
Abstract
Fulminant hepatitis can cause acute liver failure and death in both humans and mice. However, the cellular and molecular mechanisms underlying the acute disease are still not well understood. Here, we examine the role of Th17 response in the development of the acute hepatitis following infection with mouse hepatitis virus (MHV). We show that IL-17 levels in serum are rapidly elevated and positively correlated to liver damage and death of the mice. In IFN-γR−/− mice, Th17 response is enhanced and the elevated IL-17 production contributes to severe liver damage as well as detrimental inflammation because neutralization of IL-17 effectively suppresses inflammation and protects mice from liver injury. We further show that IFN-γ facilitates antigen-induced apoptosis of Th17 cells and adoptive transferred IFN-γR−/−, but not IFN-γR+/+; CD4+ T cells promotes an enhanced liver damage in wild-type mice. The results demonstrate an essential role of Th17 cells in MHV-induced immunopathology and the importance of IFN-γ in maintaining immune balance between Th1 and Th17 responses during acute viral infection.
Similar content being viewed by others

Introduction
MHV is a coronavirus and can induce a variety of diseases, including hepatitis, enteritis, and encephalitis in mice, depending on the virus strain, infection route, and age, genetic background, and immune status of the mice [1]. Intraperitoneal infection of susceptible strains of mice with hepatotropic MHV, such as MHV-A59, results in severe acute liver failure, and therefore, has served as a useful model for fulminant hepatitis in humans [2].
Accumulating evidence suggests that the immune response to virus infection is a double-edged sword with the opposing outcome of immunoprotection and immunopathology. Effector T cells are critical for the defense against pathogens, but inappropriate or poorly regulated T cell responses also induce inflammatory disorders and pathological damage. Thus, T cell response is stringently controlled under physiological conditions. Notably, IFN-γ is an effector cytokine produced by Th1 cells and CD8 T cells and plays a critical role in immune responses to microbial infection. IFN-γ also plays an essential role in the homeostatic control of effector T cells in order to minimize immunopathology [3, 4]. There is a progressive expansion of T cells in IFN-γ−/− mice infected with mycobacteria [5]. After Listeria infection, the frequency of antigen-specific T cells is higher in IFN-γ−/− mice than in wild-type mice [6]. Severe T cell-mediated immunopathology is observed in IFN-γR−/− mice infected by lymphocytic choriomeningitis virus (LCMV) [7]. Failure to suppress the expansion of the activated T cells in the absence of IFN-γ pathway leads to more severe encephalitis in mice [8]. Similarly, IFN-γ or its receptor deficient mice exhibit an increased susceptibility to MHV-induced hepatitis [9–11]. It has been hypothesized that the increased susceptibility is due to uncontrolled response to virus infection. However, the molecular and cellular basis underlying the impaired responses is largely unknown.
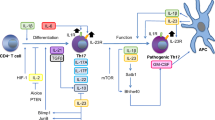
Th17 cells, a novel IL-17-secreting CD4+ T cell subset, have been shown to be involved in various inflammatory diseases. Although IL-17 was initially identified as a pathogenic cytokine in autoimmunity [12–15], increasing evidence suggests that IL-17 also plays a critical role in controlling microbial infection, especially intracellular bacterial infection [16–18]. IL-17 promotes recruitment of neutrophils to the site of infection, inducing proliferation of enterocytes and production of antibacterial defensins [19, 20]. Indeed, IL-17 is known to be induced and required for the host protection against Klebsiella pneumoniae [21], Citrobacter rodentium [22], and Candida albicans [23]. Hence, Th17 cells are an integral part of an antibacterial immunity. In contrast, the role of Th17 cells in viral infection is less clear. Previous studies indicate that simian immunodeficiency virus (SIV) infection results in depletion of Th17 cells in the ileal mucosa [24]. Similarly, IL-17 is negatively regulated by Epstein–Barr virus-induced gene 3 [25]. Th17 cells are suggested to correlate with the severity of liver damage during chronic HBV infection [26]. These previous studies suggest possible connections between IL-17 and virus infection.
In this study, we investigated the role of Th17 response during MHV-induced acute hepatitis. Immunity and pathology to hepatotropic MHV infection is specifically analyzed in the absence of IFN-γ pathway. Our results show that MHV-A59-induced Th17 response is associated with immunopathology in acute hepatitis. Increased severity of hepatitis in IFN-γR−/− mice correlates with exaggerated reactivity of Th17 cells. A blockade of IL-17 significantly decreases MHV-induced inflammation as well as liver damage. We further show that IFN-γ induces apoptosis of Th17 cells. These findings suggest a pathogenic role of Th17 cells in MHV-A59 infection, which is normally under the control of IFN-γ.
Materials and methods
Mice and infection
C57BL/6 mice deficient in IFN-γR (IFN-γR−/−) were originally from the Jackson Laboratories (Bar Harbor, ME). Sex- and age (6–8 weeks)-matched wild-type control mice were purchased from Vital River (Beijing, China). Mice were infected by i.p. injection of 5 × 105 plaque-forming unit (PFU) of MHV-A59 or MHV-A59/GOS in 0.2 ml PBS and observed daily for weight loss and mortality. For adoptive transfer experiments, naive wild-type and IFN-γR−/− mice were transferred with 5 × 106 CD4+ T cells and then with 5 × 105 PFU MHV-A59 1 day later. To neutralize IL-17, mice were i.p. injected with 100 μg of anti-mouse IL-17 mAb (TC11-18H10, Southern Biotechnology) 1 and 4 dpi. All mice were housed under specific pathogen-free condition at the animal facility of the Institute of Biophysics, Chinese Academy of Sciences, and the animal experimentation conforms to protocols approved by the institutional animal care committee.
Construction of recombinant MHV-A59/GOS
To construct a recombinant MHV-A59/GOS that expresses eGFP, the CD4 epitope OVA (SQAVHAAHAEINEAGR) and the CD8 epitope SIY (SIYRYYGL), the coding sequences of eGFP/OVA/SIY was amplified from pHS-eGFP/OVA/SIY vector and inserted into pMH54 by Sal I and Not I sites. A targeted RNA recombination was carried out between RNAs of pMH54/GOS and the recipient virus fMHV (feline MHV) in AKD cells. Recombinant viruses were selected by replication in 17Cl-1cells and were plaque purified twice (Fig. S1).
Virus titer assay
MHV-A59 titers in livers of infected mice were determined by the plaque assay on monolayer of 17Cl-1 cells. After 24 h incubation with serially diluted supernatants of tissue homogenate, plaques were counted on the cell layers. All titrations were performed in duplicates, and the average PFU per gram of tissue was calculated.
Alanine aminotransferase (ALT) activity
Serum ALT levels were measured with a kit according to the manufacturer’s instructions (Biosino).
Histological analysis
Paraffin-embedded sections (5 um thick) of liver of infected mice were stained with hematoxylin and eosin using standard techniques. Pictures were taken using an Olympus BX51 microscope.
Intrahepatic lymphocytes isolation
Hepatic lymphocytes were isolated as previously described [27]. Briefly, livers were perfused through the portal vein with PBS, excised, thoroughly dissected, and gently passed through a 100-gauge stainless mesh. The cell suspension was centrifuged at 500×g for 6 min. Then the pellet was resuspended in 40% Percoll solution and loaded on the 70% Percoll solution followed by centrifugation at 900×g for 20 min. The cell layer between 40% and 70% Percoll was collected for hepatic lymphocytes.
Flow cytometry
Cell suspensions were stained with fluorochrome-conjugated mouse-specific Abs against CD4 (BD Pharmingen), CD25 (BD Pharmingen), Gr1 (BD Pharmingen), IL-17 (eBioscience), Foxp3 (Miltenyi Biotec), IFN-γR (eBioscience), or isotype-matched control Abs. To detect SIY peptide-specific CD8+ T cells, cells were first blocked by CD16/CD32 (BD, Pharmingen) and stained with SIY-loaded Kb-Fc dimmer (BD Pharmingen). To identify IL-17-producing cells, cells were activated for 4 h with 50 ng/ml PMA and 500 ng/ml ionomycin in the presence of brefeldin A (GolgiPlug; BD Bioscience). Cells were fixed and permeabilized using the Cytofix/Cytoperm kit (BD Bioscience) before staining with anti-IL17 antibody. The apoptotic cells were detected by a TUNEL assay Kit (Beyotime) according to the manufacturer’s instructions. All samples were then analyzed with a BD FACSCalibur cytometer (BD, Biosciences).
Cytokine assays
At indicated time points post MHV-A59 infection, serum and liver homogenates were prepared from infected mice. The amount of cytokines was quantified using the cytometric bead array kit for mouse inflammatory cytokines (CBA; BD Biosciences) on a FACSCalibur cytometer equipped with CellQuestPro and CBA software (Becton Dickinson). The concentration of IL-17 in serum was determined by Elisa kit (Dakewe), according to the manufacturer’s instruction.
Quantitative PCR
RNA was extracted from infected livers using the TRI REAGENT Kit per the manufacturer’s protocols (Molecular Research Center, Inc). cDNA was synthesized from 2 μg of total RNA using the Reverse Transcription System (Promega). IL-17 transcript was detected with primers: (sense) 5′-GCT TCA TCT GTG TCT CTG AT-3′ and (antisense) 5′-GGT CTT CAT TGC GGT GGA GA-3′ [28]. The amount of β-actin transcript was determined using primers (5′-GAA GTG TGA CGT TGA CAT CCG TA-3′ and 5′-CTC AGG AGG AGC AAT GAT CTT GA-3′) (Invitrogen). IL-17 transcript level was expressed as a relative copy number normalized to β-actin level.
Th17 differentiation and induction of apoptosis
CD4+ T cells were purified from spleen with anti-CD4 beads (Miltenyi Biotech). Purified CD4+ T cells were stimulated with plastic-bound anti-CD3 and anti-CD28 supplemented with IL-6 (20 ng/ml, Prospec) and human TGF-β (5 ng/ml, R&D system) for 4 days to induce Th17 cells. To analyze antigen-induced apoptosis of Th17 cells, CD4+ T cells from DO11.10 mice were stimulated with 500 ng/ml OVA323-339 peptide plus irradiated splenocytes from BALB/c mice in the presence of IL-6 and human TGF-β for 5 days. IFN-γ (50 ng/ml) or R46A2 (anti-IFN-γ, 10 μg/ml) were supplemented on day 2 as indicated.
Statistical analysis
All experiments were repeated at least three times. Data are expressed as mean ± SE. Statistical analysis was performed by the Wilcoxon signed-ranks test, Student’s t test, or one-way analysis of variance. The data were considered to be significantly different when p < 0.05.
Results
IL-17 is associated with severe disease during acute MHV-A59 infection
To investigate the role of IL-17 in MHV-A59-induced hepatitis, we infected wild-type C57BL/6 mice with 5 × 105 PFU MHV-A59 (Fig. S2), determined the serum cytokine level before infection (day 0) and 3 and 5 days post infection (dpi), and monitored the survival of the infected mice. IL-17 level was increased following MHV-A59 infection. Notably, the level of IL-17 was significantly higher in mice that eventually died than in those that survived (Fig. 1a). In the surviving mice, IL-17 level was 112.3 ± 9.3 ng/ml 3 dpi and decreased slightly 5 dpi. In contrast, IL-17 level was 370.3 ± 30.2 ng/ml 3 dpi and increased to 749.6 ± 49.1 ng/ml 5 dpi in mice that died between days 5 and 10. In parallel, serum ALT levels were also significantly higher in mice that eventually died than in those that survived (Fig. 1b). These results suggest a positive correlation among the serum IL-17 level, liver damage, and poor survival following MHV-A59 infection.

IL-17 is positively associated with liver damage and death post MHV-A59 infection. a Seventeen C57BL/6 mice were i.p. infected with 5 × 105 PFU MHV-A59/mouse. The concentration of IL-17 in serum was determined at indicated time points. Shown are individual and mean values of mice which died between days 5 and 10 (open square) or survived during the experiment period of 15 days (closed square). The serum IL-17 levels are statistically higher in dead than that in survived mice both 3 and 5 dpi. **P < 0.01, *P < 0.05. b The serum ALT activities were determined
MHV-A59 infection induces an exaggerated IL-17 response in IFN-γR−/− mice
Studies have shown that IFN-γ−/− and IFN-γR−/− mice were much more susceptible to MHV-induced hepatitis [9–11]. Consistently, we observed that all IFN-γR−/− mice succumbed to death with 5 × 105 PFU MHV-A59 infection between 5 and 10 dpi while less than half of the wild-type mice died (Fig. S3). The infected IFN-γR−/− mice rapidly lost weight and never recovered, whereas wild-type mice only lost weight during the first few days post infection and recovered eventually (Fig. S3). We analyzed the serum level of IL-17 before the mice were dead. Compared with the wild-type mice, serum IL-17 level was significantly elevated in IFN-γR−/− mice 5 dpi (Fig. 2a). Furthermore, the level of IL-17 transcript in the liver was higher in IFN-γR−/− mice than in wild-type mice (Fig. 2b). These results suggest that, in the absence of the IFN-γ receptor, IL-17 production is exaggerated during MHV-A59 infection.

Exaggerated IL-17 induction in infected IFN-γR−/− mice. a Groups of IFN-γR−/− (open bar) and wild-type mice (closed bar) were i.p. infected with 5 × 105 PFU of MHV-A59/mouse. Serum samples were collected 5 dpi, and the concentration of IL-17 was determined by an Elisa kit. b Livers of IFN-γR−/− (open bar) and wild-type mice (closed bar) were collected at indicated time points. Local IL-17 expression was determined on total RNA by quantitative RT-PCR. Data were expressed as a relative copy number normalized to β-actin content
Neutralization of IL-17 protects IFN-γR−/− mice from death
To evaluate MHV-A59-induced liver damage in IFN-γR−/− mice, we measured the serum ALT after infection. Serum ALT activities were elevated at comparable levels in both wild-type and IFN-γR−/− mice 3 dpi. However, significantly higher serum ALT was observed in IFN-γR−/− mice than wild-type mice 5 dpi, when IL-17 induction was elevated (Fig. 3a). Similarly, histological examination revealed more extensive necrosis in the livers of IFN-γR−/− mice than in that of wild-type mice 5 dpi (Fig. 3b).

IL-17 contributes to severe liver damage in IFNγR−/− mice. a IFN-γR−/− (open bar) and wild-type mice (closed bar) were bled 0, 3, and 5 dpi, and the serum ALT activities was analyzed. b Liver tissues at 5 dpi were harvested. Paraffin-embedded sections were prepared and stained with hematoxylin and eosin. More necrotic area could be seen in the liver of infected IFN-γR−/− mice (as indicated by arrows). Scale bar = 250 μm. c IFNγR−/− (open bar) and wild-type mice (closed bar) were killed 3 or 5 dpi, and virus titers in the liver were determined. d Systemic IL-17 neutralization was performed by i.p. injection of 100 μg/200 ul/mouse anti-IL-17Ab at 1 and 4 dpi. ALT activities in serum of IFN-γR−/− mice with (closed bar) or without IL-17 blockade (open bar) was analyzed at 3 and 5 dpi. e. Survival of infected IFN-γR−/− mice in the absence (open triangle) or presence (closed square) of anti-IL-17 Abs was monitored. These mice were monitored daily until 14 dpi, and there was no change from 8 dpi
Increased virus titer in IFN-γR−/− mice might be responsible for immunopathology, we therefore quantified the virus titers in the livers of infected mice. As expected, IFN-γR−/− mice had higher virus titers than wild-type mice. However, a rapid clearance of the virus was observed in both groups at 5 dpi when severe liver damage occurred in IFN-γR−/− mice, indicating that liver injury was not merely due to virus replication at this time point (Fig. 3c). Additionally, we analyzed the frequencies and numbers of virus-specific CD8+ T cells in the spleen and liver of mice infected by recombinant MHV, RA59/GOS. No significant difference was observed between IFN-γR−/− and wild-type mice, indicating that CD8 T cells are likely capable of clearing the virus in IFN-γR−/− mice (Fig. S4).
To determine the specific contribution of IL-17 to liver pathology in IFN-γR−/− mice, mice were treated with a specific IL-17 neutralizing mAb. Anti-IL-17 treatment significantly decreased the ALT level (Fig. 3d) and delayed MHV-A59-induced death in IFN-γR−/− mice (Fig. 3e). In contrast, IL-17 neutralization affected little on the viral load (data not shown). The results above further show that not the virus itself, but the augmented IL-17 production contributes to MHV-A59-induced liver damage and increased mortality in IFN-γR−/− mice.
IL-17 is responsible for the uncontrolled inflammation in IFN-γR−/− mice
IL-17 is known to be able to elicit the production of other inflammatory cytokines and chemokines such as IL-6, TNF-α and MCP-1 [29, 30]. Therefore we measured these inflammatory mediators in the serum of mice at different time points post MHV-A59 infection. Compared to the wild-type mice, the serum levels of IL-6, TNF-α and MCP-1 were significantly elevated in IFN-γR−/− mice (Fig. 4a). Consistently, TNF-α and IL-6 in the liver were dramatically increased in infected IFN-γR−/− mice especially 5 dpi (Fig. 4b). MCP-1 was higher 3 dpi but lower 5 dpi in liver of IFN-γR−/− mice than wild-type mice. An increased influx of Gr1+ neutrophilic granulocytes was observed in IFN-γR−/− liver 5 dpi (Fig. 4c), which is consistent with the previous report that IL-17 mediates recruitment, activation and proliferation of neutrophils [31]. Importantly, neutralization of IL-17 greatly reduced the level of these inflammatory mediators (Fig. 4d). These results suggest that exacerbated IL-17 production accounts for, at least partly, the uncontrolled inflammation in IFN-γR−/− mice during MHV-A59 infection.

IL-17 is involved in severe inflammation in IFN-γR−/− mice. a IFN-γR−/− (open bar) and wild-type mice (closed bar) were bled 0, 3, and 5 dpi, and levels of cytokines in serum were quantified by CBA mouse inflammation kit (BD). In general, the inflammatory cytokines, including TNF-α, IL-6, and MCP-1, were obviously increased in IFN-γR−/− mice compared with wild-type controls. b Livers of IFN-γR−/− (open bar) and wild-type mice (closed bar) were collected at indicated time points, and levels of cytokines in local livers were quantified by CBA mouse inflammation kit (BD). Consistent with that in serum, the inflammatory cytokines were significantly elevated in the infected livers of IFN-γR−/− mice. c Hepatic lymphocytes were isolated 5 dpi and stained with PE-conjugated Abs against Gr1. The frequencies of Gr1+ cells in liver were measured by FACS. d Systemic IL-17 neutralization was performed in IFN-γR−/− mice as mentioned above. Livers of these mice with (closed bar) or without IL-17 neutralization (open bar) were collected 5 dpi, and levels of local cytokines were quantified by CBA mouse inflammation kit (BD). Systemic treatment with Abs against IL-17 significantly reduced the production of inflammatory cytokines TNF-α, IL-6, and MCP-1
The number of Th17 cells is elevated in IFN-γR−/− mice
To assay Th17 response, we determined the frequencies and numbers of Th17 cells in spleen at different time points post infection by intracellular staining and flow cytometry. The frequency of Th17 cells in the wild-type mice increased following the infection, but the increase was much more dramatic in IFN-γR−/− mice with the highest level at 5 dpi (Fig. 5a; Table 1). Similarly, the frequencies of Th17 cells in the liver were also significantly higher in IFN-γR−/− mice (5.15 ± 0.56%) than in wild-type mice (1.74 ± 0.08%) 5 dpi (Fig. 5b). We also determined the frequencies and numbers of regulatory T cells (Tregs) at different time points post infection. However, no significant difference was observed in splenic Tregs between IFN-γR−/− and wild-type mice (Fig.S5). These results further support an exacerbated Th17 response in the absence of IFN-γR.

Exacerbated Th17 response in IFN-γR−/− mice. a Splenocytes of IFN-γR−/− (open bar) and wild-type mice (closed bar) were isolated at indicated time points post infection. After stimulation with PMA/ionomycin, cells were subjected to intracellular cytokine staining. The frequencies of IL-17-expressing CD4+ T cells in total CD4+ T cells of spleen were determined by FACS. b Hepatic lymphocytes of IFN-γR−/− (open bar) and wild-type mice (closed bar) were isolated at indicated time points post infection. Following the treatment described in Fig. 5a, the frequencies of IL-17-expressing CD4+ T cells in total CD4+ T cells of liver were determined by FACS
IFN-γ induces apoptosis of Th17 cells
To investigate how IFN-γ controlled the virus-induced Th17 response, we determined whether IFN-γ had a direct effect on IL-17 producing CD4+ cells. CD4+ T cells from IFN-γR−/− and wild-type mice were purified and differentiated into Th17 cells by stimulation with anti-CD3 and anti-CD28 in the presence of TGF-β1 and IL-6 for 4 days [32, 33]. Flow cytometry analysis showed that Th17 cells from wild-type mice, but not IFN-γR−/− mice, expressed IFN-γR (Fig. 6a). Because IFN-γ was known to induce apoptosis of activated T cells [3, 4, 34, 35], we analyzed whether IFN-γ might have a direct role in inducing apoptosis of Th17 cells. Thus, OVA-specific Th17 cells were generated from DO11.10 transgenic CD4+ T cells by stimulation with OVA peptide for 5 days. Following OVA stimulation, a large fraction of Th17 cells differentiated and then underwent apoptosis. Addition of IFN-γ increased their apoptosis (Fig. 6b). Neutralization of IFN-γ slightly decreased Th17 cells apoptosis (Fig. 6b). Because little IFN-γ was secreted by Th17 cells, IFN-γ-induced apoptosis of Th17 cells may happen through a paracrine effect. The ex vivo experiment showed that there was more apoptosis of Th17 cells in wild-type mice than that in IFN-γR−/− mice (Fig. 6c). These results suggest that IFN-γ could restrict Th17 cell expansion by inducing their apoptosis, and the dysfunction of this pathway is probably responsible for the exacerbated Th17 induction during MHV-A59 infection in IFN-γR−/− mice.

IFN-γ promotes apoptosis of Th17 cells in vitro and ex vivo. a Sorted CD4+ T cells from normal IFN-γR−/− (right) and wild-type individuals (left) were stimulated with 0.3 μg/ml plastic-bound anti-CD3 and 0.3 μg/ml soluble anti-CD28 in presence of 20 ng/ml recombinant mouse IL-6 and 3 ng/ml human TGF-β. Four days after activation, cells were restimulated with PMA/ionomycin and subjected to intracellular cytokine staining for IL-17. IFN-γR expression on Th17 cells was detected by staining with a PE-labeled Ab against CD119 (black line). A PE-labeled isotype-matched Ab was used as a control (filled). b Sorted CD4+ T cells from Do11.10 mice were stimulated with or without OVA peptide in presence of 20 ng/ml recombinant mouse IL-6 and 3 ng/ml human TGF-β plus recombinant mouse IL-2. 50 ng/ml mouse recombinant IFN-γ or 10 μg/ml neutralizing rat Abs R46A2 (anti-IFN-γ) was applied as indicated 1 day post stimulation. Five days later, cells were restimulated with PMA/ionomycin and subjected to intracellular cytokine staining for IL-17. Apoptosis of Th17 cells was determined by FITC-labeled TUNEL staining. A FITC-labeled isotype-matched Ab was used as a control. Ratios of apoptotic to nonapoptotic Th17 cells were statistically analyzed. c Hepatic lymphocytes were isolated from MHV-A59 infected IFN-γR−/− mice (open bar) and wild-type mice (closed bar) at different time points post infection. After stimulation with PMA/ionomycin, cells were subjected for IL-17 staining and TUNEL assay. Apoptosis of Th17 cells was determined by FITC-labeled TUNEL staining as described above. Ratios of apoptotic Th17 cells were statistically analyzed
Adoptive transfer of IFN-γR−/− CD4+ T cells leads to enhanced immunopathology in wild-type mice
To confirm the hypothesis that the immunopathology in IFN-γR−/− mice was caused by exaggerated Th17 response, IFN-γR−/− and wild-type CD4+ T cells were purified from mice infected with the recombinant MHV-A59 (RA59/GOS), which induced virus specific immune response without causing the death of the mice. Total CD4+ T cells, including the antigen-primed CD4+ T cells, were adoptively transferred into naive wild-type recipients and then challenged with MHV-A59. At 4 dpi, the proportions of Th17 cells in liver were increased in wild-type recipients receiving IFN-γR−/− CD4+ T cells (Fig. 7a). Consistently, wild-type mice given IFN-γR−/− CD4+ T cells had a more visible weight loss and liver damage than mice given wild-type CD4+ T cells (Fig. 7b). In addition, adoptive transfer of IFN-γR−/− CD4+ T cells led to increased production of inflammatory cytokines TNF-α, MCP-1, and IL-6 in the liver (Fig. 7c). These results further support the notion that augmented Th17 response is a major contributor to MHV-induced immunopathology in IFN-γR−/− mice.

Adoptive transfer of IFN-γR−/− CD4+ T cells induces immunopathology and inflammation. a IFN-γR−/− and wild-type mice were i.p. infected with RA59/GOS 5 × 105 PFU/mouse. Twenty days later, the mice were sacrificed and immunized CD4+ T cells were isolated from spleens. Naive wild-type mice received 5 × 106 immunized CD4+ T cells 1 day prior to infection and then challenged with 5 × 105 PFU MHV-A59/mouse. Hepatic lymphocytes from the IFN-γR−/− ≥ WT and WT ≥ WT recipients were isolated 4 dpi. Following stimulation with PMA/ionomycin, cells were subjected to intracellular cytokine staining. The frequencies of IL-17-expressing CD4+ T cells in liver were measured. b Body weight of the IFN-γR−/− ≥ WT (open triangle) and WT ≥ WT mice (closed square) was monitored daily post MHV-A59 infection. ALT activities in serum of the recipients on days 2 and 4 were analyzed. More severe immunopathology and liver disease were observed in IFN-γR−/− ≥ WT mice (open bar) than that in WT ≥ WT ones (closed bar). c Livers of the recipients were collected 4 dpi, and levels of local cytokines were quantified by CBA mouse inflammation kit (BD). The inflammatory cytokines TNF-α, MCP-1, and IL-6 were significantly elevated in the infected livers of IFN-γR−/− ≥ WT mice (open bar) compared with WT ≥ WT ones (closed bar)
Discussion
Fulminant viral hepatitis is a rare but potentially fatal disease in clinic. Mortality without supportive management and/or liver transplantation is over 70%, especially in children. The disease is characterized by massive hepatic necrosis associated with failure of hepatic regeneration. Using the model of MHV-A59-induced fulminant hepatitis in this study, we investigated the involvement and potential function of Th17 cells during MHV infection. Although Th17 cells have been implicated in chronic inflammation, here we show that MHV-A59 triggers Th17 response during acute infection, which is associated with pathogenesis. Without IFN-γR signaling pathway, Th17 cells are exaggerated and their effector cytokine IL-17 stimulated production of proinflammatory mediators, leading to pathogenesis and hepatic injury. In addition to antagonizing Th17 development, we find that IFN-γ also induces apoptosis of Th17 cells both in vitro and ex vivo. Thus, IFN-γ negatively regulates Th17 cells generation and survival during MHV-A59 infection.
HIV-specific Th17 cells are detectable especially in early infection, suggesting that Th17 cells may exert in acute viral infection [36]. In addition, IL-17R signaling modulates the host response to herpes simplex virus corneal infection at early time-points post infection, actively promoting corneal inflammation [37]. Respiratory syncytial virus infection in the absence of STAT1 results in augmented IL-17 levels as well as enhanced airway dysfunction [38]. Mice lacking both of the transcription factors T-bet and eomesodermin develop high levels of IL-17-secreting CD8+ T cells following LCMV infection, leading to a progressive inflammatory and wasting syndrome [39]. Th17 cells also contributed to the liver damage during chronic HBV infection [26]. Therefore, it is conceivable that Th17 cells could cause pathogenesis in addition to the protective property in many viral infections [40]. Consistently, in our study MHV-A59-induced Th17 cells contribute to detrimental inflammation and liver disease, implying the involvement of Th17 cells in fulminant viral hepatitis in clinical settings. In particular, MHV-A59 infection drives exacerbated Th17 response in IFN-γR−/− mice, correlating with prolonged viral persistence. This may be mediated by inhibition of T cell cytotoxicity by IL-17, permitting sustained viral infection, similar to what has been observed in the pathogenesis of Theiler’s murine encephalomyelitis virus-induced demyelinating disease [41].
Against the destructive Th17 response, host may downregulate or even deplete virus-induced Th17 cells with a protective strategy. The induction of type I IFN-s by viral infection may constrain Th17 development [42]. In addition, innate anti-inflammatory cytokines IL-10 and TGFβ elicited by HCV suppress Ag-specific Th17 cells [43]. Notably, immunity to viruses is ascribed to Th1 responses whose production of IFN-γ could antagonize Th17 cells [44, 45]. Extensive studies have indicated the inhibitory effect of IFN-γ on differentiation processes of Th17 cells [46]. Herein, our results demonstrate IFN-γ also affects Th17 apoptosis. It is also observed that Th17 cells are markedly depleted in SIV-infected rhesus macaques [24]. In our study, compared with susceptible IFN-γR−/− mice that display vigorous Th17 induction and severe liver pathology, resistant wild-type mice have limited Th17 response and disease progression. IL-17 neutralization significantly delayed death of IFNγR−/− mice, although it had little effect on the death of wild-type mice. This suggests a stringent restriction of Th17 cells during viral infection in wild-type mice.
In summary, our results show that Th17 cells are involved in acute MHV-A59 infection and negatively regulated by IFN-γ. The enhanced Th17 response in the absence of IFN-γ stimulates inflammation and therefore, severe liver damage. This finding may shed light on the pathogenesis of acute viral infection, especially fulminant viral hepatitis, in clinical settings. The notion that IFN-γ modulates pathogenic Th17 response during viral infection may help to develop new diagnosis and therapy for virus-induced inflammatory diseases.
References
Homberger FR (1997) Enterotropic mouse hepatitis virus. Lab Anim 31:97–115
Lavi E, Gilden DH, Highkin MK, Weiss SR (1986) The organ tropism of mouse hepatitis virus A59 in mice is dependent on dose and route of inoculation. Lab Anim Sci 36:130–135
Liu Y, Janeway CA Jr (1990) Interferon gamma plays a critical role in induced cell death of effector T cell: a possible third mechanism of self-tolerance. J Exp Med 172:1735–1739
Refaeli Y, Van Parijs L, Alexander SI, Abbas AK (2002) Interferon gamma is required for activation-induced death of T lymphocytes. J Exp Med 196:999–1005
Dalton DK, Haynes L, Chu CQ, Swain SL, Wittmer S (2000) Interferon gamma eliminates responding CD4 T cells during mycobacterial infection by inducing apoptosis of activated CD4 T cells. J Exp Med 192:117–122
Badovinac VP, Tvinnereim AR, Harty JT (2000) Regulation of antigen-specific CD8+ T cell homeostasis by perforin and interferon-gamma. Science 290:1354–1358
Henrichsen P, Bartholdy C, Christensen JP, Thomsen AR (2005) Impaired virus control and severe CD8+ T-cell-mediated immunopathology in chimeric mice deficient in gamma interferon receptor expression on both parenchymal and hematopoietic cells. J Virol 79:10073–10076
Chu CQ, Wittmer S, Dalton DK (2000) Failure to suppress the expansion of the activated CD4 T cell population in interferon gamma-deficient mice leads to exacerbation of experimental autoimmune encephalomyelitis. J Exp Med 192:123–128
Kyuwa S, Tagawa Y, Shibata S, Doi K, Machii K, Iwakura Y (1998) Murine coronavirus-induced subacute fatal peritonitis in C57BL/6 mice deficient in gamma interferon. J Virol 72:9286–9290
Schijns VE, Wierda CM, van Hoeij M, Horzinek MC (1996) Exacerbated viral hepatitis in IFN-gamma receptor-deficient mice is not suppressed by IL-12. J Immunol 157:815–821
Kyuwa S, Shibata S, Tagawa Y, Iwakura Y, Machii K, Urano T (2002) Acute hepatic failure in IFN-gamma-deficient BALB/c mice after murine coronavirus infection. Virus Res 83:169–177
Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y (2006) IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol 177:566–573
Pickens SR, Volin MV, Mandelin AM 2nd, Kolls JK, Pope RM, Shahrara S (2010) IL-17 contributes to angiogenesis in rheumatoid arthritis. J Immunol 184:3233–3241. doi:jimmunol.0903271
Wildbaum G, Zohar Y, Karin N (2010) Antigen-specific CD25-Foxp3-IFN-{gamma}highCD4+ T cells restrain the development of experimental allergic encephalomyelitis by suppressing Th17. Am J Pathol. doi:ajpath.2010.090855
Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L (2008) Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol 172:146–155. doi:ajpath.2008.070690
Bai H, Cheng J, Gao X, Joyee AG, Fan Y, Wang S, Jiao L, Yao Z, Yang X (2009) IL-17/Th17 promotes type 1T cell immunity against pulmonary intracellular bacterial infection through modulating dendritic cell function. J Immunol 183:5886–5895. doi:jimmunol.0901584
Sellge G, Magalhaes JG, Konradt C, Fritz JH, Salgado-Pabon W, Eberl G, Bandeira A, Di Santo JP, Sansonetti PJ, Phalipon A (2010) Th17 cells are the dominant T cell subtype primed by Shigella flexneri mediating protective immunity. J Immunol 184:2076–2085. doi:jimmunol.0900978
Riol-Blanco L, Lazarevic V, Awasthi A, Mitsdoerffer M, Wilson BS, Croxford A, Waisman A, Kuchroo VK, Glimcher LH, Oukka M (2010) IL-23 receptor regulates unconventional IL-17-producing T cells that control bacterial infections. J Immunol 184:1710–1720. doi:jimmunol.0902796
Kolls JK, Linden A (2004) Interleukin-17 family members and inflammation. Immunity 21:467–476
Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA (2006) Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 203:2271–2279
Happel KI, Dubin PJ, Zheng M, Ghilardi N, Lockhart C, Quinton LJ, Odden AR, Shellito JE, Bagby GJ, Nelson S, Kolls JK (2005) Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J Exp Med 202:761–769
Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT (2006) Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441:231–234
Huang W, Na L, Fidel PL, Schwarzenberger P (2004) Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis 190:624–631
Raffatellu M, Santos RL, Verhoeven DE, George MD, Wilson RP, Winter SE, Godinez I, Sankaran S, Paixao TA, Gordon MA, Kolls JK, Dandekar S, Baumler AJ (2008) Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes salmonella dissemination from the gut. Nat Med 14:421–428
Yang J, Yang M, Htut TM, Ouyang X, Hanidu A, Li X, Sellati R, Jiang H, Zhang S, Li H, Zhao J, Ting AT, Mayer L, Unkeless JC, Labadia ME, Hodge M, Li J, Xiong H (2008) Epstein–Barr virus-induced gene 3 negatively regulates IL-17, IL-22 and RORgamma t. Eur J Immunol 38:1204–1214
Zhang JY, Zhang Z, Lin F, Zou ZS, Xu RN, Jin L, Fu JL, Shi F, Shi M, Wang HF, Wang FS (2010) Interleukin-17-producing CD4(+) T cells increase with severity of liver damage in patients with chronic hepatitis B. Hepatology 51:81–91. doi:10.1002/hep.23273
Watanabe H, Ohtsuka K, Kimura M, Ikarashi Y, Ohmori K, Kusumi A, Ohteki T, Seki S, Abo T (1992) Details of an isolation method for hepatic lymphocytes in mice. J Immunol Methods 146:145–154
Xiao M, Wang C, Zhang J, Li Z, Zhao X, Qin Z (2009) IFNgamma promotes papilloma development by up-regulating Th17-associated inflammation. Cancer Res 69:2010–2017
Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, Pin JJ, Garrone P, Garcia E, Saeland S, Blanchard D, Gaillard C, Das Mahapatra B, Rouvier E, Golstein P, Banchereau J, Lebecque S (1996) T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med 183:2593–2603
Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, Mineau F, Pelletier JP (1998) IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol 160:3513–3521
Miyamoto M, Prause O, Sjostrand M, Laan M, Lotvall J, Linden A (2003) Endogenous IL-17 as a mediator of neutrophil recruitment caused by endotoxin exposure in mouse airways. J Immunol 170:4665–4672
Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK (2006) Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441:235–238
Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR (2006) The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126:1121–1133
Bach EA, Szabo SJ, Dighe AS, Ashkenazi A, Aguet M, Murphy KM, Schreiber RD (1995) Ligand-induced autoregulation of IFN-gamma receptor beta chain expression in T helper cell subsets. Science 270:1215–1218
Maggi E, Parronchi P, Manetti R, Simonelli C, Piccinni MP, Rugiu FS, De Carli M, Ricci M, Romagnani S (1992) Reciprocal regulatory effects of IFN-gamma and IL-4 on the in vitro development of human Th1 and Th2 clones. J Immunol 148:2142–2147
Yue FY, Merchant A, Kovacs CM, Loutfy M, Persad D, Ostrowski MA (2008) Virus-specific interleukin-17-producing CD4+ T cells are detectable in early human immunodeficiency virus type 1 infection. J Virol 82:6767–6771
Molesworth-Kenyon SJ, Yin R, Oakes JE, Lausch RN (2008) IL-17 receptor signaling influences virus-induced corneal inflammation. J Leukoc Biol 83:401–408
Hashimoto K, Durbin JE, Zhou W, Collins RD, Ho SB, Kolls JK, Dubin PJ, Sheller JR, Goleniewska K, O'Neal JF, Olson SJ, Mitchell D, Graham BS, Peebles RS Jr (2005) Respiratory syncytial virus infection in the absence of STAT 1 results in airway dysfunction, airway mucus, and augmented IL-17 levels. J Allergy Clin Immunol 116:550–557
Intlekofer AM, Banerjee A, Takemoto N, Gordon SM, Dejong CS, Shin H, Hunter CA, Wherry EJ, Lindsten T, Reiner SL (2008) Anomalous type 17 response to viral infection by CD8+ T cells lacking T-bet and eomesodermin. Science 321:408–411
Crowe CR, Chen K, Pociask DA, Alcorn JF, Krivich C, Enelow RI, Ross TM, Witztum JL, Kolls JK (2009) Critical role of IL-17RA in immunopathology of influenza infection. J Immunol 183:5301–5310. doi:jimmunol.0900995
Hou W, Kang HS, Kim BS (2009) Th17 cells enhance viral persistence and inhibit T cell cytotoxicity in a model of chronic virus infection. J Exp Med 206:313–328
Guo B, Chang EY, Cheng G (2008) The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J Clin Invest 118:1680–1690
Rowan AG, Fletcher JM, Ryan EJ, Moran B, Hegarty JE, O'Farrelly C, Mills KH (2008) Hepatitis C virus-specific Th17 cells are suppressed by virus-induced TGF-beta. J Immunol 181:4485–4494
Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT (2005) Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 6:1123–1132
Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C (2005) A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6:1133–1141
Desvignes L, Ernst JD (2009) Interferon-gamma-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity 31:974–985. doi:S1074-7613(09)00509-3
Acknowledgments
We thank Chunchun Liu for flow cytometry analysis and Xueqiang Zhao, Lijie Rong, and Lin Chen for the helpful discussion. This work was supported by Grants 30771972 from the National Natural Science Foundation of China, Grants 2006CB504304, 2006CB910901, 2009CB522505, and 2009CB918901 from the Ministry of Science and Technology of China and by the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Conflicts of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Wei Yang, Xilai Ding, and Jingjing Deng contributed equally to this work.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 975 kb)
Rights and permissions
About this article
Cite this article
Yang, W., Ding, X., Deng, J. et al. Interferon-gamma negatively regulates Th17-mediated immunopathology during mouse hepatitis virus infection. J Mol Med 89, 399–409 (2011). https://doi.org/10.1007/s00109-010-0711-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-010-0711-5