
Abstract
Fifteen new benzothiophene-based compounds were designed, synthesized, and evaluated as potential anti-Alzheimer agents. Most of the synthesized compounds exhibited remarkable AChE inhibitory activity and effectively inhibited self-mediated β-amyloid protein in vitro. Compound 3g (IC50 = 72.488 ± 3.69 μM) showed a significant β-amyloid inhibitory effect exceeding that of donepezil (IC50 = 87.414 ± 4.46 μM). Furthermore, compound 3j (IC50 = 0.498 ± 0.02 μM) showed the best inhibitory activity comparable to that of donepezil (IC50 = 0.404 ± 0.03 μM). The in vivo evaluation of the promising compounds (3g and 3j) confirmed a significant memory improvement in scopolamine-induced memory impairment model in mice. The molecular docking simulation of compounds 3g and 3j in Torpedo californica-AChE (TcAChE) active site showed a good agreement with the obtained screening results. The in silico ADMET and other physicochemical parameters were also reported.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
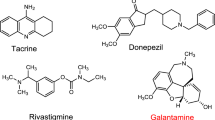
Alzheimer’s disease (AD) is considered as a slow-progressive neurodegenerative illness affecting the elderly people. It starts with a gradual loss of the short-memory and ends with a complete loss of the individual’s identity [1]. Actually, the uncertainty of the exact cause of AD pathogenesis is the main issue of the disease complexity [2]. For this reason, many factors are attributed to its pathogenesis including the deficiency in the levels of the neurotransmitter acetylcholine (ACh), the formation of amyloid-beta (Aβ) peptide plaques, the precipitation of neurofibrillary tangles (NFTs), the disruption of metals homeostasis and over production of reactive oxygen species [3,4,5]. Consequentially, the amyloid hypothesis is based on the abnormal aggregation and deposition of amyloid-β-proteins (AβP) in brain leading to toxic fibrils depositions and consequently neuronal cell death [6, 7]. Another crucial hypothesis is the acetylcholine (ACh) one that suggested the loss of the cholinergic neurons (ACh) and subsequently the reduction in its levels are the most important causative agent of AD [8, 9]. Actually, ACh is an important neurotransmitter in brain for maintaining memory functions which can be terminated by acetylcholinesterase enzymes (AChE) into choline and acetate. Thus, Inhibition of AChE by using acetylcholinesterase inhibitors (AChEIs) results in the accumulation of ACh in synapse, enhancing cholinergic transmission and finally alleviating memory functions in AD patients (Fig. 1) [10, 11]. Actually, AChEIs like tacrine, donepezil, rivastigmine and galantamine were approved in clinics for the symptomatic treatment of AD [12]. Tacrine or 9-amino-1,2,3,4-tetrahydroaminoacridine (THA) was the first FDA-approved AChEI. Unfortunately, it was withdrawn in 2013 due to its hepatotoxicity [13]. Meanwhile, donepezil (Aricept®) is considered as a dual-acting anti Alzheimer medication. It is a very potent and selective AChEI in addition to its positive impact on memory impairment induced by Aβ peptide [14]. The main pharmacophoric features in donepezil structure are highlighted in Fig. 2 [15].

The role of AChEI in the cholinergic neurons

The cardinal pharmacophore features of donepezil
The X-ray crystal structure of donepezil with Torpedo californica-AChE (TcAChE) (PDB Code: IEVE) reveals the dual interaction of donepezil with its two main subsites: the catalytic active site (CAS) and the peripheral anionic site (PAS). In fact, AChEI may inhibit AChE via a competitive mechanism, by interacting with the CAS of the enzyme, via a non-competitive mechanism, by binding PAS, or via both mechanisms, by exerting a dual binding AChE inhibition [16]. In particular, the indanone moiety of donepezil stacks against Trp279 residue in PAS, while the benzyl ring moiety interacts with Trp 84 residue in CAS [17, 18]. Nevertheless, the multifactorial hitting strategy in Alzheimer disease necessitates adapting the multitargeting drug design one [19, 20].
Several heterocyclic systems have been used as a basis to find new ligands with anti-Alzheimer’s potential. Benzothiophene scaffold is one of the privileged structures in drug discovery as this core exhibits various biological activities such as antimicrobial, anticancer, anti-inflammatory, antioxidant, antidiabetic, anticonvulsant agents and many more [21]. Accordingly, this scaffold was used as a building block in many new in anti-Alzheimer leads. Chang et al. developed benzothiophene derivatives and then labeled them with 18F for the potential diagnostic imaging. Compounds I and II (Fig. 3) showed excellent binding affinities for Aβ aggregates and high initial brain uptakes in normal mice [22]. Moreover, Ismail and his team designed and synthesized the tetrahydrothiophene derivatives in the aim of discovering new anti-Alzheimer leads. Compounds III showed 60% AChE inhibition better than donepezil itself (Fig. 3) [23]. Finally, Jeyachandran and co-workers synthesized the dihydrobenzo[b]thiophene-4,6-dicarbonitriles derivative IV which exhibited AChE inhibition with IC50 = 4.16 μM L−1 compared to donepezil 0.12 μM L−1 (Fig. 3) [24].

Reported benzothiophene derivatives with AChE inhibitory activity
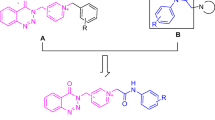
Based on the above-mentioned findings and with the aim of designing and synthesizing new donepezil analogs, we synthesized series 3 and series 4. Accordingly, the indanone aromatic head in donepezil was replaced by either benzothiophene ring or by the rigid benzothienopyimidinone structure including acetamido or propanamido linker. Meanwhile, the terminal benzyl moiety was replaced by a phenyl or by a substituted phenyl moiety (Fig. 4). The new compounds were in vitro evaluated for both their AChE and Aβ aggregation inhibitory activities compared with donepezil as a reference drug and only the most active compounds were further subjected to an in vivo behavioral assessment. The molecular docking studies of the most potent inhibitors within AChE active site were applied to explore their binding mode and justify their high affinity.

The rational of the newly synthesized compounds
Results and discussion
Chemistry
The synthetic pathways employed for the preparation of target benzothiophene derivatives are outlined in Scheme 1. Compounds 1a,b were synthesized according to the reported Gewald’s method [25,26,27]. The reaction of 1a,b with chloroacetylchloride or 2-chloropropionylchloride was carried out by overnight stirring at room temperature to yield the cornerstone intermediates 2a–d. The structures of 2a–d were confirmed by spectral data and elemental analysis. Accordingly, the appearance of an extra carbonyl stretching band at 1680–1638 cm−1 confirms the success of the acylation reaction in the IR spectra. Moreover, both the 1H NMR and 13C NMR spectra revealed a singlet signal of (NH) proton with the accurate integration and D2O-exchangeable behavior in addition to the increase in the number of the aliphatic carbons, respectively. Refluxing compounds 2a–d with different piperazine derivatives for 14 h in dry acetone in the presence of potassium carbonate and potassium iodide afforded series 3a–l. The extra aromatic moiety inserted was like a golden stamp for the success of the reaction. This was detected at the ranges of 3100–3000 cm−1 and 6.74–7.56 ppm in both IR and 1H NMR spectra, sequentially. Moreover, the extra piperazine carbons’ peaks in the range of δ 48.54–49.90 and 49.40–53.40 ppm in 13C NMR spectra were another proof. More specifically, compounds 3c, 3f, 3i, and 3l all exhibited the very significant (singlet) peak at δ 3.69 ppm with the accurate 3 protons integration corresponding to their methoxy group (OCH3) in their 1H NMR spectra. It is worth mentioning that the choice of the reaction solvent and the nature of the acyl derivative were quite crucial for the determination of the final products. The use of the absolute ethanol promotes the occurrence of a self-condensation reaction between both carbonyl and amino groups to yield the cyclized pyrimidinone derivatives 4a–c rather than affording series 3. The IR spectra of compounds 4a–c revealed the presence of only one carbonyl stretching band at 1666, 1670, and 1678 cm−1, sequentially which confirms the success of cyclization process. Unlike the 1H NMR of their respective opened compounds 3a–c, only one singlet signal corresponding to (NH) proton appeared at δ12.00 ppm. Besides, their 13C NMR spectra exhibited only one carbonyl carbon signal at a range of δ 162.96–163.03 ppm and not two signals as in their respective opened compounds 3a–c. On the other hand, applying the same reaction conditions between 2b and different piperazine derivatives in absolute ethanol as a solvent yielded the opened derivatives 3d–f not the cyclized one. The identical spectral data from this reaction with the originally prepared derivatives denied any chance of cyclization. This could clearly be attributed to the steric hindrance effect of the methyl group in the propanamido moiety in 2b.

The synthetic routes for the newly synthesized donepezil analogs
Pharmacology
In vitro AChE inhibition assay
The target compounds were evaluated for their AChE inhibitory activity by applying the improved Ellman’s method [28]. Donepezil was assigned as the reference drug of choice. Table 1 disclose the in-vitro activity results in terms of IC50 ± standard deviation (SD). In the light of our findings, the synthesized compounds showed wide range of AChE inhibition (IC50 = 0.498 ± 0.02–52.462 ± 2.67 μM) compared to donepezil as a reference standard (IC50 = 0.404 ± 0.03 μM) (Fig. 5). The close analysis of the exhibited inhibitory profile revealed that the activity of the benzothiophene derivatives 3a–3l is sharply dependent on the aromatic head group. The carboxamide derivatives 3a–3f showed less AChE inhibitory activity (IC50 range = 6.503 ± 0.34–52.462 ± 2.67 μM) and upon its replacement with the bioisosteric carboxylate ester moiety, compounds 3g–3l, better activity was achieved (IC50 range = 0.498 ± 0.02–44.868 ± 2.29 μM). Regarding the impact of linker on the inhibitory activity of carboxylate derivatives 3g–3l; the propanamido linker in the members 3j–3l showed better inhibitory activity (IC50 range = 0.498 ± 0.02–17.831 ± 0.91 μM) than the acetamido linker as in the members 3g–3i (IC50 range = 4.914 ± 0.25–44.868 ± 2.29 μM). Meanwhile, the impacts of the substituents on the terminal phenyl moiety were also dramatic. The unsubstituted phenylpiperazine analogs 3g (IC50 = 4.914 ± 0.25 μM) and 3j (IC50 = 0.498 ± 0.02 μM) were superior in their inhibitory activity relative to the substituted rings. Comparing the nature of substituents revealed that the electron withdrawing Fluro substituent as in 3h (IC50 = 16.141 ± 0.83 μM) and 3k (IC50 = 8.847 ± 0.44 μM) are better than the electron-donating methoxy substituent as in 3i (IC50 = 44.868 ± 2.29 μM) and 3l (IC50 = 17.831 ± 0.91 μM). Concisely, 3j showed the best inhibitory activity (almost like donepezil). On the other hand, the tricyclic 5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4(1H)-one derivatives 4a–c (IC50 range = 3.422 ± 0.18–25.132 ± 1.27 μM) possessed better activity compared to their corresponding opened analogs 3a–c (IC50 range = 6.503 ± 0.34–52.462 ± 2.67 μM). Among series 4a–c, compound 4a (IC50 = 3.422 ± 0.18 μM) competes in its inhibitory activity with donepezil itself.

Bar chart presentations for the results of both AChE and Aβ1-42 IC50 inhibition values. A AChE IC50 values of the synthesized analogs against donepezil; B Aβ1-42 IC50 values of the synthesized analogs against donepezil. Data are presented as mean ± S.D.
In vitro inhibition of self-mediated Aβ1-42 aggregation
The extracellular aggregation and deposition of Aβ proteins in brain represents the critical cause and distinct marker in AD patients. Moreover, Aβ1-40 and Aβ1-42 are two Aβ isoform but Aβ1-42 is more fibrillogenic and less soluble than Aβ1-40. Therefore, the inhibition of Aβ aggregates is one of the challenging approaches for the treatment of AD [29]. Accordingly, the ability of the target compounds to inhibit the self-mediated Aβ1-42 aggregation was assessed by a thioflavin T (ThT) fluorometric assay using donepezil as a reference drug [30]. The results in terms of IC50 ± standard deviation (SD) were demonstrated in Table 1. Results showed wide range of Aβ inhibition (IC50 = 72.488 ± 3.69–486.764 ± 24.73 μM) compared to donepezil as a reference standard (IC50 = 87.414 ± 4.46 μM) (Fig. 5). Compound 3g showed significant inhibitory effect exceeding that of donepezil with IC50 = 72.488 ± 3.69 μM. Compounds 3c, 3j and 4a showed comparable activity to donepezil with IC50 range = 111.221 ± 5.68–127.741 ± 6.52 μM. On the other hand, compounds 3a, 3b, 3d, 3e, 3f, 3h, 3i, 3k, 3l, 4b and 4c exhibited inhibitory activity of Aβ with IC50 range = 144.757 ± 7.38–486.764 ± 24.73 μM. Briefly, compounds 3g and 4a were the most potent Aβ1-42 inhibitors with IC50 = 72.488 ± 3.69 and 111.221 ± 5.68, respectively.
In vivo behavioral studies on scopolamine-induced dementia model
According to the findings shown in Table 1 both 3g and 3j were the best leads in comparison to donepezil. 3g exceeds donepezil as an anti-amyloidogenic agent while 3j showed a comparable in vitro AChE inhibitory activity to donepezil. Both compounds 3g and 3j were further investigated for their ability to enhance the scopolamine-induced memory impairment in mice. Scopolamine was administrated to induce dementia in the animal model then two behavioral tests were performed naming; Y- maze test [31] and step-through passive avoidance test [32]. Donepezil was used as the positive control. The results of both behavioral tests were exhibited in Fig. 6. In the Y- maze test, the scopolamine model group showed mean % alternations = 45.16 that is lower than the mean % alternations of the control group = 78.57% with P = < 0.0001. Besides, donepezil-treated group showed a significant increase in mean % alternations from 45.16% to 62.63% with P = 0.0072. Furthermore, compound 3g-treated group showed a significant increase in mean % alternations from 45.16% to 86.97% with P = 0.0051 while compound 3j-treated group showed a significant increase in mean % alternations from 45.16% to 61.86% with P = 0.0035. In the step-through passive avoidance test, the scopolamine model group showed a mean transfer latency time (TLT) = 208.4 s shorter than the mean TLT of the control group = 293.4 s with p ≤ 0.0001. In addition, donepezil-treated group showed a significant increase in mean TLT from 208.4 s to 272.0 s with P ≤ 0.0001. Furthermore, compound 3g-treated group showed a significant increase in mean TLT from 208.4 s to 240.8 s with P = 0.0071 while compound 3j-treated group showed a significant increase in mean TLT from 208.4 s to 262.6 s with P = 0.0008. Concisely, the in vivo assessment of both compounds 3g and 3j exhibited a significant memory improvement in scopolamine-induced impairment model.

Bar chart presentations for the results of the most promising compounds 3g and 3j in both Y-maze and passive avoidance tests. A % of spontaneous alternations in the Y-maze test; B The transfer latency time in seconds for step-through passive avoidance test. Data are presented as mean ± S.D. (n = 5; #p < 0.01 vs control group, and *p < 0.01 vs model group)
Structure activity relationship (SAR)
Regarding AChE inhibitory activity, the following SAR can be concluded: 1) The AChE inhibitory activity was better achieved by adopting the carboxylate ester aromatic head instead of the carboxamide one. 2) The propanamido linker was better in its AChE inhibitory activity than that of acetamido one. 3) The unsubstituted phenyl ring is privileged on any substitution. 4) The rigidification model in the pyrimidinone ring with unsubstituted terminal phenyl moiety was quite beneficial for the AChE inhibitory activity. In accordance, compounds 3j and 4a can be considered as leads with good AChE inhibitory profile comparable to that of donepezil. Regarding Aβ1-42 inhibitory effect, we can conclude that the Aβ1-42 inhibitory effect was better achieved by the carboxylate aromatic head with acetamido linker and terminal unsubstituted phenyl ring as in either the free rotating compound 3g or the rigid pyrimidinone ring as in case of compound 4a.
Molecular docking study
In the aim of rationalizing the biological data findings and exploring the possible binding interactions between the ligand drug and the target enzyme active sites, a brief in silico docking study was conducted using Molecular Operating Environment (MOE 10. 2008) software [33]. The X-ray crystallographic structure of Torpedo californica-AChE (TcAChE) complexed with donepezil was obtained from Protein Data Bank (PDB: 1EVE) [34]. The AChE active site consists mainly of three subsites; a peripheral anionic site (PAS) including Trp279, Tyr121, Asp72 and Phe290, a mid-aromatic gorge and a catalytic active site (CAS) composed of Trp84, Glu299 and Tyr334 residues. The Interaction between donepezil with the active site gorge of TcAChE showed that all structural main moieties occupied both CAS and PAS where the benzyl ring of donepezil displayed π- π stacking with Trp84 in CAS while the indanone ring of donepezil displayed π- π stacking with Trp-279 in PAS. Besides, in the middle of the receptor gorge, the charged nitrogen of donepezil piperidine ring showed π-cation interaction with the phenyl ring of Phe-330. Moreover, the carbonyl of the indanone ring formed a water mediated H-bond with Phe-288 [35]. The key interactions reproduced by the co-crystallized ligand donepezil with the AChE active site are showed in Fig. 7.

2D diagram of donepezil in the AChE binding site
Meanwhile, the docking study using donepezil as a reference ligand was performed for the most potent newly synthesized compounds (3g, 3j and 4a). Generally, compounds 3g, 3j and 4a showed a comparable binding pattern in the binding site of TcAChE with a predicted docking energy score of −16.2693 kcal/mol, −16.7301 Kcal/mol and −15.8631 Kcal/mol, respectively in comparison to the native donepezil ligand binding score of −17.2254 Kcal/mol. All compounds (3g, 3j and 4a). were able to occupy both PAS and CAS sites like donepezil where their benzothiophene rings stacked against Trp-279 in PAS while their phenyl rings stacked against Trp84 in CAS. Besides, the docked compound 3g displayed three new water mediated hydrogen bonding interactions as the following: 1) water mediated hydrogen bond between oxygen of acetamido linker and Asp72. 2) Water mediated hydrogen bond between oxygen of acetamido linker moiety and Tyr121. 3) Water mediated hydrogen bond between oxygen of ester moiety and Phe288. The docked compound 3j exhibited new water mediated hydrogen bonding interactions where the Phe288 residue formed a hydrogen bonding interaction with the two oxygens of either acetamido linker moiety or carboxylate ester moiety. These H-bonds may play an important role in stabilization of the ligands inside the receptor site and supported our hypothesis that the incorporation of a carboxylate group to the benzothiophene head may be a promising approach for enhancing the AChE inhibitory activity. Regarding compound 4a, the NH of the rigid pyrimidinone ring formed a water mediated hydrogen bonding interaction with Asp72. The nitrogen in piperazine showed water mediated hydrogen bonding interaction with Tyr121. The docking interactions of the three compounds are illustrated in Fig. 8. Concisely, the most active compounds 3g, 3j and 4a showed a common predicted binding pattern in the AChE binding site with the ligand donepezil in (CAS) and (PAS) subsites. Moreover, the hydrogen bonding interactions with different aromatic residues in the receptor active site enhanced the ligand stabilization and binding which can justify their biological potency.

I. The 2D presentation of the binding interaction between compound 3g, 3j and 4a with the TcAChE active site; II. The 3D representation of the compound 3g, 3j and 4a (violet) with donepezil (gray) in the TcAChE active site (PDB ID: 1EVE)
In silico pharmacokinetics prediction
In silico pharmacokinetics parameters prediction was performed in order to establish a relationship between physicochemical data of the synthesized compounds and their in vivo performance. So, the Pharmacokinetic properties of the most potent compounds (3g and 3j) were calculated using the free accessible ADMET predictor web and The Swiss ADME web platforms compared with donepezil as the standard drug. The results from ADMET predictor web server are outlined in Table 2. Both compounds 3g and 3j exhibited good lipophilicity logP values of 3.81 and 4.19, respectively compared to 4.36 for donepezil. Regarding to the absorption, both 3g and 3j showed moderate water solubility with values of −4.77 and −5.16, respectively compared to −4.49 for donepezil. Moreover, they possess high intestinal absorption with expected high oral bioavailability. The percentages absorbed of both 3g and 3j are 91.68% and 91.67% compared to 94.77% for donepezil. Concerning the distribution, the good values of blood brain barrier (BBB) and CNS permeability assign their efficient ability to reach and bind to their central receptor active site which is quite essential for their biological efficacy. Accordingly, the result showed that both 3g and 3j possess moderate BBB penetration with values of −0.15 and −0.16, respectively compared to 0.42 for donepezil. Besides, the tested compounds possess good CNS permeability values of −2.12 and −2.04 compared to −1.46 for donepezil. Regarding the metabolism and the excretion, both compounds 3g and 3j possess similar behavior compared to that of donepezil. Regarding the toxicity, both 3g and 3j showed oral rat acute toxicity (LD50) values of 2.34 and 2.68, respectively compared to 2.99 for donepezil and showed oral rat chronic toxicity (LOEAL) values of 1.16 and 0.82, respectively compared to 1.51 for donepezil.
In accordance, Swiss ADME web tool simply predicts the drugs bioavailability through the bioavailability radar graph where six physicochemical properties are taken into consideration: lipophilicity (LIPO), size (SIZE), polarity (POLAR), solubility (INSOLU), flexibility (FLEX), and saturation (INSATU). The pink colored zone indicates the suitable physicochemical space for oral bioavailability and in which the radar plot of the drug must fall entirely to be considered bioavailable drug. Both compounds 3g and 3j demonstrated a promising bioavailability radar where each compound radar plot entirely fall in the pink colored zone that indicates a good bioavailability (Fig. 9). On the other hand, the drug-likeness generated in accordance with the major pharmaceutical companies; Lipinski (Pfizer) [36], Veber’s (GSK) [37] and Egan’s (Pharmacia) [38] filters support their promising bioavailability with zero violation.

Oral bioavailability radar plots of the designed ligands (3g and 3j)
Experimental
All reagents and solvents were purchased from commercial suppliers and were used without further purification. All the recorded melting points were taken in an open glass capillary on a Griffin apparatus and the values given were uncorrected. Microanalyses for C, H, and N were carried out at the Regional Center for Mycology and Biotechnology, Faculty of Pharmacy, Al-Azhar University. C, H, and N analysis values were accepted within a range of ±0.4% of theoretical calculated percentages. IR spectra were determined using potassium bromide discs and values were represented in cm−1. IR spectra were recorded on Shimadzu IR 435 spectrophotometer (Shimadzu Corp., Kyoto, Japan), Faculty of Pharmacy, Cairo University. 1H NMR spectra were carried out on Bruker 400 MHz (Bruker Corp., Billerica, MA, USA) spectrophotometer, Faculty of Pharmacy, Cairo University. Chemical shifts were recorded in ppm on δ scale, coupling constants (J) were given in Hz and peak multiplicities are designed as follows: s, singlet; d, doublet, t, triplet; q, quartet; m, multiplet. 13C NMR spectra were carried out on Bruker 100 MHz spectrophotometer, Faculty of Pharmacy, Cairo University. Mass spectrum was carried out on Direct Inlet part to mass analyzer in Thermo Scientific GCMS model ISQ at the Regional Center for Mycology and Biotechnology (RCMB), Faculty Of pharmacy, Al-Azhar University. The reaction progress was monitored by TLC using aluminum sheets precoated with UV fluorescent silica gel (MERCK 60F 254), spots were visualized using UV Lamp. The used eluting system was methanol: ethyl acetate: toluene with the ratio 1:2:3, respectively.
2-Amino-6-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxamide (1a)
1a was prepared according to the reported procedure [25, 26].
Yield: 60% (reported 62%), M.P. 187−189 °C as reported. IR (cm−1): 3402, 3375, 3182, 2947, 1631.
Ethyl 2-amino-6-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxylate (1b)
1b was prepared according to the reported procedure [27].
Yield: 61% (reported 63%), M.P. 112−114 °C as reported. IR (cm−1): 3425, 3313, 2943, 1647.
General procedure for preparation of compounds 2a–d
The appropriate chloroacyl chloride (0.011 mol) was added to a solution of either 2a or 2b (0.01 mol) in glacial acetic acid (20 ml). The reaction mixture was stirred at room temperature overnight then poured on ice/cold water. The formed precipitate was filtered, dried, and crystallized from ethanol.
2-(2-Chloroacetamido)-6-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxamide (2a)
Yield: 65%; M.P. 200–202 °C; IR (cm−1): 3414, 3325, 3263 2947, 2916, 1680, 1643;1H NMR (400 MHz, DMSO-d6) δ 1.04 (d, J = 4.0 Hz, 3H, CH3 at C6), 1.29–1.36 (m, 1H, CH, C5), 1.83–1.85 (m, 2H, CH2, C7), 2.20–2.27 (m, 1H, CH, C6), 2.70–2.71 (m, 1H, CH, C5), 2.74–2.75 (m, 2H, CH2, C4), 4.50 (s, 2H, CO-CH2-Cl), 7.02,7.53 (brs, 2H, NH2, D2O exchangeable), 12.28 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO) δ 21.6, 25.3, 29.1, 31.0, 32.3, 43.0, 117.4, 126.8, 129.2, 141.7, 163.9, 167.6; Anal. Calcd. for C12 H15Cl N2 O2 S (286.78): C, 50.26; H, 5.27; N, 9.77; Found: C, 50.48; H, 5.41; N, 10.03.
2-(2-Chloropropanamido)-6-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxamide (2b)
Yield: 60%; M.P. 184–185 °C; IR (cm−1): 3383, 3329, 3244 2947, 2920, 1680, 1639;1H NMR (400 MHz, DMSO-d6) δ 1.04 (d, J = 8.0 Hz, 3H, CH3 at C6), 1.28–1.39 (m, 1H, CH, C5), 1.65 (d, J = 8.0 Hz, 3H, CH3-CH-Cl), 1.83–186 (m, 2H, CH2, C7), 2.21–2.27 (m, 1H, CH, C6), 2.71–2.72 (m, 1H, CH, C5), 2.73–275 (m, 2H, CH2, C4), 4.97 (q, J = 8.0 Hz, 1H, CH3-CH-Cl), 7.04–7.66 (brs, 2H, 2H, NH2, D2O exchangeable), 12.28 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO) δ 21.6, 22.0, 25.3, 29.2, 31.0, 32.4, 55.1, 117.6, 126.9, 129.3, 141.8, 166.6, 167.6; Anal. Calcd. for C13 H17Cl N2 O2 S (300.80): C, 51.91; H, 5.70; N, 9.31; Found: C, 52.18; H, 5.83; N, 9.55.
Ethyl 2-(2-chloroacetamido)-6-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxylate (2c)
2c was prepared according to the reported procedure [39].
Yield: 70%, M.P. 118–119 °C as reported. IR (cm−1): 3224, 2954, 2924, 1666.
Ethyl 2-(2-chloropropanamido)-6-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxylate (2d)
Yield: 64%; M.P. 78–80 °C; IR (cm−1): 3410, 2989, 2870, 1662; 1H NMR (400 MHz, DMSO-d6) δ 1.02 (d, J = 8.0 Hz, 3H, CH3 at C6), 1.32 (t, J = 8 Hz, 3H, OCH2CH3), 136–1.37 (m, 1H, CH, C5), 1.68 (d, J = 8.0 Hz, 3H, CH3-CH-Cl), 1.81–1.83 (m, 2H, CH2, C7), 2.18–2.25 (m, 1H, CH, C6), 2.62–2.74 (m, 2H, CH2, C4), 2.86–2.90 (m, 1H, CH, C5), 4.31 (q, J = 8.0 Hz, 2H, OCH2CH3), 5.08 (q, J = 8.0 Hz, 1H, CH3-CH-Cl), 11.69 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO) δ 14.6, 21.6, 21.9, 26.0, 29.1, 30.9, 32.2, 55.1, 61.1, 112.8, 127.0, 130.9, 145.7, 165.5, 167.0; Anal. Calcd. for C15 H20 Cl N O3 S (329.84): C, 54.62; H, 6.11; N, 4.25; Found: C, 54.81; H, 6.34; N, 4.51.
General procedure for preparation of compounds 3a–l
A solution of the respected tetrahydrobenzothiophene derivative 2a–d (0.01 mol) in dry acetone (25 ml) was mixed with the appropriate piperazine (0.02 mol) and potassium carbonate (0.01 mol) in the presence of a few speaks of potassium iodide. The mixture was heated under reflux for 14 hours. The separated solid was filtered while hot, dried and crystallized from ethanol.
6-Methyl-2-(2-(4-phenylpiperazin-1-yl)acetamido)-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxamide (3a)
Yield: 70%; M.P. 240–241 °C; IR (cm−1): 3506, 3302, 3155, 3070, 2947, 2827, 1670, 1647; 1H NMR (400 MHz, DMSO-d6) δ 1.04 (d, J = 8.0 Hz, 3H, CH3 at C6), 1.29–1.35 (m, 1H, CH, C5), 1.82–1.85 (m, 2H, CH2, C7), 2.20–2.26 (m, 1H, CH, C6), 2.67 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 2.69–2.70 (m, 1H, CH, C5), 2.71–2.73 (m, 2H, CH2, C4), 3.25–3.27 (m, 6H, 2CH2 piperazine and CO-CH2-N), 6.77–7.25 (m, 5H, aromatic H), 7.55 (brs, 2H, NH2, D2O exchangeable), 12.42 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO) δ 21.7, 25.4, 29.1, 31.1, 32.4, 48.5, 53.2, 60.7, 115.8, 116.5, 119.2, 125.8, 129.0, 129.4, 142.4, 151.4, 167.6, 167.8; MS (m/z, %): 412.47 [M+, 9.60], 175.37 [100.00]. Anal. Calcd. for C22 H28 N4 O2 S (412.55): C, 64.05; H, 6.84; N, 13.58; Found: C, 63.89; H, 7.02; N, 13.82.
2-(2-(4-(4-Fluorophenyl)piperazin-1-yl)acetamido)-6-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxamide (3b)
Yield: 68%; M.P. 244–245 °C; IR (cm−1): 3506, 3305, 3163, 2947, 2823, 1670, 1647; 1H NMR (400 MHz, DMSO-d6) δ 1.03 (d, J = 8.0 Hz, 3H, CH3 at C6), 1.26–1.36 (m, 1H, CH, C5), 1.82–1.84 (m, 2H, CH2, C7), 2.19–2.25 (m, 1H, CH, C6), 2.66 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 2.68–2.70 (m, 1H, CH, C5), 2.72–2.74 (m, 2H, CH2, C4), 3.19–3.22 (m, 4H, 2CH2, piperazine), 3.27 (s, 2H, CO-CH2-N), 6.94–7.08 (m, 4H, aromatic H), 7.56 (brs, 2H, NH2, D2O exchangeable), 12.42 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO) δ 21.7, 25.4, 29.2, 31.1, 32.4, 49.3, 53.2, 60.7, 115.6, 117.4, 125.8, 129.0, 142.5, 148.3, 155.2, 157.6, 167.6, 167.8; MS (m/z, %): 430.72 [M+, 10.96], 43.19 [100.00]. Anal. Calcd. for C22 H27 F N4 O2 S (430.54): C, 61.37; H, 6.32; N, 13.01; Found: C, 61.50; H, 6.49; N, 13.29.
2-(2-(4-(4-Methoxyphenyl)piperazin-1-yl)acetamido)-6-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxamide (3c)
Yield: 67%; M.P. 260–262 °C; IR (cm−1): 3495, 3309, 3147, 2951, 2827, 1678, 1647; 1H NMR (400 MHz, DMSO-d6) δ 1.03 (d, J = 8.0 Hz, 3H, CH3 at C6), 1.30–1.34 (m, 1H, CH, C5), 1.82–1.84 (m, 2H, CH2, C7), 2.19–2.25 (m, 1H, CH, C6), 2.65 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 2.68–2.70 (m, 1H, CH, C5), 2.73–2.74 (m, 2H, CH2, C4), 3.13–3.16 (m, 4H, 2CH2, piperazine), 3.25 (s, 2H, CO-CH2-N), 3.69 (s, 3H, OCH3), 6.82–6.91 (m, 4H, aromatic H), 7.54 (brs, 2H, NH2, D2O exchangeable), 12.39 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO) δ 21.7, 25.4, 29.2, 31.1, 32.4, 49.8, 53.4, 55.7, 60.7, 114.7, 116.5, 117.7, 125.8, 129.0, 142.4, 145.8, 153.2, 167.6, 167.9; MS (m/z, %): 442.67 [M+, 32.36], 45.21 [100.00]. Anal. Calcd. for C23 H30 N4 O3 S (442.57): C, 62.42; H, 6.83; N, 12.66; Found: C, 62.31; H, 6.97; N, 12.88.
6-Methyl-2-(2-(4-phenylpiperazin-1-yl)propanamido)-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxamide (3d)
Yield: 65%; M.P. 235–338 °C; IR (cm−1): 3491, 3414, 3167, 3035 2951, 2831, 1680, 1647; 1H NMR (400 MHz, DMSO-d6) δ 1.03 (d, J = 8.0 Hz, 3H, CH3 at C6), 1.21 (d, J = 8.0 Hz, 3H, CH3-CH-N), 1.31–1.35 (m, 1H, CH, C5), 1.81–1.84 (m, 2H, CH2, C7), 2.19–2.25 (m, 1H, CH, C6), 2.64 (t, J = 8.0 Hz, 4H, 2CH2, piperazine), 2.69–2.70 (m, 1H, CH, C5), 2.72–2.74 (m, 2H, CH2, C4), 3.23–3.27 (m, 4H, 2CH2, piperazine), 3.50 (q, J = 4.0 Hz,1H, CH3-CH-N), 6.76–7.22 (m, 5H, aromatic H), 7.53 (brs, 2H, NH2, D2O exchangeable), 12.51 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO) δ 10.2, 21.7, 25.4, 29.2, 31.1, 32.5, 48.6, 49.4, 62.8, 115.7, 116.4, 119.1, 125.6, 128.9, 129.4, 142.8, 151.5, 167.6, 171.0; MS (m/z, %): 426.14 [M+, 26.51], 41.1 [100.00]. Anal. Calcd. for C23 H30 N4 O2 S (426.21): C, 64.76; H, 7.09; N, 13.13; Found: C, 64.95; H, 7.24; N, 13.41.
2-(2-(4-(4-Fluorophenyl)piperazin-1-yl)propanamido)-6-methyl-4,5,6,7-tetrahydro-1- benzothiophene-3-carboxamide (3e)
Yield: 63%; M.P. 227–229 °C; IR (cm−1): 3491, 3305, 3155, 3051 2920, 2819, 1678, 1647; 1H NMR (400 MHz, DMSO-d6) δ 1.03 (d, J = 8.0 Hz, 3H, CH3 at C6), 1.20 (d, J = 8.0 Hz, 3H, CH3-CH-N), 1.28–1.34 (m, 1H, CH, C5), 1.81–1.83 (m, 2H, CH2, C7), 2.18–2.24 (m, 1H, CH, C6), 2.63 (t, J = 8.0 Hz, 4H, 2CH2, piperazine), 2.67 (m, 1H, CH, C5), 2.72 (m, 2H, CH2, C4), 3.16–3.23 (m, 4H, 2CH2, piperazine), 3.45 (q, J = 8.0 Hz, 1H, CH3-CH-N), 6.93–7.07 (m, 4H, aromatic H), 7.47 (brs, 2H, NH2, D2O exchangeable), 12.48 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO) δ 10.2, 21.7, 25.4, 29.2, 31.1, 32.4, 49.3, 49.5, 62.7, 115.6, 117.4, 125.6, 129.0, 142.7, 148.4, 155.2, 157.5, 167.6, 170.9; MS (m/z, %): 444.73 [M+, 27.58], 207.17 [100.00]. Anal. Calcd. for C23 H29 F N4 O2 S (444.57): C, 62.14; H, 6.58; N, 12.60; Found: C, 61.97; H, 6.69; N, 12.89.
2-(2-(4-(4-Methoxyphenyl)piperazin-1-yl)propanamido)-6-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxamide (3f)
Yield: 66%; M.P. 256–258 °C; IR (cm−1): 3498, 3441, 3414, 3051 2958, 2816, 1681, 1647; 1H NMR (400 MHz, DMSO-d6) δ 1.04 (d, J = 4.0 Hz, 3H, CH3 at C6), 1.21 (d, J = 8.0 Hz, 3H, CH3-CH-N), 1.28–1.34 (m, 1H, CH, C5), 1.81–1.84 (m, 2H, CH2, C7), 2.19–2.25 (m, 1H, CH, C6), 2.63 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 2.67–2.70 (m, 1H, CH, C5), 2.73 (m, 2H, CH2, C4), 3.12–3.21 (m, 4H, 2CH2, piperazine), 3.50 (q, J = 8.0 Hz, 1H, CH3-CH-N), 3.69 (s, 3H, OCH3), 6.74–6.91 (m, 4H, aromatic H), 7.56 (brs, 2H, NH2, D2O exchangeable), 12.46 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO) δ 10.2, 21.7, 25.4, 29.2, 31.1, 32.4, 49.5, 49.9, 55.7, 62.8, 114.7, 116.4, 117.6, 125.6, 129.0, 142.7, 145.9, 153.2, 167.5, 171.0; MS (m/z, %): 456.65 [M+, 15.10], 57.39 [100.00]. Anal. Calcd. for C24 H32 N4 O3 S (456.60): C, 63.13; H, 7.06; N, 12.27; C, 63.40; H, 7.21; N, 12.49.
Ethyl 6-methyl-2-(2-(4-phenylpiperazin-1-yl)acetamido)-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxylate (3g)
Yield: 71%; M.P. 229–230 °C; IR (cm−1): 3332, 3039, 2943, 2831, 1680, 1670; 1H NMR (400 MHz, DMSO-d6) δ 1.02 (d, J = 4.0 Hz, 3H, CH3 at C6), 1.27 (t, J = 8.0 Hz, 3H, OCH2CH3), 1.32–1.36 (m, 1H, CH, C5), 1.81–1.83 (m, 2H, CH2, C7), 2.18–2.24 (m, 1H, CH, C6), 2.57–2.63 (m, 2H, CH2, C4), 2.68 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 2.86–2.90 (m, 1H, CH, C5), 3.25 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 3.36 (s, 2H, CO-CH2-N), 4.24 (q, J = 8.0 Hz, 2H, OCH2CH3), 6.77–6.81 (m, 1H, aromatic H), 6.95–6.97 (m, 2H, aromatic H), 7.21–7.25 (m, 2H, aromatic H), 12.08 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO) δ 14.6, 21.6, 26.1, 29.1, 31.0, 32.3, 48.6, 53.3, 60.7, 60.8, 111.8, 115.8, 119.3, 126.0, 129.4, 130.6, 146.2, 151.3, 165.3, 168.4; MS (m/z, %): 4441.63 [M+, 27.33], 277.28 [100.00]. Anal. Calcd. for C24 H31 N3 O3 S (441.59): C, 65.28; H, 7.08; N, 9.52; Found: C, 65.12; H, 7.30; N, 9.71.
Ethyl 2-(2-(4-(4-fluorophenyl)piperazin-1-yl)acetamido)-6-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxylate (3h)
Yield: 69%; M.P. 221–223 °C; IR (cm−1): 3178, 3039, 2947, 2831, 1670, 1666; 1H NMR (400 MHz, DMSO-d6) δ 1.02 (d, J = 8.0 Hz, 3H, CH3 at C6), 1.27 (t, J = 8.0 Hz, 3H, OCH2CH3), 1.33–1.36 (m, 1H, CH, C5), 1.81–1.83 (m, 2H, CH2, C7), 2.17–2.24 (m, 1H, CH, C6), 2.61–2.62 (m, 2H, CH2, C4), 2.68 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 2.85–2.90 (m, 1H, CH, C5), 3.19 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 3.34 (s, 2H, CO-CH2-N), 4.24 (q, J = 8.0 Hz, 2H, OCH2CH3), 6.98–6.99 (m, 2H, aromatic H), 7.04–7.08 (m, 2H, aromatic H), 12.06 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO) δ 14.6, 21.6, 26.1, 29.1, 31.0, 32.3, 40.4, 49.3, 53.3, 60.7, 60.73, 111.8, 115.7, 117.6, 126.0, 130.6, 138.2, 146.2, 148.2, 165.3, 168.3: MS (m/z, %): 459.37 [M+, 3.66], 193.27 [100.00]. Anal. Calcd. for C24 H30 F N3 O3 S (459.58): C, 62.72; H, 6.58; N, 9.14; Found: C, 62.95; H, 6.71; N, 9.32.
Ethyl 2-(2-(4-(4-methoxyphenyl)piperazin-1-yl)acetamido)-6-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxylate (3i)
Yield: 65%; M.P. 181–183 °C; IR (cm−1): 3217, 3039, 2943, 2835, 1680, 1662; 1H NMR (400 MHz, DMSO-d6) δ 1.02 (d, J = 8.0 Hz, 3H, CH3 at C6), 1.27 (t, J = 8.0 Hz, 3H, OCH2CH3), 1.32–1.36 (m, 1H, CH, C5), 1.81–1.83 (m, 2H, CH2, C7), 2.17–2.24 (m, 1H, CH, C6), 2.58–2.61 (m, 2H, CH2, C4), 2.67 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 2.85–290 (m, 1H, CH, C5), 3.13 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 3.36 (s, 2H, CO-CH2-N), 3.69 (s, 3H, OCH3), 4.25 (q, J = 8.0 Hz, 2H, OCH2CH3), 6.82–6.84 (m, 2H, aromatic H), 6.90–6.92 (m, 2H, aromatic H), 12.05 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO) δ 14.6, 21.6, 26.1, 29.1, 31.0, 32.3, 49.9, 53.4, 55.6, 60.7, 60.8, 111.8, 114.8, 117.8, 126.0, 130.6, 145.7, 146.2, 153.4, 165.2, 168.4; MS (m/z, %): 471.43 [M+, 4.16], 70.20 [100.00]. Anal. Calcd. for C25 H33 N3 O4 S (471.61): C, 63.67; H, 7.05; N, 8.91; Found: C, 63.51; H, 7.23; N, 9.14.
Ethyl 6-methyl-2-(2-(4-phenylpiperazin-1-yl)propanamido)-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxylate (3j)
Yield: 55%; M.P. 187–188 °C; IR (cm−1): 3186, 3051, 2954, 2823, 1597, 1573; 1H NMR (400 MHz, DMSO-d6) δ 1.02 (d, J = 8.0 Hz, 3H, CH3 at C6), 1.22–127 (m, 6H, 2CH3 (OCH2CH3 and CH3-CH-N)), 1.32–1.38 (m, 1H, CH, C5), 1.80–1.83 (m, 2H, CH2, C7), 2.17–2.21 (m, 1H, CH, C6), 2.59–2.61 (m, 2H, CH2, C4), 2.65 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 2.84–2.89 (m, 1H, CH, C5), 3.25 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 3.56 (q, J = 8.0 Hz, 1H, CH3-CH-N), 4.22 (q, J = 8.0 Hz, 2H, OCH2CH3), 6.77–6.80 (m, 1H, aromatic H), 6.94–6.96 (m, 2H, aromatic H), 7.20–7.24 (m, 2H, aromatic H), 12.21 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO) δ 10.3, 14.6, 21.6, 26.1, 29.1, 31.0, 32.3, 48.7, 49.4, 60.6, 63.0, 111.6, 115.8, 119.3, 125.9, 129.4, 130.6, 146.6, 151.4, 165.3, 171.5; MS (m/z, %): 455.06 [M+, 8.77], 84.21 [100.00]. Anal. Calcd. for C25 H33 N3 O3 S (455.61): C, 65.90; H, 7.30; N, 9.22; Found: C, 65.73; H, 7.46; N, 9.49.
Ethyl 2-(2-(4-(4-fluorophenyl)piperazin-1-yl)propanamido)-6-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxylate (3k)
Yield: 50%; M.P. 194–195 °C; IR (cm−1): 3221, 3051, 2947, 2835, 1670, 1666; 1H NMR (400 MHz, DMSO-d6) δ 1.01 (d, J = 4.0 Hz, 3H, CH3 at C6), 1.21–1.27 (m, 6H, 2CH3 (OCH2CH3 and CH3-CH-N)), 1.32–1.34 (m, 1H, CH, C5), 1.80–182 (m, 2H, CH2, C7), 2.16–2.23 (m, 1H, CH, C6), 2.58–2.60 (m, 2H, CH2, C4), 2.64 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 2.84–2.88 (m, 1H, CH, C5), 3.20 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 3.56 (q, J = 8.0 Hz, 1H, CH3-CH-N), 4.22 (q, J = 8.0 Hz, 2H, OCH2CH3), 6.94–6.97 (m, 2H, aromatic H), 7.03–7.08 (m, 2H, aromatic H), 12.20 (s, 1H, NH, D2O exchangeable); 13C NMR (101 MHz, DMSO) δ 10.3, 14.6, 21.6, 26.1, 29.1, 31.0, 32.3, 40.4, 49.4, 49.5, 60.6, 62.9, 111.6, 115.7, 117.5, 125.9, 130.6, 146.6, 148.3, 155.3, 165.3, 171.5; MS (m/z, %): 473.53 [M+, 9.23], 60.19 [100.00]. Anal. Calcd. for C25 H32 F N3 O3 S (473.60): C, 63.40; H, 6.81; N, 8.87; Found: C, 63.26; H, 6.97; N, 9.10.
Ethyl 2-(2-(4-(4-methoxyphenyl)piperazin-1-yl)propanamido)-6-methyl-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxylate (3l)
Yield: 58%; M.P. 179–180 °C; IR (cm−1): 3240, 3043, 2947, 2831, 1681, 1662; 1H NMR (400 MHz, DMSO-d6) δ 1.02 (d, J = 4.0 Hz, 3H, CH3 at C6), 1.22–1.29 (m, 6H, 2CH3 (OCH2CH3 and CH3-CH-N), 1.31–1.36 (m, 1H, CH, C5), 1.81–1.82 (m, 2H, CH2, C7), 2.18–2.24 (m, 1H, CH, C6), 2.65 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 2.68–2.72 (m, 2H, CH2, C4), 2.85–2.90 (m, 1H, CH, C5), 3.14 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 3.56 (q, J = 8.0 Hz, 1H, CH3-CH-N), 3.69 (s, 3H, OCH3), 4.23 (q, J = 8.0 Hz, 2H, OCH2CH3,), 6.82–6.84 (m, 2H, aromatic H), 6.90–6.92 (m, 2H, aromatic H), 12.19 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO) δ 10.4, 14.6, 21.6, 26.1, 29.1, 31.0, 32.3, 49.5, 50.1, 55.6, 60.6, 62.9, 111.6, 114.8, 117.7, 125.9, 130.6, 145.8, 146.6, 153.4, 165.2, 171.6; MS (m/z, %): 485.48 [M+, 12.28], 120.14 [100.00]. Anal. Calcd. for C26 H35 N3 O4 S (485.23): C, 63.40; H, 6.81; N, 8.87; Found: C, 63.67; H, 6.95; N, 9.14.
General procedure for preparation of compounds 4a–c
A solution of 2a (0.01 mol) in absolute ethanol (25 ml) was mixed with the appropriate substituted piperazine (0.02 mol) and potassium carbonate (0.01 mol) in the presence of a few speaks of potassium iodide. The mixture was heated under reflux for 14 h. After cooling down, the separated solid was filtered, dried, and crystallized from ethanol.
7-Methyl-2-((4-phenylpiperazin-1-yl)methyl)-5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4(1H)-one (4a)
Yield: 58%; M.P. 226–228 °C; IR (cm−1): 3444, 3024, 2943, 2881, 1666; 1H NMR (400 MHz, DMSO-d6) δ 1.04 (d, J = 8.0 Hz, 3H, CH3 at C7), 1.35–1.39 (m, 1H, CH, C6), 1.83–1.87 (m, 2H, CH2, C8), 2.30–2.36 (m, 1H, CH, C7), 2.63 (t, J = 4.0 Hz,, 4H, 2CH2, piperazine), 2.74–2.83 (m, 2H, CH2, C5), 3.05–3.09 (m, 1H, CH, C6), 3.14 (t, J = 4.0 Hz,, 4H, 2CH2, piperazine), 3.48 (s, 2H, -HN-CCH2-N), 6.75–6.78 (m, 1H, aromatic H), 6.90–6.92 (m, 2H, aromatic H), 7.18–7.22 (m, 2H, aromatic H), 12.00 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO) δ 21.7, 25.5, 29.2, 30.5, 32.9, 48.6, 52.9, 60.3, 115.9, 119.3, 121.6, 129.4, 130.8, 131.9, 151.4, 154.3, 158.8, 163.0; MS (m/z, %): 394.07 [M+, 6.36], 234.27 [100.00]. Anal. Calcd. for C22 H26 N4 O S (394.53): C, 66.97; H, 6.64; N, 14.20; Found: C, 67.09; H, 6.73; N, 14.46.
2-((4-(4-Fluorophenyl)piperazin-1-yl)methyl)-7-methyl-5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4(1H)-one (4b)
Yield: 55%; M.P. 231–233 °C; IR (cm−1): 3417, 3020, 2947, 2881, 1670; 1H NMR (400 MHz, DMSO-d6) δ 1.04 (d, J = 8.0 Hz, 3H, CH3 at C7), 1.35–1.38 (m, 1H, CH, C6), 1.83–1.86 (m, 2H, CH2, C8), 2.29–2.35 (m, 1H, CH, C7), 2.62 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 2.73–2.83 (m, 2H, CH2, C5), 3.04–3.07 (m, 1H, CH, C6), 3.08 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 3.48 (s, 2H, NH-CCH2-N), 6.91–6.94 (m, 2H, aromatic H), 7.01–7.05 (m, 2H, aromatic H), 12.02 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO) δ 21.7, 25.5, 29.2, 30.4, 32.9, 49.4, 52.9, 60.3, 115.8, 117.7, 121.6, 130.8, 131.9, 148.3, 154.3, 155.3, 158.8, 163.0; MS (m/z, %): 412.44 [M+, 7.37], 162.67 [100.00]. Anal. Calcd. for C22 H25 F N4 O S (412.52): C, 64.05; H, 6.11; N, 13.58; Found: C, 64.21; H, 6.28; N, 13.72.
2-((4-(4-Methoxyphenyl)piperazin-1-yl)methyl)-7-methyl-5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4(1H)-one (4c)
Yield: 55%; M.P. 222–224 °C; IR (cm−1): 3329, 3043, 2939, 2827, 1678; 1H NMR (400 MHz, DMSO-d6) δ 1.05 (d, J = 8.0 Hz, 3H, CH3 at C7), 1.34–1.42 (m, 1H, CH, C6), 1.84–1.87 (m, 2H, CH2, C8), 2.31–2.37 (m, 1H, CH, C7), 2.62 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 2.74–2.81 (m, 2H, CH2, C5), 3.02 (t, J = 4.0 Hz, 4H, 2CH2, piperazine), 3.08–3.09 (m, 1H, CH, C6), 3.48 (s, 2H, NH-CCH2-N), 3.68 (s, 3H, OCH3), 6.79–6.82 (m, 2H, aromatic H), 6.86–6.89 (m, 2H, aromatic H), 12.04 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO) δ 21.7, 25.5, 29.2, 30.5, 32.9, 50.1, 53.0, 55.6, 60.5, 114.7, 117.9, 121.5, 130.8, 131.7, 145.8, 153.4, 154.6, 159.1, 163.0; MS (m/z, %): 424.26 [M+, 20.04], 158.15 [100.00]. Anal. Calcd. for C23 H28 N4 O2 S (424.56): C, 65.07; H, 6.65; N, 13.20; Found: C, 64.88; H, 6.79; N, 13.43.
Biology
In vitro AChE inhibition assay
The acetyl cholinesterase inhibitory potencies of the target compounds were determined invitro based on improved Ellman’s method [28]. This method is relied on the ability of AChE enzyme to hydrolyze acetylthiocholine substrate into thiocholine followed by ongoing reaction of the thiol with 5, 5′-dithiobis-2-nitrobenzoate ion (DTNB) to produce the yellow anion of 5-thio-2-nitro-benzoic acid. The intensity of the yellow color is directly proportional with the enzyme activity. The color intensity was determined at 412 nm using spectrophotometer. This assay was performed using Acetylcholinesterase Inhibitor Screening Kit from Biovision®. The assay’s procedure was performed according to the manufacturer instructions. Donepezil was used as reference drug. Four different concentrations of each compound (0.1, 1.0, 10, 100 μM) were used to determine the concentration that causes 50% inhibition of AChE activity (IC50). The IC50 was calculated by plotting inhibition curve and assessing the influence of four different concentrations. The results are reported as mean ± standard deviation (SD).
In vitro inhibition of self-mediated Aβ1-42 aggregation
In order to test the anti-amyloidogenic activity of the target compounds, our experimental approach relied mainly on in vitro ThT assays [30]. ThT is a histochemical dye that is known to bind to the beta sheets of an aggregated amyloid peptide resulting in an intense fluorescent product. In the presence of an Aβ42 ligand, this reaction is abolished resulting in decrease or total loss of fluorescence. Actually, this assay was performed for all the synthesized compounds at four different concentrations for each compound (0.1, 1.0, 10 and 100 μM) using Beta Amyloid 1–42 (Aβ42) Ligand Screening Assay Kit from Biovision®. The assay was carried according to the manufacturer instructions Donepezil was used as reference drug. The ThT fluorescence was measured at 440 nm (excitation) and 490 nm (emission). The IC50 was calculated and the results are reported as mean ± standard deviation (SD).
In vivo behavioral studies
Healthy adult mice, weighing 25–30 g were supplied from the Laboratory Animal House of VACSERA (Egypt). All animals were maintained in the animal house with well-ventilated cages with free access to standard forage. Moreover, all animals were held in standard environmental regulated conditions: temperature (25 ± 2 °C), relative humidity (60 ± 10%), room air change (12–18 times/h) and light/dark cycle (12/12). Food and water were available ad libitum. The study was carried out in accordance with the APA ethical standards and with the approval of the Ethics Committee for Animal Experimentation at Faculty of Pharmacy, Cairo University. All mice were divided into five groups containing five mice in each: i) control or vehicle (normal saline) group, ii) model or scopolamine group (3.0 mg/kg), iii) scopolamine plus donepezil (2.0 mg/kg), iv) scopolamine plus compound 3g (2.0 mg/kg) and v) scopolamine plus compound 3j (2.0 mg/kg). The route of drug administration was intraperitoneal injection (i.p.) for all the groups. Donepezil and test compounds were administered once daily for 7 seven days to the respective group of animals. Scopolamine-group of animals was given normal saline only. On the seventh day, all animal groups except control group were administrated scopolamine i.p. to induce amnesia 30 min. after test or donepezil administration to the respective group of animals. The different animal groups were adopted for Y-maze and step-through passive avoidance experiments. The experimental results are presented as Mean ± SD. The significance of difference between groups was calculated using unpaired student t-test using GraphPad Prism 8 software [40]. P value less than 0.05 denoted the presence of a statistically significant difference [41]. In the Y- maze test, Y-maze composed of three equally spaced horizontal arms (120°, 45 cm long, 14 cm wide and 16 cm high). Training session of 15 min. was carried out in which animals were placed in Y-maze and allowed to move freely between the arms of the maze. Normal mice will prefer to experience a different arm of the maze than the one they visited. Main study was performed after scopolamine injection where the animal was placed in the middle of the arm to explore all three of those arms. The experiment was performed for 15 min. where the sequence of arm entries and number of arm entries were recorded by camera. Further, consecutive arm choices (ABC, BCA, CAB not BAB) and novel arm entry were considered as the memory improvement. The memory improvement score was calculated using the following equation: % alternation = (No. of alternations/Total arm entries − 2) × 100 [31].
In the step-through passive avoidance test, the test apparatus consisted of two-compartment chambers separated by a sliding door. An illuminated chamber, free from any electric stimuli, was connected by a hole to a large dark chamber with electrifiable grid floor. The mice adopted for two separate trials: a training trial and a test trial. For the training trial, each mouse was first placed in a light chamber and after 10 s, the door between the light/dark compartments was opened so that the mouse could move freely into the dark chamber. Upon entering the dark chamber, the door immediately closed and an electric foot shock (0.5 mA, 24 V, 3 s, once) was delivered through the floor grid. The mice were then returned to its original cage. The time interval expressed in seconds between the placement of the mouse in the light chamber and the entry into the dark chamber was recorded which called TLT. For the test trial, 24 h later, each animal was again placed into the light chamber, and TLT was measured again to indicate the memory improvement [32].
In silico molecular studies
Molecular docking study provides a focused insight about the various ligand interactions with the enzyme’s active site to identify the molecular features responsible for a certain biological activity. Accordingly, molecular docking study was applied for the most potent compounds (3g, 3j and 4a) into TcAChE active site using donepezil as a reference ligand. All the docking studies were performed using Molecular Operating Environment (MOE 10.2008) software [33]. The X-ray crystal structure of donepezil-TcAChE complex (PDB ID: 1EVE) was downloaded from Protein Data Bank (PDB) [34]. First, protons and partial charges were added to the protein structure. To ensure the accuracy of docking study, validation was carried by re-docking the co-crystallized ligand (donepezil) into TcAChE active site. The docking validation results showed a near perfect alignment with the original ligand with rmsd of 0.9052 and with an energy score of −17.2254 kcal/mol. Target compounds were protonated and energy minimized by Merk Molecular Force Field (MMFF94x) to a gradient of 0.05 kcal mol−1Å−1. Docking of the most stable conformers was carried using Triangle Matcher Replacement and London dG scoring function.
In silico pharmacokinetics prediction
Pharmacokinetic properties such as absorption, distribution, metabolism, excretion, and toxicity (ADMET) play a crucial role in the drug development process. Therefore, pharmacokinetic properties’ prediction was conducted and calculated using the free accessible web server pkCSM (http://biosig.unimelb.edu.au/pkcsm/prediction) and using Swiss ADME predictor. The Swiss Institute of Bioinformatics (SIB) invented such a free web tool to predict the pharmacokinetics and the drug likeness for newly investigated leads [42]. Accordingly, this study was applied for the most potent newly synthesized compounds (3g and 3j) compared with donepezil as standard drug. The compounds structures’ smiley were entered then choose the prediction mode to run the prediction study [43].
Conclusion
The new benzothiophenes were evaluated for their in vitro AChE and β-amyloid protein inhibitory activities. The results emitted the light on both 3g and 3j possessing the highest inhibitory effect compared to donepezil. 3g and 3j significantly improved the cognition impairment in scopolamine-induced dementia model in Y maze and step-through passive avoidance tests. From the SAR study point of view the carboxylate fragment to benzothiophene head was superior to the amidic one. The molecular docking investigation revealed that AChE inhibitory activity is enhanced by incorporating a carboxylate fragment to benzothiophene head mainly due to H-bonds formation with different aromatic residues in the receptor active site. Finally, the predicted physicochemical and pharmacokinetic properties revealed that both 3g and 3j possessed a good water solubility, a high human intestinal absorption, a good lipophilicity, and a moderate BBB penetration. 3g and 3j could be considered as promising lead candidates for further optimization and development of new anti-Alzheimer’s agents.
References
Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, et al. Alzheimer’s disease. Lancet. 2021;397:1577–90. https://doi.org/10.1016/S0140-6736(20)32205-4.
Chaves S, Resta S, Rinaldo F, Costa M, Josselin R, Gwizdala K, et al. Hydroxybenzimidazole-Donepezil analogues as multitarget-directed ligands for the treatment of Alzheimer’ s disease. Molecules. 2020;25:985.
Gong CX, Liu F, Iqbal K. Multifactorial hypothesis and multi-targets for Alzheimer’s disease. J Alzheimer’s Dis. 2018;64:S107–17. https://doi.org/10.3233/JAD-179921.
Santos MA, Chand K, Chaves S. Recent progress in repositioning Alzheimer’s disease drugs based on a multitarget strategy. Future Med Chem. 2016;8:2113–42. https://doi.org/10.4155/fmc-2016-0103.
Patil P, Thakur A, Sharma A, Flora SJS. Natural products and their derivatives as multifunctional ligands against Alzheimer’s disease. Drug Dev Res. 2020;81:165–83. https://doi.org/10.1002/ddr.21587.
Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–85.
Pimplikar SW. Reassessing the amyloid cascade hypothesis of Alzheimer’s disease. Int J Biochem Cell Biol. 2009;41:1261–8. https://doi.org/10.1016/j.biocel.2008.12.015.
Bartus RT, Dean RL, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217:408–17. https://doi.org/10.1126/science.7046051.
Derabli C, Boualia I, Abdelwahab AB, Boulcina R, Bensouici C, Kirsch G, et al. A cascade synthesis, in vitro cholinesterases inhibitory activity and docking studies of novel Tacrine-pyranopyrazole derivatives. Bioorg Med Chem Lett. 2018;28:2481–84. https://doi.org/10.1016/j.bmcl.2018.05.063.
Chalupova K, Korabecny J, Bartolini M, Monti B, Lamba D, Caliandro R, et al. Novel tacrine-tryptophan hybrids: Multi-target directed ligands as potential treatment for Alzheimer’s disease. Eur J Med Chem. 2019;168:491–514. https://doi.org/10.1016/j.ejmech.2019.02.021.
Godyń J, Jończyk J, Panek D, Malawska B. Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol Rep. 2016;68:127–38. https://doi.org/10.1016/j.pharep.2015.07.006.
Huang L, Su T, Shan W, Luo Z, Sun Y, He F, et al. Inhibition of cholinesterase activity and amyloid aggregation by berberine-phenyl-benzoheterocyclic and tacrine-phenyl-benzoheterocyclic hybrids. Bioorg Med Chem. 2012;20:3038–48. https://doi.org/10.1016/j.bmc.2012.02.059.
Suwanhom P, Saetang J, Khongkow P, Nualnoi T, Tipmanee V, Lomlim L. Synthesis, biological evaluation, and in silico studies of new acetylcholinesterase inhibitors based on quinoxaline scaffold. Molecules. 2021;26. https://doi.org/10.3390/molecules26164895.
Gomaa AA, Makboul RM, El-Mokhtar MA, Abdel-Rahman EA, Ahmed EA, Nicola MA. Evaluation of the neuroprotective effect of donepezil in type 2 diabetic rats. Fundam Clin Pharmacol. 2021;35:97–12. https://doi.org/10.1111/fcp.12585.
El-Sayed NAE, Farag AES, Ezzat MAF, Akincioglu H, Gülçin İ, Abou-Seri SM. Design, synthesis, in vitro and in vivo evaluation of novel pyrrolizine-based compounds with potential activity as cholinesterase inhibitors and anti-Alzheimer’s agents. Bioorg Chem. 2019;93. https://doi.org/10.1016/j.bioorg.2019.103312.
Francis PT, Palmer AM, Snape M, Wilcock GK, Neurol J. the cholinergic hypothesis of Alzheimer. Neurosurg Psychiatry. 1999;60:137–47.
Piemontese L, Tomás D, Hiremathad A, Capriati V, Candeias E, Cardoso SM, et al. Donepezil structure-based hybrids as potential multifunctional anti-Alzheimer’s drug candidates. J Enzym Inhib Med Chem. 2018;33:1212–24. https://doi.org/10.1080/14756366.2018.1491564.
Costa M, Josselin R, Silva DF, Cardoso SM, May NV, Chaves S, et al. Donepezil-based hybrids as multifunctional anti-Alzheimer’s disease chelating agents: Effect of positional isomerization. J Inorg Biochem. 2020;206:111039.
Arya A, Chahal R, Rao R, Rahman MH, Kaushik D, Akhtar MF, et al. Acetylcholinesterase inhibitory potential of various sesquiterpene analogues for alzheimer’s disease therapy. Biomolecules. 2021;11:1–30. https://doi.org/10.3390/biom11030350.
Sukumaran SD, Nasir SB, Tee JT, Buckle MJC, Othman R, Rahman NA, et al. Analogues of 2′-hydroxychalcone with modified C4-substituents as the inhibitors against human acetylcholinesterase. J Enzym Inhib Med Chem. 2021;36:130–7. https://doi.org/10.1080/14756366.2020.1847100.
Keri RS, Chand K, Budagumpi S, Balappa Somappa S, Patil SA, Nagaraja BM. An overview of benzo[b]thiophene-based medicinal chemistry. Eur J Med Chem. 2017;138:1002–33. https://doi.org/10.1016/j.ejmech.2017.07.038.
Chang YS, Jeong JM, Lee Y, Kima HW, Rai G. Synthesis and evaluation of benzothiophene derivatives as ligands for imaging β-amyloid plaques in Alzheimer’s disease. Nuc Med Bio. 2006;33:811–20.
Ismail MM, Kamel MM, Mohamed LW, Faggal SI, Galal MA. Synthesis and biological evaluation of thiophene derivatives as acetylcholinesterase inhibitors. Molecules. 2012;17:7217–31. https://doi.org/10.3390/molecules17067217.
Jeyachandran V, Kumar RR, Ali MA, Choon TS. A one-pot domino synthesis and discovery of highly functionalized dihydrobenzo[b]thiophenes as AChE inhibitors. Bioorg Med Chem Lett. 2013;23:2101–5. https://doi.org/10.1016/j.bmcl.2013.01.122.
Shynkarenko PE, Vlasov SV, Kovalenko SM, Shishkina SV, Shishkin OV, Chernykh VP. Convenient synthesis of substituted 2-(2-iminocoumarin-3-yl)-thieno[2,3-d] pyrimidin-4-ones. J Heterocycl Chem. 2010;47:800–6. https://doi.org/10.1002/jhet.219.
Kranz M, Wall M, Evans B, Miah A, Ballantine S, Delves C, et al. Identification of PDE4B Over 4D subtype-selective inhibitors revealing an unprecedented binding mode. Bioorg Med Chem. 2009;17:5336–41. https://doi.org/10.1016/j.bmc.2009.03.061.
Romagnoli R, Prencipe F, Oliva P, Cacciari B, Balzarini J, Liekens S, et al. Synthesis and biological evaluation of new antitubulin agents containing 2-(30,40,50-trimethoxyanilino)-3,6-disubstituted-4,5,6,7-tetrahydrothieno[2,3-c]pyridine Scaffold. Molecules. 2020;25. https://doi.org/10.3390/molecules25071690.
Ellman GL, Courtney KD, Andres V, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. https://doi.org/10.1016/0006-2952(61)90145-9.
Marr R, Hafez D. Amyloid beta and Alzheimer’s disease: the role of neprilysin-2 in amyloid-beta clearance. Front Aging Neurosci. 2014;6. https://doi.org/10.3389/fnagi.2014.00187.
Levine H. Thioflavine T interaction with synthetic Alzheimer’s disease @-amyloid peptides: detection of amyloid aggregation in solution. USA: Cambridge University Press; 1993.
Kumar D, Gupta SK, Ganeshpurkar A, Gutti G, Krishnamurthy S, Modi G, et al. Development of Piperazinediones as dual inhibitor for treatment of Alzheimer’s disease. Eur J Med Chem. 2018;150:87–101. https://doi.org/10.1016/j.ejmech.2018.02.078.
Nam SO, Park DH, Lee YH, Ryu JH, Lee YS. Synthesis of aminoalkyl-substituted coumarin derivatives as acetylcholinesterase inhibitors. Bioorg Med Chem. 2014;22:1262–7. https://doi.org/10.1016/j.bmc.2014.01.010.
Molecular Operating Environment (MOE), 2022.02 Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2022.
Kryger G, Silman I, Sussman JL. Structure of acetylcholinesterase complexed with E2020 (Aricept ®): implications for the design of new anti-Alzheimer drugs. 1999. http://biomednet.com/elecref/0969212600700297.
Chaves S, Resta S, Rinaldo F, Costa M, Josselin R, Gwizdala K, et al. Design, synthesis, and in vitro evaluation of hydroxybenzimidazole-donepezil analogues as multitarget-directed ligands for the treatment of Alzheimer’s disease. Molecules. 2020;25. https://doi.org/10.3390/molecules25040985.
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2012;64:4–17. https://doi.org/10.1016/j.addr.2012.09.019.
Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–23. https://doi.org/10.1021/jm020017n.
Egan WJ, Merz KM, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J Med Chem. 2000;43:3867–77. https://doi.org/10.1021/jm000292e.
Perrissin, Duc, Narcisse, et al. 4,5,6,7,-tetrahydrobenzo (b) and 5,6,7,8,-tetrahydro 4H cyclohepta (b) thiophene. Eur J Med Chem. 1980;15:413–8.
GraphPad Prism version 8.00 for Windows, GraphPad Software, La Jolla California USA; 2018. www.graphpad.com.
Wang D, Hu M, Li X, Zhang D, Chen C, Fu J, et al. Design, synthesis, and evaluation of isoflavone analogs as multifunctional agents for the treatment of Alzheimer’s disease. Eur J Med Chem. 2019;168:207–20. https://doi.org/10.1016/j.ejmech.2019.02.053.
Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:1–13. https://doi.org/10.1038/srep42717.
Pires DEV, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58:4066–72. https://doi.org/10.1021/acs.jmedchem.5b00104.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mahmoud, Z., Sayed, H.S., Mohamed, L.W. et al. Development of new donepezil analogs: synthesis, biological screening and in silico study rational. Med Chem Res 31, 1754–1770 (2022). https://doi.org/10.1007/s00044-022-02941-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-022-02941-8