
Abstract
During the last years, the progression to control malaria disease seems to be slowed and WHO (World Health Organization) reported a modeling analysis with the prediction of the increase in malaria morbidity and mortality in sub-Saharan Africa during the COVID-19 pandemic. A rapid way to the discovery of new drugs could be carried out by performing investigations to identify drugs based on repurposing of “old” drugs. The 5-nitrothiazole drug, Nitazoxanide was shown to be active against intestinal protozoa, human helminths, anaerobic bacteria, viruses, etc. In this work, Nitazoxanide and analogs were prepared using two methodologies and evaluated against P. falciparum 3D7. A bithiazole analog, showed attractive inhibitory activity with an EC50 value of 5.9 μM, low propensity to show toxic effect against HepG2 cells at 25 μM, and no cross-resistance with standard antimalarials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Malaria is caused by Plasmodium spp. parasites and transmitted to human by the Anopheles vector mosquito. The WHO 2020 report, estimated 229 million malaria cases in 2019, principally in the sub-Saharan region [1]. During the last years, the progression to control the disease seems to be slowed as the estimated deaths in 2019 were 409,000, compared with 405,000 deaths in 2018 and 585,000 in 2010, with 67% of infants under-five. The COVID-19 pandemic has impacted on the investigation and control of infectious diseases [2,3,4]. As a result of 75% reduction in routine malaria control measures, WHO (World Health Organization) reported a modeling analysis with the prediction of the increase in malaria morbidity and mortality in sub-Saharan Africa during the COVID-19 pandemic [5]. Moreover, the emergence of Anopheles vector resistance to the currently used insecticides and of malaria parasite resistance to the available drugs, including artemisinin and its derivatives [6,7,8], jeopardize malaria control achievements. Thus, there is an urgent need of new and safe drugs to treat this illness.
It is well known that drug discovery and development take about 12–15 years and the process presents a substantial cost. However, an attractive and rapid way to the discovery of new drugs could be carried out by performing investigations to identify drugs to treat rare or common diseases based on repurposing of “old” drugs [9, 10]. Thiazole is an important class of five-membered heterocyclic compounds with diverse biological properties, thus it is found in many drugs such as sulfathiazol (antimicrobial drug), ritonavir (antiretroviral drug), abafungin (antifungal drug), and tiazofurin (antineoplastic drug) [11].
The 5-nitrothiazole drug, nitazoxanide (Ntz, 1 Fig. 1) presents various biological activities. Ntz was first synthesized by Rossignol et al. [12, 13], in the early 1970s as a veterinary anthelminthic agent. Then, this drug was shown to be active against intestinal protozoa, human helminths, and anaerobic bacteria [14,15,16]. In humans, Ntz is rapidly metabolized to tizoxanide (Tiz), which is as effective against these parasites and bacteria as the parent drug (Fig. 1). The initial indication for nitazoxanide was for cryptosporidiosis and giardiasis, but subsequent investigations have demonstrated that the drug is effective for amoebiasis [17]. In addition, Ntz is active against nonprotozoan parasites, including the intestinal tapeworm Hymenolepis nana [18], and Trichuris trichiura [19]. Moreover, many researchers concluded that it presents antiviral activity against enteric viruses, [20,21,22] MERS virus [23], and that Ntz and analogs are promising compounds to treat hepatitis C infection [24,25,26]. Furthermore, several investigations were focused on the mechanisms of action [27,28,29,30].

Antiplasmodial heterocycles: nitazoxanide (1), tizoxanide (2), thiazole derived amino acids (3), and aminomethylthiazole carboxamide derivatives (4)
During the last year, vast investigations to find drugs repositioning efficient against SARS-CoV2 were performed. As part of these studies, Ntz has demonstrated in vitro antiviral activity against SARS-CoV2 [30, 31]. In addition, it was suggested that this drug amplifies host innate immune antiviral responses by triggering foreign cytoplasmic RNA sensing and the type 1 interferon axis [32].
Navarrete-Vazquez et al. [33] synthesized two benzologues of Ntz and Tiz and compared their in vitro antiparasitic activities against Giardia intestinalis, Entamoeba histolytica, Trichomonas vaginalis, Trypanosoma cruzi, Leishmania mexicana and the rodent malaria parasite Plasmodium berghei. Ntz and Tiz showed IC50 values in the low micromolar range against P. berghei, (IC50s of 3.9 μM and 5.2 μM, respectively). Furthermore, in a recent review, Sharma and Prashe highlighted several thiazole derived amino acids (3) [34], and aminomethylthiazole carboxamide derivatives (4) (Fig. 1) [35], as promising antimalarials with EC50 values in the low micromolar or submicromolar range, respectively [36]. Considering these results, we decided to explore the “old” and versatile drug Ntz, its metabolite Tiz and bis-heterocycles analogs as part of a research program for the synthesis of potential antimalarials [37,38,39,40,41,42,43,44,45,46,47].
In this work, we present the synthesis and the antiplasmodial activity of Ntz, Tiz, and 2-aminothiazole analogs (Fig. 2). These compounds and six 2-aminothiazole-4-carboylate derivatives, previously obtained by us [41, 45, 47], were evaluated against P. falciparum 3D7. Compounds of A series do not present the nitro substituent in the thiazole ring in order to study the influence of that group in the antimalarial activity.

Structures of Ntz analogs of Series A and B
Results and discussion
Analogs synthesis
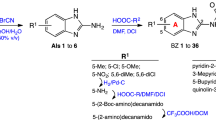
The synthesis was started with thiazole preparation from thiourea and bromoacetaldehyde diethyl acetal employing Hanstzch reaction to obtain compound 5, Scheme 1 [48]. Different ratios of EtOH and AcOH were assayed and the best yield was obtained with AcOH 100%. Nitration using Dickey et al. methodology [49], resulted in compound 6 in good yield.

Synthesis of Ntz and analogs of type A and B
Aware of the low nucleophilicity of the aromatic amine of 2-aminothiazole [41, 47], and consequently, of the low or moderate yield for preparation of the corresponding amide, we paid special attention to the reported methodologies capable of performing this reaction.
Several groups reported the synthesis of Ntz analogs using acyl chloride derivatives; in other cases EDCI and HOBt or aminocarbonylation followed by iodobenzene substituted reaction were used. All of the methods reported moderate to good yields [50,51,52,53]. Recently, our group reported the successful use of oxyma (ethylcianoacetate oxime), in combination with DIC for solid phase peptide synthesis (SPPS) or with EDCI for solution macrocylization [54]. Moreover, Albericio et al. described the impressive coupling efficiency of this coupling agent surpassing HOAt in more demanding peptide models [55]. In addition, the in situ acyl chloride formation using triphosgene is a simple and effective methodology that allowed amide bond formation using aromatic amines [54]. Taking into account these previous results, we decided to explore the use of oxyma/EDCI or triphosgene to obtain amides from 2-aminothiazole compounds 5 or 6, Scheme 1. The thiazole and pyrrole carboxylic acids used to obtain 7, 10, 11, and 13 were prepared as was described by our group [41].
Compounds 7 and 9 of A series, Scheme 1, were obtained using oxyma/EDCI in low or moderate yield, respectively. As analog 8 was obtained in a higher yield using triphosgene methodology, we explored this methodology for the synthesis of 7 and 9. Thus, using triphosgene, the yields for these amides derivatives were improved, from 7 to 63% for 7 and from 34 to 60% for 9. In the case of compounds of B series, Scheme 1, the use of oxyme allowed to obtain 10 and 12 in good (41%) and very good yield (68%), respectively, considering that 12 was obtained by Ballard and co-workers [53], using 4-chlorobenzoyl chloride in 57%. It is important to note that in our case the reaction mixture of 12 was stirred for 4 h at reflux and then at room temperature for 24 h.
Amides 11 and 1 were prepared by both strategies, (i) and (ii) Scheme 1. In the case of 11, the results were similar, but Ntz (1) was obtained only in traces from the reaction mixture using oxyma/EDCI. Nevertheless, the corresponding activated acetylsalycilic acid with oxyma was isolated and its structure was confirmed by NMR and MS analysis. In contrast, using triphosgene Ntz was obtained in 54% yield. Taking into account these results, the strategy (ii) was selected to obtain compound 13 in 40% yield based on the recovered starting material.
In order to obtain the Ntz metabolite, (2, Tiz), first, we tried hydrolysis using basic conditions, 20% NaOH/EtOH (1:1) or 20% LiOH/ EtOH (1:1), but instead of the desired compound, salicylic acid and acetylsalicylic acid were isolated from the reaction mixture. These results allowed us to conclude that in those conditions, the amide bond of 1 is labile. To overcome this difficulty, we intended acid conditions to hydrolyze 1. Fortunately, the use of HCl 36% at 50 °C during 6 h rendered Tiz (2) in 70% yield, Scheme 2. In the case of the Ntz analog (8), which does not present a nitro group, the hydrolysis was performed using basic conditions to obtain 14 in excellent yield (94%).

Hydrolysis of the ester group of Ntz (1) and analog 8
In addition, two derivatives of 7 and 11 were prepared by deprotection of Boc with HCl(g)/Dioxane to obtain 15 and 16, Scheme 3.

Deprotection of compounds 7 and 11
Antimalarial evaluation
The synthesized compounds and six analogs previously obtained by our group (17–22, Table 1) [41, 45, 47] were assayed at 10 μM for inhibitory activity against the chloroquine-sensitive 3D7 strain of P. falciparum using the SYBR Green assay [56]. The inhibitory activities in Table 1 are the average values determined from two independent experiments.
The amides of 2-aminothiazole and the metabolite Tiz (2) showed poor inhibitory activity at 10 μM (% inhibition <5%). The same results were obtained for the derivatives of 2-aminothiazole-4-carboxylate, compounds 17, 19–22. However, some 5-nitro-2-aminothiazole derivatives (10–13) and compound 18 containing 1-methyl-4-nitropyrrole showed moderate growth inhibition of P. falciparum 3D7 at 10 μM (% inhibition >27%), suggesting that the presence of the NO2 substituent was favorable for inhibitory activity. Ntz (1) exhibited moderate activity against P. falciparum at 10 μM (% inhibition = 30%), whereas Tiz (2) was a poor inhibitor of P. falciparum 3D7 strain (% inhibition = 4%). By contrast, Ntz and Tiz showed IC50 values in the low micromolar range against P. berghei (3.9 and 5.2 μM, respectively) [33]. The parasites’ susceptibility to Ntz and Tiz might be related to the genomic and proteomic differences between the P. falciparum (human malaria parasite) [57], and P. berghei (rodent parasite) [58], species. Indeed, a comparative analysis of the complete proteomes of primate- and rodent-Plasmodium species, including P. falciparum and P. berghei, identified 30 proteins well conserved in the primate parasites that are putatively absent in rodent parasites [59].
The Boc carbamate 11 and pivalic amide 13, exhibited greater inhibitory activity, 88% and 85% at 10 μM, respectively. In contrast, the amine derivative 16 showed decreased effect against P. falciparum 3D7 strain. Compounds 11 and 13 were tested in twofold serial dilutions, in two independent experiments. Table 2 displays the assessed EC50 values for 11, 13, and artesunate, tested in parallel as a positive control. As both compounds 11 and 13 showed EC50 values in the low micromolar range, 5.9 and 8 μM, respectively, they were submitted to cytotoxic assessment against HepG2 cells (hepatocyte carcinoma) and the selectivity indexes (SI) were calculated. In the case of 11, the cytotoxicity determination was limited by the solubility of the compound under the assay conditions (25 μM). By contrast, compound 13 had the cytotoxic effect assessed (IC50 = 81 µM), thereby allowing the determination of the SI. The SI value of 10 indicates a reasonable selectivity for the parasite cells (Table 2).
To further investigate the antiplasmodial activity of the nitazoxanide series, the cross-resistance profile of the representative nitazoxanide analog 11 was assessed against a panel of drug-resistant parasites (Table 3). The panel consisted of representative resistant strains including Dd2, TM90C6B, and 3D7R_848. Dd2 is a chloroquine-, cycloguanil-, and pyrimethamine-resistant strain, while the TM90C6B strain shows resistance against atovaquone. The 3D7R_848 strain was generated in our laboratory and is resistant to MMV692848, a potent PfPI4K inhibitor. To verify the resistant profile of the selected strains, standard antimalarial drugs (artesunate, pyrimethamine, and atovaquone) and MMV692848 were assessed and used as positive controls for inhibition. The pyrimethamine, atovaquone, and MMV692848 showed greater EC50 values against the Dd2, TM90C6B, and 3D7R_848 strains with resistance indexes (RI) of >222, >1250, and 20, respectively (Table 3). By contrast, the strains in the panel were sensitive to artesunate (EC50s = 7–12 nM), confirming their susceptibility to inhibition by small molecules.
The inhibitory activity assessment of 11 against Dd2 and TM90C6B strains indicated that the EC50 values were three and fourfold greater, respectively than the EC50 values against 3D7 sensitive strain, while the EC50 value against 3D7R_848 strain was similar to the EC50 value against the sensitive strain (Fig. 3). These findings indicate that 11 has comparable potency against the resistant strains in the panel. Moreover, the RI values varied from 0.9 to 3.9 (Table 3), which were considerably smaller than the RI values observed for the standard antimalarials against the resistant strains (RI = 20 – >1250). These findings suggest no cross-resistance between the bis thiazole derivative (11) and the antimalarials pyrimethamine, atovaquone, and MMV692848.

Half-maximal inhibitory concentration of 11 against sensitive (3D7) and resistant (Dd2, TM90C6B and 3D7R_848) strains, normalized by the IC50 value against the sensitive strain (*P < 0.05)
The main molecular target of nitazoxanide in protozoa and anaerobic bacteria is pyruvate:ferredoxin oxidoreductase (PFOR), a biomolecule involved in the enzyme-dependent electron transfer reaction and essential to the anaerobic energy metabolism, especially, in anaerobic protozoan parasites [60, 61]. Nevertheless, to the best of our knowledge, no genes encoding PFOR have been isolated from P. falciparum, suggesting that the antiplasmodial activity of the nitazoxanide derivatives is via other mechanism of action. In line with this, organisms that lack the PFOR and resistant to metronidazole (a nitroimidazole derivative that targets PFOR catalytic reaction) are sensitive to nitazoxanide and nitazoxanide derivatives [62, 63]. Furthermore, compound 11 showed comparable potencies against the sensitive and resistant strains of the parasite, indicating that the mechanism of action underlying the antiplasmodial activity is not related to the inhibition of the dihydrofolate reductase (DHFR), cytochrome bc1 complex (cyt bc1), and phosphatidylinositol 4-OH kinase (PI4K) enzymes. Therefore, the mechanism of action of 11 against P. falciparum is unknown and shall be investigated.
Conclusion
Two methodologies were studied to prepare NTz and analogs. The use of triphosgene rendered higher yield than the use of oxyma to obtain the desired amides. All the compounds of A series and all the amide derivatives of 2-aminothiazole-4-carboxylate, which not present nitro group showed poor inhibitory activity against P. falciparum (3D7 strain) at 10 μM. Ntz showed moderate inhibition with 30% of growth reduction of parasite at 10 μM and Tiz was a poor inhibitor of P. falciparum 3D7 strain. As the previous evaluation of Ntz and Tiz, carried out by Navarrete-Vazquez et al. [33], was performed against schizonts of the rodent P. berghei, our investigations unveiled weak activity for Ntz against the human parasite P. falciparum. However, the bis thiazole analogs (11 and 13), showed attractive inhibitory activity with EC50 values of 5.9 μM and 8 μM, and low propensity to show toxic effect against HepG2 cells. Moreover, the representative compound of the series (11) showed no cross-resistance against a panel of sensitive and resistant parasites. We concluded that the nitro group of 11 and 13 play an important role for their bioactivity against the human malaria parasite. Taking into account that the amine derivative 16 is a poor inhibitor, it could be concluded that the presence of an amide or carbamate in the second thiazole ring, derived from 2-aminothiazole-4-carboxylate, improve the antimalarial activity.
Experimental
Most of the reagents were purchased from commercial suppliers and used without further purification, some of them were synthetized by part of the group.
1H and 13C NMR spectra were recorded on Brucker Avance Neo 400 MHz spectrometer using the indicated deuterated solvents and TMS as an internal standard. Chemical shifts (δ) are given in parts per millon (ppm), multiplicities according to: singlet (s), broad singlet (ba), doublet (d), triplet (t), (doublet of doublets (dd), doublet of triplets (dt), doublet of doublet of doublets (ddd), multiplet (m). Coupling constants (J) are expressed in Hz.
All mass spectra were acquired with a Shimadzu 8040 HPLC-MS-MS equipment, with LC-20AD pumps, a SIL-20A autosampler, electrospray ionization and triple-quadrupole mass detector. Flash column chromatography was carried out with silica gel 60 (J.T. Baker, 40 μm average particle diameter). All reactions and chromatographic separations were monitored by TLC, conducted on 0.25-mm silica gel plastic sheets (Macherey/Nagel, Polygram_ SIL G/UV 254). TLC plates were analyzed under 254 nm UV light, iodine vapor, or ninhydrine spray. Yields are reported for chromatographically and spectroscopically (1H and 13C NMR) pure compounds.
All solvents were purified according to literature procedures [64]. All reactions were carried in dry, freshly distilled solvents under anhydrous conditions unless otherwise stated.
General procedure for amide bond formation using oxyma pure
Oxyma pure (1.2 mmol), EDCI (1.2 mmol), DIPEA (1.2 mmol), 4-DMAP (catalytic) were added to a stirred solution of the respective acid (1.2 mmol) and amine (1.0 mmol) under N2 atmosphere in dry solvent (CH2Cl2 or THF) at 0 °C. The resulting mixture was stirred at room temperature for 12–48 h and then evaporated in vacuo. The crude was dissolved in EtOAc, washed with 5% v/v HCl and with a saturated solution of NaHCO3, dried with Na2SO4, filtered, and evaporated in vacuo. The crude was purified by flash chromatography using the corresponding eluent to give the amide.
General procedure for amide bond formation using triphosgene
To a stirred solution of (1.0 mmol) acid, (0.4 mmol) triphosgene were added in dry THF under N2 atmosphere at room temperature. After 10 min 2.4 mmol of 2,4,6-colidine were added, and a white precipitate was formed. After 30 min, 0.9 mmol of the respective amine and 0.9 mmol of DIPEA were added. The resulting mixture was stirred at reflux for 12–24 h. The mixture is concentrated to dryness and the crude was dissolved in EtOAc, washed with 5% v/v HCl and a saturated solution of NaHCO3, dried with Na2SO4, filtered and evaporated in vacuo. The crude was purified by flash chromatography using the corresponding eluent to give the amide.
2-[(5-Nitro-1,3-thiazol-2-yl)carbamoyl]phenyl acetate (1)
Compound 1 was obtained following the general procedure for in situ acid chloride formation from 6 (236 mg, 1.63 mmol) and acetylsalicylic acid, in 19 mL of dry THF. Reaction was refluxed 6 h and stirred 14 h at 40–50 °C. Yield 42%. Brown powder. Rf = 0.35 using a mixture of CHCl3:MeOH (3:0.2) as eluent. 1H NMR (400 MHz, DMSO) δ (ppm): 13.61 (ba, 1H), 8.68 (s, 1H), 7.87 (dd, J = 7.9, 1.6 Hz, 1H), 7.67 (td, J = 7.9, 1.7 Hz, 1H), 7.43 (td, J = 7.6, 1.0 Hz, 1H), 7.30 (dd, J = 8.17, 0.9 Hz, 1H), 2.26 (s, 3H). 13C NMR (100 MHz, Acetona-d6) δ (ppm): 168.7, 164.7, 162.3, 151.3, 148.9, 141.4, 133.7, 129.7, 126.1, 124.2, 123.7, 20.0. MS (ESI + ) calcd for C12H9N3O5S, [M + H]+ 308.02, found 308.05.
tert-Butyl (4-((5-nitrothiazol-2-yl)carbamoyl)thiazol-2-yl)carbamate (11)
Compound 11 was obtained following the general procedure for amide bond formation using oxyma pure from 6 (80 mg, 0.55 mmol) and 2-((tert-butoxycarbonyl)amino)thiazole-4-carboxylic acid in 8 mL of dry THF. Reaction was refluxed 3 h and stirred 14 h at 40–50 °C. Yield 35%. White powder. Rf = 0.24 with a mixture of CHCl3:MeOH (3:0.1) as eluent.1H NMR (400 MHz, CDCl3) δ (ppm): 10.51 (s, 1H), 8.38 (s, 1H), 8.0 (s, 1H), 3.91 (s, 1H), 1.58 (s, 9H). 13C NMR (100 MHz, CDCl3) δ (ppm): 176.4, 162.3, 160.8, 160.6, 146.4, 141.3, 141.0, 121.8, 84.2, 28.1. MS (ESI -) m/z calcd for C12H13N5O5S2 [M–H]− 370.03, found 370.10.
SYBR green I growth inhibition assay against P. falciparum asexual forms
Continuous in vitro cultures of P. falciparum (strains 3D7, TM90C6B, and 3D7R_848) were kept using an adaptation of the method described by Trager and Jansen [65]. P. falciparum cultures were synchronized for assays by treatment with a solution of 5% sorbitol for 10 min, as described by Lambros and Vanderberg [66]. Compounds were diluted to a stock concentration of 20 mM in 100% DMSO before the experiments and maintained at −20 °C. Compound inhibitory potencies were determined using the SYBR Green I phenotypic assay (Smilkstein et al.) [67]. Briefly, in 96-well plates, 180 μL of a parasite suspension in the early trophozoite (ring) form at 0.5% parasitemia and 2% hematocrit was incubated for 72 h with 20 μL of 10× concentrated serial dilutions of each compound. The antimalarial artesunate, pyrimethamine, atovaquone, and MMV692848 were used as positive controls of inhibition for the cross-resistance experiments. The plates were kept at 37 °C and in a low oxygen atmosphere (5% O2, 5% CO2, 90% N2). Positive and negative growth controls were added to each independent plate. The growth medium was then removed, and the deposited RBCs were resuspended in PBS buffer (116 mM NaCl, 10 mM Na2HPO4, 3 mM KH2PO4). A solution of SYBR Green I DNA Stain diluted was added in a lysis buffer (20 mM Tris, 5 mM EDTA, 0.008% (m/v) saponin, and 0.08% (m/v) Triton X-100, at pH 7.5) to induce hemolysis. The plates were incubated for an additional 30 min, after which the fluorescence of the plate was measured (absorption and emission wavelengths of 485 nm and 535 nm, respectively). Fluorescence intensity was analyzed in terms of parasite viability as compared to controls, using the Origin 9.0 software (OriginLab, Northampton, MA, USA). Concentration-response curves were built, and half-maximal inhibitory concentration (IC50) values were determined for each compound using nonlinear regression analysis.
Cultivation of human hepatocellular carcinoma cells (HepG2 cell line)
A culture of HepG2 cells was kept in a flask in a humidified atmosphere of 5% CO2 at 37 °C. The culture medium used was RPMI 1640 supplemented with 25 mM HEPES (pH 7.4), 24 mM sodium bicarbonate, 11 mM D-glucose, 40 μg/mL penicillin-streptomycin, and 10% (v/v) bovine fetal serum. Every 3–4 days, treatment with a 0.25% trypsin solution was used to release cells from the flask walls and a 1:4 proportion of the cells were maintained in culture.
MTT assay for cytotoxicity evaluation
An adaptation of the MTT assay described by Denizot and Lang [68] was employed to determine cytotoxic activity. The HepG2 cells were counted and distributed in a 96-well plate, in a proportion of 5 × 106 cells per well. The plate was incubated overnight at 37 °C, in a humidified atmosphere of 5% CO2. After that, serial dilutions of the compounds were added in a 1:9 proportion to each well, and the plates were incubated for another 24 h. Positive (no compound) controls were added to each plate for normalization of results. The supernatant was then removed, and a solution of MTT salt (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was added to each well. After 2–4 h of incubation, the formazan crystals formed by MTT reduction were solubilized in DMSO. The absorbance of the plate at 570 nm was measured, and the intensity values obtained were converted to viability values using the equation below. Dose-response curves were crafted for each compound using the OriginPro 9.0 software (OriginLab), and the minimum inhibitory concentration for 50% of cells (IC50HepG2) was determined for each compound. The selectivity index (SI) was calculated by the ratio of IC50HepG2 to IC503D7.
Data availability
NMR and MS spectra are depicted in the Supplementary Material.
References
World Health Organization, World Malaria Report 2020, World Health Organization, Geneva, Switzerland. https://apps.who.int/iris/rest/bitstreams/1321872/retrieve.
Zawawi A, Alghanmi M, Alsaady I, Gattan H, Zakai H, Couper K. The impact of COVID-19 pandemic on malaria elimination. Parasite Epidemiol Control. 2020;11:e00187. https://doi.org/10.1016/j.parepi.2020.e00187.
Rogerson SJ, Beeson JG, Laman M, Poespoprodjo JR, William T, Simpson JA, et al. Identifying and combating the impacts of COVID-19 on malaria. BMC Medicine. 2020;18:239. https://doi.org/10.1186/s12916-020-01710-x
Sherrard-Smith E, Hogan AB, Hamlet A, Watson OJ, Whittaker C, Winskill P, et al. The potential public health consequences of COVID-19 on malaria in Africa. Nat Med. 2020;26:1411–6. https://doi.org/10.1038/s41591-020-1025-y
World Health Organization. The potential impact of health service disruptions on the burden of malaria: a modelling analysis for countries in sub-Saharan Africa. 2020. https://www.who.int/publications/i/item/9789240004641
Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, et al. Artemisinin resistance in Plasmodium falciparum malaria. New Engl J Med. 2009;361:455–67. https://doi.org/10.1056/NEJMoa0808859
Leang R, Taylor WR, Bouth DM, Song L, Tarning J, Char MC, et al. Evidence of Plasmodium falciparum malaria multidrug resistance to artemisinin and piperaquine in western Cambodia: dihydroartemisinin-piperaquine open-label multicenter clinical assessment. Antimicrob Agents Chemother. 2015;59:4719–26. https://doi.org/10.1128/AAC.00835-15
Noedl H, Se Y, Schaecher K, Smith BL, Socheat D, Fukuda M. Evidence of artemisinin-resistant malaria in western Cambodia. N Engl J Med. 2008;359:2619–20. https://doi.org/10.1056/NEJMc0805011
Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A, Doig A, Guilliams T, Latimer J, McNamee C, Norris A, Sanseau P, Cavalla D, Pirmohamed M. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. 2019;18:41–58. https://doi.org/10.1038/nrd.2018.168
Shanmugam A, Muralidharan N, Velmurugan D, Gromiha MM. Therapeutic targets and computational approaches on drug development for COVID-19. Curr Top Med Chem. 2020;20:2210–20. https://doi.org/10.2174/1568026620666200710105507
Chhabria MT, Patel S, Modi P, Brahmkshatriya PK. Thiazole: a review on chemistry, synthesis and therapeutic importance of its derivatives. Curr Top Med Chem. 2016;16:2841–62. https://doi.org/10.2174/1568026616666160506130731.
Rossignol JF, Cavier R. New derivatives of 2-benzamido 5-nitrothiazoles. United States Patent. 1976;950:351. April 13
Cavier R, Rossignol JF. Etude de diverses associations d’anthelminthiques chez la souris. Rev Méd Vét. 1982;133:779–83.
Aslam S, Musher DM. Nitazoxanide: clinical studies of a broad-spectrum anti-infective agent. Future Microbiol. 2007;2:583–90. https://doi.org/10.2217/17460913.2.6.583
Dubreuil L, Houcke I, Mouton Y, Rossignol JF. In vitro evaluation of activities of nitazoxanide and tizoxanide against anaerobes and aerobic organisms. Antimicrob Agents Chemother. 1996;40:2266–70. https://doi.org/10.1128/AAC.40.10.2266
Anderson VR, Curran MP. Nitazoxanide: a review of its use in the treatment of gastrointestinal infections. Drugs. 2007;67:1947–67. https://doi.org/10.2165/00003495-200767130-00015
Rossignol JF, Kabil SM, El-Gohary Y, Younis AM. Nitazoxanide in the treatment of amoebiasis. Trans R Soc Trop Med Hyg. 2007;101:1025–31. https://doi.org/10.1016/j.trstmh.2007.04.001
Chero JC, Saito M, Bustos JA, Blanco EM, Gonzalvez G, García H, et al. Hymenolepis nana infection: symptoms and response to nitazoxanide in field conditions. Trans R Soc Trop Med Hyg. 2007;101:203–5. https://doi.org/10.1016/j.trstmh.2006.04.004
Speich B, Ame SM, Ali SM, Alles R, et al. Efficacy and safety of nitazoxanide, albendazole, and nitazoxanide-albendazole against Trichuris trichiura infection: a randomized controlled trial. PLOS Negl Trop Dis. 2012;6:e1685. https://doi.org/10.1371/journal.pntd.0001685
Rossignol JF, El-Gohary M. Nitazoxanide in the treatment of viral gastroenteritis: a randomized double-blind placebo-controlled clinical trial. Aliment Pharmacol Ther. 2006;24:1423–30. https://doi.org/10.1111/j.1365-2036.2006.03128.x.
Rossignol JF, Abou Zekry M, Hussein A, Santoro MG. Effect of nitazoxanide in treating rotavirus diarrhea: a randomized, double-blind, placebo-controlled trial. Lancet. 2006;368:124–9. https://doi.org/10.1016/s0140-6736(06)68852-1
Siddiq DM, Koo HL, Adachi JA, Viola GM. Norovirus gastroenteritis successfully treated with nitazoxanide. J Infection. 2011;63:394–7. https://doi.org/10.1016/j.jinf.2011.08.002
Rossignol JF. Nitazoxanide, a new drug candidate for the treatment of Middle East respiratory syndrome coronavirus. J Infect Public Health. 2016;9:227–30. https://doi.org/10.1016/j.jiph.2016.04.001
Rossignol JF, Elfert A, Keeffe EB. Treatment of chronic hepatitis C using a 4-week lead-in with nitazoxanide before peginterferon plus nitazoxanide. J Clin Gastroenterol. 2010;44:504–9. https://doi.org/10.1097/mcg.0b013e3181bf9b15
Korba BE, Montero AB, Farrar K, Gaye K, et al. Nitazoxanide, tizoxanide and other thiazolides are potent inhibitors of hepatitis B virus and hepatitis C virus replication. Antiviral Research. 2008;77:56–63. https://doi.org/10.1016/j.antiviral.2007.08.005
Rossignol JF. Nitazoxanide: a first-in-class broad-spectrum antiviral agent. Antiviral Research. 2014;110:94–103. https://doi.org/10.1016/j.antiviral.2014.07.014
Hoffman PS, Sisson G, Croxen MA, Welch K, et al. Antiparasitic drug nitazoxanide inhibits the pyruvate oxidoreductases of Helicobacter pylori, selected anaerobic bacteria and parasites, and Campylobacter jejuni. Antimicrobial Agents and Chemotherapy. 2007;51:868–76. https://doi.org/10.1128/aac.01159-06
Elazar M, Liu M, McKenna SA, Liu P, et al. The anti-hepatitis c agent nitazoxanide induces phosphorylation of eukaryotic initiation factor 2α via protein kinase activated by double-stranded RNA activation. Gastroenterology. 2009;137:1827–35. https://doi.org/10.1053/j.gastro.2009.07.056
Rossignol JF, La Frazia S, Chiappa L, Ciucci A, Santoro MG. Thiazolides, a new class of anti-influenza molecules targeting viral hemagglutinin at the post- translational level. J Biol Chemistry. 2009;284:29798–808. https://doi.org/10.1074/jbc.m109.029470
Stachulski AV, Santoro MG, Piacentini S, Belardo G, et al. Second-generation nitazoxanide derivatives: thiazolides are effective inhibitors of the influenza A virus. Future Medicinal Chemistry. 2018;10:851–62. https://doi.org/10.4155/fmc-2017-0217.
Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–71. https://doi.org/10.1038/s41422-020-0282-0.
Jasenosky LD, Cadena C, Mire CE, et al. The FDA-approved oral drug nitazoxanide amplifies host antiviral responses and inhibits Ebola virus. iScience. 2019;19:1279–1290. https://doi.org/10.1016/j.isci.2019.07.003
Navarrete-Vazquez G, Chávez-Silva F, Argotte-Ramos R, Rodríguez-Gutiérrez MC, et al. Synthesis of benzologues of nitazoxanide and tizoxanide: a comparative study of their in vitro broad-spectrum antiprotozoal activity. Bioorg Med Chem Lett. 2011;21:3168–71. https://doi.org/10.1016/j.bmcl.2011.02.100
Karade HN, Acharya BN, Sathe M, Kaushik MP. Design, synthesis, and antimalarial evaluation of thiazole-derived amino acids. Med Chem Res. 2008;17:19–29. https://doi.org/10.1007/s00044-008-9089-0
Cabrera DG, Douelle F, Feng T-S, et al. Novel orally active antimalarial thiazoles. J Med Chem. 2011;54:7713–9. https://doi.org/10.1021/jm201108k
Sharma M, Prasher P. An epigrammatic status of the ‘azole’-based antimalarial drugs. RSC Med Chem. 2020;11:184–211. https://doi.org/10.1039/C9MD00479C
Peña S, Scarone L, Medeiros A, Manta E, et al. Synthesis of precursors and macrocycle analogs of aerucyclamides as anti-trypanosomal agents. Med Chem Commun. 2012;3:1443–8. https://doi.org/10.1039/C2MD20218B
Peña S, Scarone L, Manta E, Serra G. First total synthesis of aerucyclamide B. Tetrahedron Letters. 2013;54:2806–8. https://doi.org/10.1016/j.tetlet.2013.03.060
Peña S, Fagundez C, Medeiros A, Comini M, et al. Synthesis of cyclohexapeptides as antimalarial and anti-trypanosomal agents. Med Chem Commun. 2014;5:1309–16. https://doi.org/10.1039/C4MD00135D
Peña S, Scarone L, Serra G. Macrocycles as potential therapeutic agents in neglected diseases. Future Med Chem. 2015;7:355–82. https://doi.org/10.4155/fmc.14.133
Franco J, Medeiros A, Benítez D, Perelmuter K, et al. In vitro activity and mode of action of distamycin analogues against African trypanosomes. Eur J Med Chem. 2017;126:776–88. https://doi.org/10.1016/j.ejmech.2016.12.002
Fagundez C, Sellanes D, Serra G. Synthesis of cyclic peptides as potential anti-malarials. ACS Combinatorial Sci. 2018;20:212–9. https://doi.org/10.1021/acscombsci.7b00154
Posada L, Serra G. First total synthesis of versicotide D and analogs. Tetrahedron Lett. 2019;60:151281. https://doi.org/10.1016/j.tetlet.2019.151281
Fagundez C, Sellanes D, Peña S, Scarone L, et al. Synthesis, profiling, and in vivo evaluation of cyclopeptides containing N-methyl amino acids as antiplasmodial agents. ACS Med Chem Lett. 2019;10:137–41. https://doi.org/10.1021/acsmedchemlett.8b00543
Franco J, Scarone L, Comini MA. Novel distamycin analogues that block the cell cycle of African trypanosomes with high selectivity and potency. Eur J Med Chem. 2020;189:112043. https://doi.org/10.1016/j.ejmech.2020.112043
Serra G, Posada L, Hojo H. On-resin synthesis of cyclic peptides via tandem N-to-S acyl migration and intramolecular thiol additive-free native chemical ligation. Chem Comm. 2020;56:956–9. https://doi.org/10.1039/C9CC07783A
Alvarez N, Velluti F, Guidali F, Serra G, et al. New BI and TRI-Thiazole copper (II) complexes in the search of new cytotoxic drugs against breast cancer cells. Inorg Chim Acta. 2020;508:119622. https://doi.org/10.1016/j.ica.2020.119622
Hohmann K, Mohr R, Hahnke M. Process for preparing an azo compound from a 2-aminothiazole diazo component.United States Patent 4046752. Washington, DC: EE.UU; 1974.
Dickey JB, Towne EB, Wright GF. Nitration of 2-aminothiazoles. J Org Chem. 1955;20:499–510. https://doi.org/10.1021/jo01122a013.
Odingo J, Bailey MA, Files M, Early JV, et al. In vitro evaluation of novel Nitazoxanide derivatives against Mycobacterium tuberculosis. ACS Omega. 2017;2:5873–90. https://doi.org/10.1021/acsomega.7b00892
Gergely M, Kollar L. Aminothiazoles and aminothiadiazoles as nucleophiles in aminocarbonylation of iodobenzene derivatives. Tetrahedron. 2018;74:2030–40. https://doi.org/10.1016/j.tet.2018.03.007
Stachulski AV, Pidathala C, Row EC, Sharma R, et al. Thiazolides as novel antiviral agents. 2. Inhibition of hepatitis C virus replication. J Med Chem. 2011;54:8670–80. https://doi.org/10.1021/jm201264t
Ballard TE, Wang X, Olekhnovich I, Koerner T, et al. Synthesis and antimicrobial evaluation of Nitazoxanide-based analogues: identification of selective and broad spectrum activity. Chem Med Chem. 2010;6:362–77. https://doi.org/10.1002/cmdc.201000475
Posada L, Davyt D, Serra G. First total synthesis of versicotide A, B and C. RSC Adv. 2020;10:43653. https://doi.org/10.1039/D0RA09635K
Subirós-Funosas R, Prohens R, Barbas R, et al. Oxyma: an efficient additive for peptide synthesis to replace the benzotriazole-based HOBt and HOAt with a lower risk of explosion. Chem Eur J. 2009;15:9394–403. https://doi.org/10.1002/chem.200900614
Smilkstein M, Sriwilaijaroen N, Kelly JX, et al. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother. 2004;48:1803–6. https://doi.org/10.1128/aac.48.5.1803-1806.2004
Gardner MJ, Hall N, Fung E, White O, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. https://doi.org/10.1038/nature01097
Hall N, Karras M, Raine JD, Carlton JM. et alA comprehensive survey of the Plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science. 2005;307:82–86. https://doi.org/10.1126/science.1103717
Frech C, Chen N. Genome comparison of human and non-human malaria parasites reveals species subset-specific genes potentially linked to human disease. PLoS Comput Biol. 2011;7:e1002320 https://doi.org/10.1371/journal.pcbi.1002320
Anderson VR, Curran MP. Nitazoxanide: a review of its use in the treatment of gastrointestinal infections. Drugs. 2007;67:1947–67. https://doi.org/10.2165/00003495-200767130-00015
Wang CC. Validating targets for antiparasite hemotherapy. Parasitology. 1997;114:S31–S44.
Fox LM, Saravolatz LD. Nitazoxanide: a new thiazolide antiparasitic agent. Clin Infect Dis. 2005;40:1173–80. https://doi.org/10.1086/428839
Müller J, Wastling J, Sanderson S, Müller N, Hemphill A. A novel Giardia lamblia nitroreductase, GlNR1, interacts with nitazoxanide and other thiazolides. Antimicrob Agents Chemother. 2007;51:1979–86. https://doi.org/10.1128/AAC.01548-06
Armarego WLF, Chai C. Purification of laboratory chemicals, sixth edn. Butterworth-Heinemann: Elsevier Inc.; 2009.
Trager W, Jensen J. Human malaria parasites in continuous culture. Science. 1976;193:673–5. https://doi.org/10.1126/science.781840
Lambros C, Vanderberg JP. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol. 1979;65:418–20.
Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother. 2004;48:1803–6. https://doi.org/10.1128/AAC.48.5.1803-1806.2004
Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods. 1986;89:271–7. https://doi.org/10.1016/0022-1759(86)90368-6
Acknowledgements
This work was supported by Grants from CSIC Grupos 2006 (Universidad de la República) and PEDECIBA (Uruguay). We thank the Sao Paulo Research Foundation—FAPESP for funding the research (CEPID grant 2013/07600–3, 2020/12904–5, 2018/07287-7). This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
About this article

Cite this article
Irabuena, C., Scarone, L., de Souza, G.E. et al. Synthesis and antiplasmodial assessment of nitazoxanide and analogs as new antimalarial candidates. Med Chem Res 31, 426–435 (2022). https://doi.org/10.1007/s00044-021-02843-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-021-02843-1