
Abstract
The kidney is sensitive to changes in oxygen delivery. This sensitivity has the merit of facilitating the kidneys in their adjustment of erythropoietin (EPO) production to changes in oxygen supply. The main determinant of EPO synthesis is the transcriptional activity of its gene in kidneys, which is related to local oxygen tensions. Regulation of EPO production is mediated by hypoxia-inducible factor (HIF). When local oxygen tension decreases, accumulated HIF binds to the key sequence of the EPO gene, the hypoxia-responsive element (HRE), and activates transcription of EPO. HIF consists of a constitutive β-subunit and one of alternative oxygen-regulated HIF α-subunits (HIF-1α, HIF-2α, and HIF-3α), and HIF-2α is responsible for erythropoietin production. However, the high sensitivity to changes in oxygen tension also makes the kidney prone to hypoxic injury. Severe energy depletion and subsequent activation of a number of critical alterations in metabolism occurs under hypoxic conditions. Hypoxia is also a profibrogenic stimulus. In addition to ischemic acute renal failure, hypoxia can also play a crucial role in the development of nephrotoxic acute kidney injury, radiocontrast nephropathy, and acute glomerulonephritis. Furthermore, accumulating evidence suggests that chronic hypoxia is a final common pathway to end-stage kidney failure in chronic kidney disease. Given that renal hypoxia has pivotal roles on the development and progression of both acute and chronic kidney disease, hypoxia can be a valid therapeutic target for chronic kidney disease. Activation of HIF leads to expression of a variety of adaptive genes in a coordinated manner. Studies utilizing HIF-stimulating agents proved efficacy in various kidney disease models, suggesting that HIF activation is an ideal target of future therapeutic approaches.
Similar content being viewed by others


Oxygenation of the kidney
Although blood flow to the kidney is high, accounting for 20% of cardiac output, the presence of oxygen shunt diffusion between arterial and venous vessels that run in close parallel contact keeps renal tissue oxygen tensions comparatively low. Most of the blood supply is directed to the renal cortex, and oxygen tension in the renal cortex is around 30 to 50 mmHg, although this decreases dramatically in accordance with changes in renal perfusion. In contrast, oxygen tension in the renal medulla does not rise above 10 to 25 mmHg because of countercurrent oxygen exchange, resulting in borderline chronic oxygen deprivation. Furthermore, the cells in the S3 segment of the proximal tubule and the medullary thick ascending limbs are particularly vulnerable to oxygen deprivation because of the high metabolic activity, principally owing to the activity of the basolateral Na/K-ATPase. As a consequence, the kidney is very sensitive to changes in oxygen delivery. While this sensitivity has the merit of facilitating the kidneys in their adjustment of erythropoietin (EPO) production to changes in oxygen supply, it also renders them prone to hypoxic injury.
Mechanisms of renal erythropoietin production
Anemia is a common complication in patients with chronic kidney disease (CKD). Anemia impairs oxygen delivery to tissues, and the kidney senses hypoxia and stimulates erythropoiesis as a compensatory response. While mechanisms involved in the pathogenesis of renal anemia include chronic inflammation, iron deficiency, and shortened half-life of erythrocytes, the primary cause is insufficient production of EPO [1].
Circulating EPO in normal adults is mainly produced by the peritubular fibroblast-like interstitial cells of the kidney. When hematocrit is in the normal range, the kidney produces a low level of EPO, with EPO expression limited to a small number of these fibroblasts in the deep cortex and superficial outer medulla [2–4]. The increased production of EPO under anemic circumstances involves progressive recruitment of additional interstitial fibroblasts in a pattern that spreads outward from the deep cortex toward the capsule and the inner medulla. However, even at maximal stimulation, less than 20% of cortical fibroblast-like cells express EPO, and thus, it is likely that only a yet-unidentified subgroup of specialized interstitial cells produces the hormone.
The main determinant of EPO synthesis is the transcriptional activity of its gene in kidneys, which is related to local oxygen tensions. EPO production is inversely related to oxygen availability, so that an effective feedback loop is established, which controls erythropoiesis. More than a decade ago, a sequence in the 3′-flanking region of the EPO gene that is required for EPO gene’s upregulation by hypoxia was discovered [5]. Using large quantities of lysates from cultured Chinese hamster ovary cells exposed to a hypoxia mimetic, a heterodimeric complex of proteins that bind to this specific deoxyribonucleic acid (DNA) element to regulate hypoxia gene expression was then identified [6, 7]. The complex was designated as hypoxia-inducible factor (HIF). HIF consists of a constitutive β-subunit and one of alternative oxygen-regulated HIFα subunits (HIF-1α, HIF-2α, and HIF-3α). Under hypoxia, accumulated HIF binds to the key sequence among several regulatory DNA sequences in the neighborhood of the EPO gene, the hypoxia-responsive element (HRE), and activates transcription of EPO.
While most cell types express HIF-1α and HIF-1β, HIF-2α (also called EPAS or HIF-1α-like factor) shows a more restricted pattern of expression [8]. Localization and biological functions of HIF-3α remains relatively unknown, but a splice variant of HIF-3α serves as an inhibitor of the HIF pathway in the cornea [9]. A predominant role of HIF-2α in regulation of EPO expression was confirmed by studies employing ribonucleic acid (RNA) interference to determine the contribution of HIF-1α vs HIF-2α to the hypoxic gene induction [10]. While most genes tested were responsive only to the HIF-1α small interfering RNA, EPO showed responsiveness only to HIF-2α knockdown. High-amplification immunohistochemical analyses revealed the expression of HIF-2 in peritubular fibroblasts [11], which is consistent with its role in EPO regulation. Recent analysis of HIF-2α knockdown mice revealed that EPO gene expression was significantly affected, in parallel with HIF-2α expression [12]. Conditional knockout of HIF-2α after birth demonstrated that HIF-2α plays a critical role in adult erythropoiesis [13]. In addition, studies using mice with conditional knockout of HIF-1α and/or HIF-2α in hepatocytes revealed that the hypoxic induction of liver EPO in anemic mice was also HIF-2α dependent [14].
Furthermore, HIF activation influences genes that play important roles in iron metabolism, including transferrin [15], ferroportin [16], and possibly hepcidin [16]. Thus, in addition to increased EPO expression, HIF regulates various pathways leading to efficient erythropoiesis [16].
Hypoxia in the pathogenesis of kidney disease
Aerobic respiration requires oxygen to generate energy and produces 36–38 mol adenosine triphosphate (ATP) per 1 mol glucose. In contrast, anaerobic respiration can produce only 2 mol ATP out of 1 mol glucose. Therefore, it is no wonder that severe energy depletion and subsequent activation of a number of critical alterations in metabolism occurs under hypoxic conditions [17]. Cytoskeletal disruption leads to derangements and mislocation of various polarized proteins such as integrins and Na/K-ATPase. ATP depletion activates harmful proteases and phospholipases. Induction of adhesion molecules by hypoxia initiates inflammatory reactions.
In a long term, renal tubular cells subjected to hypoxia develop functional deficits in their mitochondria, which aggravate energy deficits, subsequently causing them to undergo apoptosis [18]. Hypoxia is also a profibrogenic stimulus for tubular cells, interstitial fibroblasts, and renal microvascular endothelial cells. Tubular cells under hypoxic conditions undergo epithelial–mesenchymal transdifferentiation to become myofibroblasts [19]. Hypoxia can also activate fibroblasts and change the extracellular matrix metabolism of resident renal cells [20, 21].
pH is another factor that is implicated in cell injury under hypoxia. Under anaerobic conditions, cellular metabolism is shifted to the production of acidic metabolites such as lactate with subsequent development of acidosis. However, it is controversial how acidic pH affects cell status. Regulation of HIF target genes are also inconsistent despite slight induction of HIF protein levels under acidosis, suggesting additional, yet unidentified pH-sensitive regulatory factors [22].
Acute kidney injury
Acute renal failure (ARF) is a clinical syndrome denoted by an abrupt decline in glomerular filtration rate. Impaired renal perfusion with a resultant fall in glomerular capillary filtration pressure is a common cause of ARF and can lead to acute tubular necrosis. Apart from acute ischemia, several distinct renal insults including nephrotoxic injury can also cause acute tubular necrosis [23]. Prerenal azotemia, which is considered a functional response to renal hypoperfusion, and ischemic acute tubular necrosis occur on a continuum of the same pathophysiological process and together account for 75% of the cases of ARF. In these cases, hypoxia because of hypoperfusion of the kidney plays a crucial role in the pathogenesis. While patients with ischemic ARF typically have low systemic perfusion, their blood pressure may remain within the normal range. This type of ischemic ARF is termed “normotensive ischemic ARF” and can occur as a result of various processes [24]. These processes involve increased renal susceptibility to modest reductions in perfusion pressure and include structural changes of renal arterioles because of aging, hypertension, and CKD. Selective renal vasoconstriction plays a crucial role in some types of “normotensive ischemic ARF” including sepsis. Sepsis can act through various vasoconstrictor mediators to increase afferent arteriolar resistance. This, in combination with direct toxic effects of sepsis on the tubules and low perfusion states because of systemically reduced vascular resistance, results in “normotensive ischemic ARF.”
Hypoxia also plays an important role in development of radio-contrast nephropathy. Studies on a multi-insult rat model of ARF combining the application of contrast medium with nitric oxide synthase and cyclooxygenase inhibition showed widespread induction of HIF in the outer and inner medulla that was initiated within 10 min, reached the highest levels at 2 h, and diminished 8 h, to 24 h thereafter [25]. HIF isoforms were expressed in a cell type-specific fashion with HIF-1α in tubular and HIF-2α in interstitial and endothelial cells. The isolated perfused rat kidneys serve as a well-recognized model of hypoxic renal cell injury when perfused with cell-free oxygenated medium. The pattern of both HIF activation and pimonidazole accumulation in the isolated kidneys perfused with cell-free medium was remarkably consistent and in concordance with the findings in the multi-insult rat model of ARF [26].
Various reagents cause nephrotoxic injury to the kidney. Cisplatin is a chemotherapeutic agent widely used for the treatment of solid tumors but often induces nephrotoxic kidney injury. Whether administration of cisplatin in rats also induces HIF-1 in tubular cells apparently depends on the experimental conditions [27].
In addition to ARF, studies on acute glomerulonephritis suggested hypoxia-mediated pathomechanisms. Administration of angiotensin II combined with Habu snake venom induces acute glomerulonephritis in rats, with lesions being restricted to the glomeruli 2 days after the administration of both reagents. Immunohistochemical analysis and Western blot analysis revealed that Habu snake venom caused elevated von Hippel-Lindau tumor suppressor protein (pVHL) expression in the injured glomeruli including endothelial cells and partially podocytes [28]. Administration of thrombin as an inducer of pVHL accelerated the development of glomerulonephritis even 1 day after treatment, demonstrating a protective role of HIF in this model.
Chronic kidney disease
Close pathological analysis shows that functional impairment of the kidney is better correlated with the degree of tubulointerstitial damage than with that of glomerular injury in patients with CKD, and this finding has in turn led to the broad recognition that the final common pathway of kidney failure operates principally in the tubulointerstitium. Accumulating evidence emphasizes chronic hypoxia in the tubulointerstitium as a final common pathway to end-stage kidney injury [29, 30].
A variety of methods have been successfully employed to demonstrate renal hypoxia with CKD. Pimonidazole, which binds to hypoxic cells in vivo, allowed us to demonstrate hypoxia of the kidney in association with reduction in peritubular capillary blood flow at an early stage of a model of progressive glomerulonephritis induced by uninephrectomy and repeated injection of antimesangial Thy1 antibody [31].
Pimonidazole staining also showed the existence of regional hypoxia in polycystic kidney disease (PKD) [32]. HIF-1α and HIF-2α were upregulated in cyst epithelium and cells of cyst walls, respectively, in human PKD and in a rodent PKD model. These results showed that polycystic kidneys are hypoxic and that subsequent activation of HIF may lead to EPO production and pericystic hypervascularity.
Chronic hypoxia of the diabetic kidney was demonstrated by pimonidazole [33], measurement with a microelectrode [34], and blood-oxygen level-dependent magnetic resonance imaging [35]. We established the hypoxia-sensing transgenic rats expressing the HRE-driven luciferase vector tagged with FLAG. With these rats, we observed different patterns of renal hypoxia in rats with puromycin-induced nephritic syndrome and the remnant kidney model [36], as well as in the aging kidney [37].
Hypoxia-inducible transcription factors as a potential therapeutic target for prevention and treatment of kidney disease
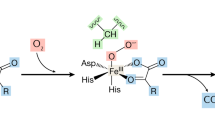
HIF-α mRNA expression is induced by hypoxia or ischemia in vivo to some extent. However, the amount of HIF is regulated mainly by the rate of degradation. The HIF-α subunit interacts with proteins that regulate its degradation, and this interaction is mediated by the pVHL. The interaction between HIFα and pVHL is triggered through post-translational HIFα hydroxylation catalyzed by specific HIF-prolyl hydroxylases (PHDs). The hydroxylated protein is then recognized by pVHL, and is rapidly degraded by the proteasome. Because HIF level is determined by its hydroxylation-induced degradation, hydroxylases involved in this reaction may be good targets for therapy against kidney disease.
The HIF hydroxylases belong to the Fe(II)- and 2-oxoglutarate-dependent dioxygenase superfamily, which is involved in a range of biosynthetic and metabolic pathways [38]. As the PHDs require iron as a cofactor to hydroxylate the critical prolines on HIF-α, the finding that some of the best-established activators of HIF-1 are chelators of iron is reasonable. Recent studies have demonstrated that l-mimosine and ethyl-3,4-dihydroxybenzoate activate the HIF pathway primarily through iron chelation and induce angiogenesis in a sponge model, while other PHD inhibitors can stabilize HIF independent of iron binding [10]. Desferoxamine and cobalt chloride are among the most well-established iron chelators in the activation of HIF. More than half a century ago, oral administration of cobaltous chloride was employed to treat anemia associated with chronic renal disease [39]. Cobalt therapy led to a significant erythropoietic response in association with improved appetite and greater tolerance for medications necessary to correct electrolyte abnormalities.
Success of studies has been reported in various models of kidney diseases as described below. However, while iron chelators may also participate post-translationally in devastating oxidant production via reactions such as Fenton chemistry, iron is a necessary cofactor for a host of important cellular functions, including oxidative phosphorylation and arachidonic acid signaling. Therefore, while these studies clearly provided rationale and promise of HIF-activating therapies in kidney diseases, potential side effects may hinder the therapeutic use of iron chelators. A systematic examination of the relative efficacy of PHD inhibitors that bind iron vs those that do not is needed.
Acute kidney injury
Renoprotective effects of chemical preconditioning with cobaltous chloride were demonstrated in an ischemic model of renal injury [40]. Administration induced upregulation of HIF-regulated genes, such as VEGF and EPO, and subsequently protected the kidney against the tubulointerstitial damage induced by ischemia and reperfusion. Activation of HIF by pretreatment with either carbon monoxide or the novel PHD inhibitor FG-4487 also ameliorated ARF induced by ischemia/reperfusion in rats [41]. Collectively, these data proved that preconditional activation of the HIF system protects the kidney against acute ischemic injury.
Activation of HIF was also effective in nephrotoxic acute kidney injury. With the in vivo administration of cobalt to activate HIF, the number of apoptotic renal tubular cells became much smaller in cisplatin nephropathy [23]. Furthermore, induction of HIF with carbon monoxide before exposure of cisplatin significantly reduced histological renal damage and tubular apoptosis in cisplatin nephropathy [42].
HIF induction therapy with cobalt chloride also ameliorated disease manifestations of an acute rat glomerulonephritis model induced by coadministration of angiotensin II and Habu snake venom, which was associated with the induction of HIF-1α in the glomeruli and renal tubules [43].
Chronic kidney disease
Given that renal hypoxia has pivotal roles on the progression of renal failure, hypoxia can be a valid therapeutic target for CKD. We tested whether cobalt attenuates tubulointerstitial injury secondary to chronic hypoxia, using a model of progressive glomerulonephritis induced by uninephrectomy and repeated injection of antimesangial Thy1 antibodies. Activation of HIF by cobalt exerted renoprotective roles in this model, and cobalt treatment had additive, positive effects to the standard therapy of renal disease namely, the blockade of the renin–angiotensin system in the hypoxic tubulointerstitium [44]. We also treated rats with the remnant kidney model, classically characterized by chronic renal failure with glomerular sclerosis and hypertension. Cobalt treatment mediated improvement in the tubulointerstitial injury as well as preservation of glomerular and peritubular capillary networks with no evidence of vascular leakage [45]. On the other hand, activation of the HIF system in podocytes induced by VHL knockout was recently reported to result activation and proliferation of podocytes and resulted in a pathology resembling rapid progressive glomerulonephritis [46]. Moreover, there is evidence that HIF may also promote renal fibrosis [47] and thus the net balance of beneficial and potential adverse effects of HIF activation may have to be carefully established under conditions of long-term intervention.
Conclusion
The kidney is sensitive to changes in oxygen delivery. While this sensitivity makes the kidney an appropriate organ to produce EPO on demand for oxygen supply, it also renders the kidney prone to hypoxic injury. Hypoxia plays a crucial role in the pathogenesis of both acute and CKD and is a good target of therapeutic approaches. HIF, the master switch of hypoxic adaptation responses, is an apparently ideal target for future therapies aiming at improving oxygen supply and hypoxia tolerance of the kidney. On the other hand, continuous activation of the HIF system in tumors promotes their growth and some other diseases in which hypervascularity plays a devastating role, however, and therapeutic utilization of HIF activation may require the pharmacological optimization.
References
Nangaku M, Eckardt KU (2006) Pathogenesis of renal anemia. Semin Nephrol 26:261–268
Koury ST, Koury MJ, Bondurant MC, Caro J, Graber SE (1989) Quantitation of erythropoietin-producing cells in kidneys of mice by in situ hybridization: Correlation with hematocrit, renal erythropoietin mRNA and serum erythropoietin concentration. Blood 74:645
Bachmann S, Le Hir M, Eckardt KU (1993) Co-localization of erythropoietin mRNA and ecto-5′-nucleotidase immunoreactivity in peritubular cells of rat renal cortex indicates that fibroblasts produce erythropoietin. J Histochem Cytochem 41:335
Maxwell PH, Osmond MK, Pugh CW, Heryet A, Nicholls LG, Tan CC, Doe BG, Ferguson DJ, Johnson MH, Ratcliffe PJ (1993) Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int 44:1149
Semenza GL, Wang GL (1992) A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol 12:5447
Wang GL, Semenza GL (1993) Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J Biol Chem 268:21513
Beck I, Weinmann R, Caro J (1993) Characterization of hypoxia-responsive enhancer in the human erythropoietin gene shows presence of hypoxia-inducible 120-kD nuclear DNA-binding protein in erythropoietin-producing and nonproducing cells. Blood 82:704
Wiesener MS, Jurgensen JS, Rosenberger C, Scholze CK, Horstrup JH, Warnecke C, Mandriota S, Bechmann I, Frei UA, Pugh CW, Ratcliffe PJ, Bachmann S, Maxwell PH, Eckardt KU (2003) Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J 17:271
Makino Y, Cao R, Svensson K, Bertilsson G, Asman M, Tanaka H, Cao Y, Berkenstam A, Poellinger L (2001) Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature 414:550
Warnecke C, Griethe W, Weidemann A, Jurgensen JS, Willam C, Bachmann S, Ivashchenko Y, Wagner I, Frei U, Wiesener M, Eckardt KU (2003) Activation of the hypoxia-inducible factor-pathway and stimulation of angiogenesis by application of prolyl hydroxylase inhibitors. FASEB J 17:1186
Rosenberger C, Mandriota S, Jurgensen JS, Wiesener MS, Horstrup JH, Frei U, Ratcliffe PJ, Maxwell PH, Bachmann S, Eckardt KU (2002) Expression of hypoxia-inducible factor-1alpha and -2alpha in hypoxic and ischemic rat kidneys. J Am Soc Nephrol 13:1721
Morita M, Ohneda O, Yamashita T, Takahashi S, Suzuki N, Nakajima O, Kawauchi S, Ema M, Shibahara S, Udono T, Tomita K, Tamai M, Sogawa K, Yamamoto M, Fujii-Kuriyama Y (2003) HLF/HIF-2alpha is a key factor in retinopathy of prematurity in association with erythropoietin. EMBO J 22:1134
Gruber M, Hu CJ, Johnson RS, Brown EJ, Keith B, Simon MC (2006) Acute postnatal ablation of Hif-2alpha results in anemia. Proc Natl Acad Sci USA 104:2301
Rankin EB, Biju MP, Liu Q, Unger TL, Rha J, Johnson RS, Simon MC, Keith B, Haase VH (2007) Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest 117:1068
Rolfs A, Kvietikova I, Gassmann M, Wenger RH (1997) Oxygen-regulated transferrin expression is mediated by hypoxia-inducible factor-1. J Biol Chem 272:20055
Peyssonnaux C, Zinkernagel AS, Schuepbach RA, Rankin E, Vaulont S, Haase VH, Nizet V, Johnson RS (2007) Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J Clin Invest 117:1926
Devarajan P (2006) Update on mechanisms of ischemic acute kidney injury. J Am Soc Nephrol 17:1503
Tanaka T, Miyata T, Inagi R, Kurokawa K, Adler S, Fujita T, Nangaku M (2003) Hypoxia-induced apoptosis in cultured glomerular endothelial cells: involvement of mitochondrial pathways. Kidney Int 64:2020
Manotham K, Tanaka T, Matsumoto M, Ohse T, Inagi R, Miyata T, Kurokawa K, Fujita T, Ingelfinger JR, Nangaku M (2004) Transdifferentiation of cultured tubular cells induced by hypoxia. Kidney Int 65:871
Norman JT, Clark IM, Garcia PL (2000) Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int 58:2351
Norman JT, Orphanides C, Garcia P, Fine LG (1999) Hypoxia-induced changes in extracellular matrix metabolism in renal cells. Exp Nephrol 7:463
Willam C, Warnecke C, Schefold JC, Kugler J, Koehne P, Frei U, Wiesener M, Eckardt KU (2006) Inconsistent effects of acidosis on HIF-alpha protein and its target genes. Pflugers Arch 451:534
Lameire N, Van Biesen W, Vanholder R (2005) Acute renal failure. Lancet 365:417
Abuelo JG (2007) Normotensive ischemic acute renal failure. N Engl J Med 357:797
Rosenberger C, Heyman SN, Rosen S, Shina A, Goldfarb M, Griethe W, Frei U, Reinke P, Bachmann S, Eckardt KU (2005) Up-regulation of HIF in experimental acute renal failure: evidence for a protective transcriptional response to hypoxia. Kidney Int 67:531
Rosenberger C, Rosen S, Shina A, Bernhardt W, Wiesener MS, Frei U, Eckardt KU, Heyman SN (2006) Hypoxia-inducible factors and tubular cell survival in isolated perfused kidneys. Kidney Int 70:60
Tanaka T, Kojima I, Ohse T, Inagi R, Miyata T, Ingelfinger JR, Fujita T, Nangaku M (2005) Hypoxia-inducible factor modulates tubular cell survival in cisplatin nephrotoxicity. Am J Physiol Renal Physiol 289:F1123
Kudo Y, Kakinuma Y, Iguchi M, Sato T, Sugiura T, Furihata M, Shuin T (2007) Modification in the von Hippel-Lindau protein is involved in the progression of experimentally induced rat glomerulonephritis. Nephron Exp Nephrol 106:e97
Nangaku M (2006) Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal failure. J Am Soc Nephrol 17:17
Eckardt KU, Bernhardt WM, Weidemann A, Warnecke C, Rosenberger C, Wiesener MS, Willam C (2005) Role of hypoxia in the pathogenesis of renal disease. Kidney Int 99(Suppl):S46
Matsumoto M, Tanaka T, Yamamoto T, Noiri E, Miyata T, Inagi R, Fujita T, Nangaku M (2004) Hypoperfusion of peritubular capillaries induced chronic hypoxia prior to progression of tubulointerstitial injury in a progressive model of rat glomerulonephritis. J Am Soc Nephrol 15:1574
Bernhardt WM, Wiesener MS, Weidemann A, Schmitt R, Weichert W, Lechler P, Campean V, Ong AC, Willam C, Gretz N, Eckardt KU (2007) Involvement of hypoxia-inducible transcription factors in polycystic kidney disease. Am J Pathol 170:830
Izuhara Y, Nangaku M, Inagi R, Tominaga N, Aizawa T, Kurokawa K, van Ypersele de Strihou C, Miyata T (2005) Renoprotective properties of angiotensin receptor blockers beyond blood pressure lowering. J Am Soc Nephrol 16:3631
Palm F, Cederberg J, Hansell P, Liss P, Carlsson PO (2003) Reactive oxygen species cause diabetes-induced decrease in renal oxygen tension. Diabetologia 46:1153
Ries M, Basseau F, Tyndal B, Jones R, Deminiere C, Catargi B, Combe C, Moonen CW, Grenier N (2003) Renal diffusion and BOLD MRI in experimental diabetic nephropathy. Blood oxygen level-dependent. J Magn Reson Imaging 17:104
Tanaka T, Miyata T, Inagi R, Fujita T, Nangaku M (2004) Hypoxia in renal disease with proteinuria and/or glomerular hypertension. Am J Pathol 165:1979
Tanaka T, Kato H, Kojima I, Ohse T, Son D, Kawakami T, Yatagawa T, Inagi R, Fujita T, Nangaku M (2006) Hypoxia and expression of hypoxia-inducible factor in the aging kidney. J Gerontol A Biol Sci Med Sci 61A:795
Schofield CJ, Ratcliffe PJ (2004) Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol 5:343
Gardner HF (1953) The use of cobaltous chloride in the anemia associated with chronic renal disease. J Lab Clin Med 41:56
Matsumoto M, Makino Y, Tanaka T, Tanaka H, Ishizaka N, Noiri E, Fujita T, Nangaku M (2003) Induction of renoprotective gene expression by cobalt ameliorates ischemic injury of the kidney in rats. J Am Soc Nephrol 14:1825
Bernhardt WM, Campean V, Kany S, Juergensen J-S, Weidemann A, Warnecke C, Arend M, Klaus S, Gunzler V, Amann K, Willam C, Wiesener MS, Eckardt K-U (2006) Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J Am Soc Nephrol 17:1970
Weidemann A, Bernhardt WM, Klanke B, Daniel C, Buchholz B, Campean V, Amann K, Warnecke C, Wiesener MS, Eckardt KU, Willam C (2007) HIF-activation protects from cisplatin-induced acute toxic kidney injury. J Am Soc Nephrol (in press)
Kudo Y, Kakinuma Y, Mori Y, Morimoto N, Karashima T, Furihata M, Sato T, Shuin T, Sugiura T (2005) Hypoxia-inducible factor-1alpha is involved in the attenuation of experimentally induced rat glomerulonephritis. Nephron Exp Nephrol 100:95
Tanaka T, Matsumoto M, Inagi R, Miyata T, Kojima I, Ohse T, Fujita T, Nangaku M (2005) Induction of protective genes by cobalt ameliorates tubulointerstitial injury in the progressive Thy1 nephritis. Kidney Int 68:2714
Tanaka T, Kojima I, Ohse T, Ingelfinger JR, Adler S, Fujita T, Nangaku M (2005) Cobalt promotes angiogenesis via hypoxia-inducible factor and protects tubulointerstitium in the remnant kidney model. Lab Invest 85:1292
Ding M, Cui S, Li C, Jothy S, Haase V, Steer BM, Marsden PA, Pippin J, Shankland S, Rastaldi MP, Cohen CD, Kretzler M, Quaggin SE (2006) Loss of the tumor suppressor Vhlh leads to upregulation of Cxcr4 and rapidly progressive glomerulonephritis in mice. Nat Med 12:1081
Higgins DF, Biju MP, Akai Y, Wutz A, Johnson RS, Haase VH (2004) Hypoxic induction of Ctgf is directly mediated by Hif-1. Am J Physiol Renal Physiol 287:F1223
Acknowledgement
This work was supported in part by research grants from Grant-in-Aid for Scientific Research from Japan Society for the Promotion of Science (Grants 19390228).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nangaku, M., Eckardt, KU. Hypoxia and the HIF system in kidney disease. J Mol Med 85, 1325–1330 (2007). https://doi.org/10.1007/s00109-007-0278-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-007-0278-y