to targeted therapies: six degrees of separation. Cancer Discov 2:876–880
Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M, Stuart DD (2013) Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 494:251–255
Becker A, Crombag L, Heideman DA, Thunnissen FB, van Wijk AW, Postmus PE, Smit EF (2011) Retreatment with erlotinib: regain of TKI sensitivity following a drug holiday for patients with NSCLC who initially responded to EGFR-TKI treatment. Eur J Cancer 47:2603–2606
Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, Rosenberg M, Goetz EM, Sullivan RJ, Farlow DN et al (2014) MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov 4:61–68
Guinney J, Ferté C, Dry J, McEwen R, Manceau G, Kao KJ, Chang KM, Bendtsen C, Hudson K, Huang E et al (2014) Modeling RAS phenotype in colorectal cancer uncovers novel molecular traits of RAS dependency and improves prediction of response to targeted agents in patients. Clin Cancer Res 20:265–272
Zhao B, Pritchard JR, Lauffenburger DA, Hemann MT (2014) Addressing genetic tumor heterogeneity through computationally predictive combination therapy. Cancer Discov 4:166–174
Acknowledgments
We thank our friends of the Laboratory of Molecular Pharmacology for the continued discussion and help. Research in the authors’ laboratory is supported by AIRC, Associazione Italiana per la Ricerca sul Cancro—2010 Special Program Molecular Clinical Oncology 5‰, project 9970, and AIRC Investigator Grants, projects 10116 and 14205.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Leto, S.M., Trusolino, L. Primary and acquired resistance to EGFR-targeted therapies in colorectal cancer: impact on future treatment strategies. J Mol Med 92, 709–722 (2014). https://doi.org/10.1007/s00109-014-1161-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-014-1161-2