
Abstract
We report a convenient and efficient synthesis of indeno[1,2-c]pyrazol-4(1H)-ones (4a‒o) by the reaction of a variety of 2-acyl-(1H)-indene-1,3(2H)-diones (1) and 2-hydrazinylbenzo[d]thiazole/2-hydrazinyl-6-substitutedbenzo[d]thiazoles (2) in the presence of glacial acetic acid in good yields. The structure of the compounds thus prepared were confirmed by analytical and spectral (FT-IR, 1H NMR, 13C NMR, and HRMS) techniques. All the synthesized indeno[1,2-c]pyrazol-4(1H)-ones (4a‒o) were assayed for their in vitro Type II diabetes inhibitory activity by using Acarbose as standard drug and in vitro antimicrobial activity utilizing Streptomycin and Fluconazole as reference drugs. Among the synthesized derivatives, 4e (IC50 = 6.71 μg/mL) was found to be more potent against α-glucosidase enzyme as compared with the standard Acarbose (IC50 = 9.35 μg/mL) and 4i (IC50 = 11.90 μg/mL) exhibited good inhibitory activity against α-amylase enzyme as compared with the standard Acarbose (IC50 = 22.87 μg/mL). Also, all the titled compounds showed good antimicrobial activity. In addition, in vitro α-glucosidase and α-amylase inhibition were supported by docking studies performed on the derivatives 4e and 4o, respectively.

Similar content being viewed by others
Introduction
Diabetes mellitus is a metabolic disorder resulting from inadequate secretion of insulin characterized by chronic hyperglycemia caused by high calorie diets rich in fat, carbohydrates and proteins (Kumar et al. 2017). The International Diabetes Foundation (IDF) reports that there were 425 million diagnosed cases of diabetes globally in 2017 which is estimated to increase to 629 million by 2045. Recently, there are more than 46 million diabetics in North America and the Caribbean, 58 million in Europe, 26 million in South and Central America, 39 million in Middle East and North Africa, 16 million in Africa and 82 million in South-East Asia. There are 352 million people at the risk of developing Type II diabetes (IDF Diabetes Atlas 2017). The emerging factors that contribute to the spread of Type II diabetes, comprising a progressively technological society, food habits with high calorie diets rich in fats and carbohydrates, and an increasingly inactive lifestyle (Wagman et al. 2017). Type II diabetes is associated with hypertension, dyslipidemia, obesity, cardiovascular disease, etc. It may also eventually cause tissue or vascular damage leading to severe diabetic complications such as retinopathy, neuropathy, and nephropathy (Keri et al. 2015). Out of several enzymes known, α-amylase and α-glucosidase are the key enzymes in the lowering of postprandial hyperglycemia observed in case of Type II diabetes mellitus (T2DM) (Patil et al. 2013). α-Amylase inhibits dietary starch from being absorbed into the body system and leads to lowering of blood glucose by the inhibition of salivary and pancreatic amylase (Ajiboye et al. 2016). α-Glucosidase inhibitors have been reported to reduce postprandial hyperglycemia in diabetic mellitus resulting in the lowering of glucose absorption by carbohydrate digestion and increases digestion time (Chaudhry et al. 2017).
Likewise, the emergence of bacterial resistance of pathogenic microorganisms is rapidly becoming a major worldwide problem (Mor et al. 2017). Therefore, the demand for new antimicrobial agents is necessary, but now days, it leads to a challenging task for chemists to synthesize new molecules with excellent activity (Kim et al. 2012).
In the recent years, indeno-fused heterocycles are recognized as important frameworks with a broad spectrum of pharmacological properties. Among them, indenopyrazoles have gained substantial attention due to their wide range of biological activities such as antitubercular (Ahsan et al. 2011), tyrosine kinase inhibitors (Khan et al. 2019), CNS agents (Lemke et al. 1978), antioxidant activity (Mor et al. 2019), non-steroidal anti-inflammatory drugs (Lemke et al. 1989), anticancer (Mor et al. 2016), antimicrobial (Shareef et al. 2019), anti-HIV and anticonvulsant activities (Ahsan et al. 2012), and cyclin-dependent kinase (CDK) inhibitors (Singh et al. 2006). Moreover, methyl 3-((6-methoxy-1,4-dihydroindeno[1,2-c]pyrazol-3-yl)amino)benzoate was the first indenopyrazole that was reported as a Tubulin Polymerization Inhibitor (Minegishi et al. 2015).
Similarly, benzothiazole is a privileged bicyclic ring system associated with numerous pharmacological activities like antitumor (Gabr et al. 2015), anticonvulsant (Amnerkar and Bhusari 2010), antimicrobial (Chugunova et al. 2015; Kamal et al. 2015), antihelmintic (Sarkar et al. 2013), antileishmanial (Keri et al. 2015), antitubercular (Patel et al. 2013), anti-inflammatory (Shafi et al. 2012), antipsychotic (Yevich et al. 1986), antioxidant (Bhat and Belagali 2016), antidiabetic (Meltzer-Mats et al. 2013; Kamal et al. 2015) activities, etc. Some of the important marketed drugs involving benzothiazole nucleus are riluzole, sibenadet hydrochloride (Viozan), and pramipexole (Scott and Njardarson 2018). Zopolrestat is another significant drug containing benzothiazole core with antidiabetic effects (Carvalho et al. 2006).
To the best of our knowledge as revealed by literature surveys (Khan et al. 2019), none of hetrocycles with indenopyrazole skeleton have been reported to exhibit antidiabetic effects. Therefore, we thought of synthesizing some new benzothiazole tethered indenopyrazoles to see the additive effect of these moieties towards the preliminary examination of in vitro antidiabetic activity (Doddaramappa et al. 2015). In this perspective and in continuation of our interest in the synthesis of heterocycles containing nitrogen (Zhou et al. 2017; Huang and Huang 2019) and sulfur as heteroatoms and their biological activities herein, we report the synthesis, characterization, α-amylase and α-glucosidase inhibition, antimicrobial evaluation and docking studies of several benzothiazole tethered indeno[1,2-c]pyrazol-4(1H)-ones (4a‒o).
Materials and methods
Chemistry
All reagents were used without any further purification. Melting points were observed using Electrothermal Melting Point apparatus, LABCO Co., India and are not corrected. The FT-IR spectra were recorded in KBr on IR affinity-1 FTIR (Shimadzu) spectrophotometer, and results are reported in cm−1. 1H NMR and 13C NMR spectra were recorded on Bruker AVANCE III NMR spectrometer operating at 400 and 100 MHz, respectively, with CDCl3 as the solvent and tetramethylsilane (TMS) as the internal standard. Chemical shifts (δ) are reported in parts per million (ppm), and coupling constants (J) are expressed in Hertz (Hz). HRMS were obtained from Waters Synapt G2-Si QTOF and SCIEX 5600+ QTOF mass analyser by using the electrospray ionization (ESI) method. The purity of synthesized compounds was checked by precoated TLC plates (SIL G/UV254, ALUGRAM) using a mixture of hexane and ethyl acetate as eluent and visualization was achieved via UV light.
General procedure for the synthesis of 2-acyl-(1H)-indene-1,3(2H)-diones (1)
2-Acyl-(1H)-indene-1,3(2H)-diones (1) needed for the purpose were prepared via Claisen condensation of diethylphthlate and appropriate aliphatic ketones in presence of freshly prepared sodium methoxide following the procedure presented in literature (Mor et al. 2016).
General procedure for the synthesis of 2-hydrazinylbenzo[d]thiazole/2-hydrazinyl-6-substitutedbenzo[d]thiazoles (2)
Benzo[d]thiazol-2-amine/6-substitutedbenzo[d]thiazol-2-amines were prepared by the reaction of aniline/4-substituted anilines and sodium thiocyanate in bromine/glacial acetic acid solution under stirring for 16 h. After completion of reaction, the salt of benzo[d]thiazol-2-amine/6-substitutedbenzo[d]thiazol-2-amines thus obtained was filtered through suction and washed with hexane. Thereafter, the salt was dissolved in water upon warming and the product was precipitated by adding dil. NaOH solution. The solid thus formed was filtered through suction and recrystallized from ethanol to afford the corresponding amines in high yields (Mor et al. 2017). To a solution of hydrazine hydrochloride in ethylene glycol was added the appropriate benzo[d]thiazol-2-amine/6-substitutedbenzo[d]thiazol-2-amines in portions with continuous stirring and the resulting mixture was heated to reflux on a heating mantle for 2 h. A fine crystalline solid was separated out on cooling which was filtered, washed with water and crystallized from rectified sprit to yield the corresponding 2-hydrazinylbenzo[d]thiazole/2-hydrazinyl-6-substitutedbenzo[d]thiazoles (2) in good yields (Mor et al. 2017).
General procedure for the synthesis of benzothiazolyl hydrazones (3)
A solution of equimolar quantities of 2-acyl-(1H)-indene-1,3(2H)-diones (1, 3 mmol) and hydrazines (2, 3 mmol) in dry methanol (15 mL) was heated on a water bath for 15 min in presence of catalytic amount of glacial acetic acid (4–5 drops). Thereafter, reaction mixture was cooled at room temperature. The solid thus separated out was filtered through suction and recrystallized from ethyl acetate-ethanol to give the corresponding benzothiazolyl hydrazones (3a‒o) as orange solids (Sawhney and Lemke 1983; Mor et al. 2019).
General procedure for the synthesis of indeno[1,2-c]pyrazol-4-ones (4a–o)
Benzothiazolyl hydrazones (3) were charged with glacial acetic acid (30 mL) and heated to reflux on a heating mantle for 7–9 h till the completion of reaction as indicated by TLC. The reaction mixture was cooled at room temperature and the solid thus obtained was filtered, and recrystallized from chloroform to furnish the target compounds 4a–o in good yields. The physical and spectral data of compounds 4a–o are as follows:
1-(Benzo[d]thiazol-2-yl)-3-methylindeno[1,2-c]pyrazol-4(1H)-one (4a) 

Yellow solid; yield: 62%; mp 238–240 °C; FTIR (KBr): νmax 798, 1093, 1381, 1498, 1543, 1598 (C=N stretch), 1707 (C=O stretch), 2850, 2960 (aliphatic C–H stretch), 3068 (aromatic C–H stretch) cm−1; 1H NMR (400 MHz, CDCl3): δ = 2.45 (3H, s, CH3), 7.35–7.44 (2H, m, Ar-H), 7.50–7.55 (2H, m, Ar-H), 7.60 (1H, d, J = 7.20 Hz, Ar-H), 7.88 (1H, d, J = 7.96 Hz, Ar-H), 8.03 (1H, d, J = 8.12 Hz, Ar-H), 8.54 (1H, d, J = 7.40 Hz, 8-H); 13C NMR (100 MHz, CDCl3): δ = 29.68 (CH3), 121.36, 121.69, 122.83, 124.09, 124.29, 124.54, 125.45, 126.75, 130.69, 132.58, 133.32, 133.58, 140.10, 148.70, 150.95, 158.73, 184.34 (C-4) ppm; HRMS: m/z (M+) Cacld. for C18H11N3OS: 317.0623, found: 318.0680 [M+H]+.
3-Methyl-1-(6-methylbenzo[d]thiazol-2-yl)indeno[1,2-c]pyrazol-4(1H)-one (4b): 

Yellow solid; yield 67%; mp 215–218 °C; FTIR (KBr): νmax 798, 1087, 1386, 1500, 1550, 1606 (C = N stretch), 1712 (C=O stretch), 2848, 2954 (aliphatic C–H stretch), 3075 (aromatic C–H stretch) cm−1; 1H NMR (400 MHz, CDCl3): δ = 2.44 (3H, s, CH3), 2.51 (3H, s, CH3), 7.32–7.38 (2H, m, Ar-H), 7.49–7.53 (2H, m, Ar-H), 7.59 (1H, d, J = 7.08 Hz, Ar-H), 7.66 (1H, s, Ar-H), 7.90 (1H, d, J = 8.28 Hz, Ar-H), 8.53 (1H, d, J = 7.40 Hz, 8-H); 13C NMR (100 MHz, CDCl3): δ= 22.70 (CH3), 31.94 (CH3), 121.49, 122.38, 123.97, 124.27, 124.54, 128.26, 130.64, 132.63, 133.45, 133.57, 135.73, 140.16, 143.67, 148.56, 148.94, 158.58, 184.38 (C-4); HRMS: m/z (M+) Cacld. for C19H13N3OS: 331.0779, found: 332.0839 [M+H]+.
1-(6-Methoxybenzo[d]thiazol-2-yl)-3-methylindeno[1,2-c]pyrazol-4(1H)-one (4c): 

Yellow solid; yield 69%; mp 210–212 °C; FTIR (KBr): νmax 761, 1097, 1373, 1500, 1556, 1606 (C=N stretch), 1716 (C=O stretch), 2918, 2962 (aliphatic C–H stretch), 3086 (aromatic C–H stretch) cm−1; 1H NMR (400 MHz, CDCl3): δ = 2.44 (3H, s, CH3), 3.90 (3H, s, OCH3), 7.11 (1H, dd, J = 2.52, J = 8.92 Hz, Ar-H), 7.31–7.40 (2H, m, Ar-H), 7.49–7.60 (2H, m, Ar-H), 7.90 (1H, d, J = 8.88 Hz, Ar-H), 8.49 (1H, d, J = 7.40 Hz, 8-H); 13C NMR (100 MHz, CDCl3): δ = 29.79 (CH3), 55.98 (OCH3), 115.94, 121.49, 121.64, 123.51, 124.36, 124.54, 130.31, 130.71, 132.70, 133.64, 134.14, 134.77, 140.27, 145.25, 148.58, 158.06, 184.46 (C-4); HRMS: m/z (M+) Calcd. for C19H13N3O2S: 347.0728, found: 348.0786 [M+H]+.
1-(6-Chlorobenzo[d]thiazol-2-yl)-3-methylindeno[1,2-c]pyrazol-4(1H)-one (4d): 

Yellow solid; yield 62%; mp 228–230 °C; FTIR (KBr): νmax 798, 1099, 1371, 1494, 1546, 1598 (C=N stretch), 1712 (C=O stretch), 2920, 2962 (aliphatic C–H stretch), 3089 (aromatic C–H stretch) cm−1; 1H NMR (400 MHz, CDCl3): δ = 2.45 (3H, s, CH3), 7.36–7.57 (4H, m, Ar-H), 7.86 (1H, d, J = 1.84 Hz, Ar-H), 7.94 (1H, d, J = 8.48 Hz, Ar-H), 8.48 (1H, d, J = 7.40 Hz, 8-H); 13C NMR (100 MHz, CDCl3): δ = 25.29 (CH3), 121.01, 121.41, 121.58, 123.59, 124.42, 127.59, 130.48, 130.85, 131.29, 132.42, 133.64, 134.23, 134.49, 140.01, 148.95, 149.50, 184.30 (C-4); HRMS: m/z (M+) Calcd. for C18H10ClN3OS: 351.0233, found: 352.0291 [M+H]+.
1-(6-Bromobenzo[d]thiazol-2-yl)-3-methylindeno[1,2-c]pyrazol-4(1H)-one (4e): 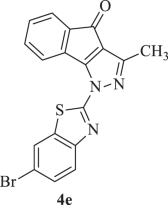

Yellow solid; yield 70 %; mp 208–212 °C; FTIR (KBr): νmax 765, 1099, 1394, 1496, 1546, 1598 (C=N stretch), 1710 (C=O stretch), 2850, 2958 (aliphatic C–H stretch), 3089 (aromatic C–H stretch) cm−1; 1H NMR (400 MHz, CDCl3): δ = 2.44 (3H, s, CH3), 7.36–7.44 (2H, m, Ar-H), 7.49–7.55 (2H, m, Ar-H), 7.76 (1H, d, J = 8.64 Hz, Ar-H), 7.88 (1H, d, J = 8.64 Hz, Ar-H), 8.47 (1H, d, J = 7.40 Hz, 8-H); 13C NMR (100 MHz, CDCl3): δ = 29.63 (CH3), 118.31, 121.50, 123.14, 123.86, 124.22, 124.39, 129.99, 130.22, 130.40, 130.77, 132.35, 133.54, 134.13, 141.53, 148.88, 150.21, 164.01, 184.72 (C-4); HRMS: m/z (M+) Cacld. for C18H10BrN3OS: 394.9728, found: 395.9782 [M+H]+.
1-(Benzo[d]thiazol-2-yl)-3-isopropylindeno[1,2-c]pyrazol-4(1H)-one (4f): 

Yellow solid; yield 67%; 200–202 °C; FTIR (KBr): νmax 756, 1039, 1398, 1502, 1548, 1598 (C=N stretch), 1701 (C=O stretch), 2929, 2972 (aliphatic C–H stretch), 3057 (aromatic C–H stretch) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.42 (6H, d, J= 6.96 Hz, –CH(CH3)2, 3.08–3.15 (1H, m,–CH(CH3)2), 7.35–7.43 (2H, m, Ar-H), 7.50–7.55 (2H, m, Ar-H), 7.60 (1H, d, J = 7.24 Hz, Ar-H), 7.87 (1H, d, J= 7.52 Hz, Ar-H), 8.03 (1H, d, J = 8.12 Hz, Ar-H), 8.56 (1H, d, J = 7.44 Hz, 8-H); 13C NMR (100 MHz, CDCl3): δ = 21.19 (–CH(CH3)2), 28.15 (–CH(CH3)2), 121.66, 122.81, 122.90, 124.25, 124.44, 125.38, 126.70, 130.66, 132.81, 133.38, 133.52, 139.20, 140.13, 150.99, 159.36, 159.52, 183.85 (C-4); HRMS: m/z (M+) Calcd. for C20H15N3OS: 345.0936, found: 346.1030 [M+H]+.
3-Isopropyl-1-(6-methylbenzo[d]thiazol-2-yl)indeno[1,2-c]pyrazol-4(1H)-one (4g): 

Yellow solid; yield 67%; mp 170–174 °C; FTIR (KBr): νmax 759, 1055, 1381, 1502, 1548, 1602 (C=N stretch), 1708 (C=O stretch), 2848, 2966 (aliphatic C–H stretch), 3061 (aromatic C–H stretch) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.42 (6H, d, J= 6.92 Hz, –CH(CH3)2, 2.51 (3H, s, CH3), 3.08–3.14 (1H, m, –CH(CH3)2), 7.32–7.37 (2H, m, Ar-H), 7.50 (1H, t, J = 7.64 Hz, Ar-H), 7.59 (1H, d, J = 6.96 Hz, Ar-H), 7.65 (1H, s, Ar-H), 7.90 (1H, d, J = 8.28 Hz, Ar-H), 8.53 (1H, d, J = 7.40 Hz, 8-H); 13C NMR (100 MHz, CDCl3): δ = 21.21 (–CH(CH3)2), 28.16 (–CH(CH3)2), 29.71 (CH3), 121.44, 122.35, 122.77, 124.21, 124.43, 128.19, 128.98, 130.58, 132.86, 133.48, 135.63, 140.19, 148.98, 158.62, 159.12, 159.23, 183.84 (C-4); HRMS: m/z (M+) Calcd. for C21H17N3OS: 359.1092, found: 360.1158 [M+H]+.
3-Isopropyl-1-(6-methoxybenzo[d]thiazol-2-yl)indeno[1,2-c]pyrazol-4(1H)-one (4h): 

Yellow solid; yield 65%; mp 212–214 °C; FTIR (KBr): νmax 759, 1089, 1398, 1504, 1550, 1608 (C=N stretch), 1703 (C=O stretch), 2837, 2962 (aliphatic C–H stretch), 3078 (aromatic C–H stretch) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.42 (6H, d, J= 6.92 Hz, –CH(CH3)2, 3.07–3.14 (1H, m,–CH(CH3)2), 3.90 (3H, s, OCH3), 7.11 (1H, dd, J = 2.36, J = 8.92 Hz, Ar-H), 7.32–7.37 (2H, m, Ar-H), 7.50 (1H, t, J = 7.56 Hz, Ar-H), 7.59 (1H, d, J = 7.24 Hz, Ar-H), 7.90 (1H, d, J = 8.92 Hz, Ar-H), 8.50 (1H, d, J = 7.44 Hz, 8-H); 13C NMR (100 MHz, CDCl3): δ = 21.22 (–CH(CH3)2), 28.14 (–CH(CH3)2), 55.94 (OCH3), 104.51, 115.71, 122.63, 123.38, 124.21, 124.33, 130.56, 132.83, 133.45, 134.72, 140.20, 145.19, 157.21, 157.91, 158.85, 159.17, 183.83 (C-4); HRMS: m/z (M+) Calcd. for C21H17N3O2S: 375.1041, found: 376.1103 [M+H]+.
1-(6-Chlorobenzo[d]thiazol-2-yl)-3-isopropylindeno[1,2-c]pyrazol-4(1H)-one (4i): 

Yellow solid; yield 70%; mp 182–184 °C; FTIR (KBr): νmax 763, 1109, 1381, 1500, 1544, 1597 (C=N stretch), 1703 (C=O stretch), 2870, 2964 (aliphatic C–H stretch), 3086 (aromatic C–H stretch) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.42 (6H, d, J= 6.92 Hz, –CH(CH3)2, 3.07–3.14 (1H, m, –CH(CH3)2), 7.36 (1H, t, J = 7.48 Hz, Ar-H), 7.45–7.51 (2H, m, Ar-H), 7.59 (1H, d, J = 7.20 Hz, Ar-H), 7.82 (1H, d, J = 1.64 Hz, Ar-H), 7.91 (1H, d, J = 8.68 Hz, Ar-H), 8.46 (1H, d, J = 7.40 Hz, 8-H); 13C NMR (100 MHz, CDCl3): δ = 21.13 (–CH(CH3)2), 28.12 (–CH(CH3)2), 121.29, 123.06, 123.49, 124.30, 127.46, 130.73, 131.17, 132.61, 133.47, 134.53, 140.01, 149.51, 159.30, 159.48, 159.80, 183.67 (C-4); HRMS: m/z (M+) Calcd. for C20H14ClN3OS: 379.0546, found: 380.0605 [M+H]+.
1-(6-Bromobenzo[d]thiazol-2-yl)-3-isopropylindeno[1,2-c]pyrazol-4(1H)-one (4j): 

Yellow solid; yield 65%; mp 190–194 °C; FTIR (KBr): νmax 738, 1078, 1390, 1502, 1539, 1593 (C=N stretch), 1707 (C=O stretch), 2870, 2960 (aliphatic C–H stretch), 3062 (aromatic C–H stretch) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.41 (6H, d, J= 6.92 Hz, –CH(CH3)2, 3.07–3.14 (1H, m,–CH(CH3)2), 7.37 (1H, t, J = 7.44 Hz, Ar-H), 7.51 (1H, t, J = 7.56 Hz, Ar-H), 7.59–7.64 (2H, m, Ar-H), 7.88 (1H, d, J = 8.64 Hz, Ar-H), 8.00 (1H, d, J = 1.76 Hz, Ar-H), 8.48 (1H, d, J = 7.28 Hz, 8-H); 13C NMR (100 MHz, CDCl3): δ = 21.15 (–CH(CH3)2), 28.14 (–CH(CH3)2), 118.70, 123.11, 123.90, 124.23, 124.34, 130.22, 130.79, 132.63, 133.53, 134.99, 140.02, 144.69, 149.89, 159.39, 159.55, 159.83, 183.74 (C-4); HRMS: m/z (M+) Calcd. for C20H14BrN3OS: 423.0041, found: 424.0100 [M+H]+.
1-(benzo[d]thiazol-2-yl)-3-isobutylindeno[1,2-c]pyrazol-4(1H)-one (4k): 

Yellow solid; yield 57%; mp 222–226 °C; FTIR (KBr): νmax 761, 1091, 1390, 1494, 1556, 1597 (C=N stretch), 1705 (C=O stretch), 2873, 2958 (aliphatic C–H stretch), 3066 (aromatic C–H stretch) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.01 (6H, d, J = 6.60 Hz, –CH2CH(CH3)2), 2.26–2.33 (1H, m, –CH2CH(CH3)2), 2.66 (2H, d, J = 7.28 Hz, –CH2CH(CH3)2), 7.34–7.43 (2H, m, Ar-H), 7.50–7.55 (2H, m, Ar-H), 7.59 (1H, d, J = 7.20 Hz, Ar-H), 7.87 (1H, d, J = 8.00 Hz, Ar-H), 8.03 (1H, d, J= 8.12 Hz, Ar-H), 8.55 (1H, d, J= 7.44 Hz, 8-H); 13C NMR (100 MHz, CDCl3): δ = 22.43 (–CH2CH(CH3)2), 27.79 (–CH2CH(CH3)2), 36.32 (–CH2CH(CH3)2), 121.63, 122.79, 123.98, 124.24, 124.43, 125.39, 126.69, 130.62, 132.67, 133.34, 133.53, 140.13, 148.32, 150.93, 152.78, 158.81, 184.14 (C-4); HRMS: m/z (M+) Calcd. for C21H17N3OS: 359.1092, found: 360.1155 [M+H]+.
3-Isobutyl-1-(6-methylbenzo[d]thiazol-2-yl)indeno[1,2-c]pyrazol-4(1H)-one (4l): 

Yellow solid; yield 54%; mp 186–190 °C; FTIR (KBr): νmax 773, 1134, 1444, 1465, 1562, 1602 (C=N stretch), 1693 (C=O stretch), 2868, 2953 (aliphatic C–H stretch), 3064 (aromatic C–H stretch) cm−1; 1H NMR (400 MHz, CDCl3): δ= 1.00 (6H, d, J = 6.60 Hz, –CH2CH(CH3)2), 1.96–2.02 (1H, m, –CH2CH(CH3)2), 2.41 (3H, s, CH3), 2.78 (2H, d, J = 7.44 Hz, –CH2CH(CH3)2), 7.31–7.40 (2H, m, Ar-H), 7.45–7.51 (2H, m, Ar-H), 7.77 (1H, t, J = 8.00 Hz, Ar-H), 8.02 (1H, d, J = 7.56 Hz, Ar-H), 8.40 (1H, d, J= 9.28 Hz, 8-H); 13C NMR (100 MHz, CDCl3): δ = 21.44 (–CH2CH(CH3)2), 22.31 (CH3), 28.00 (–CH2CH(CH3)2), 37.09 (-CH2CH(CH3)2), 119.77, 120.95, 121.95, 123.88, 125.12, 127.58, 128.98, 130.64, 132.01, 139.18, 140.27, 148.66, 150.06, 151.58, 154.85, 159.83, 182.64 (C-4); HRMS: m/z (M+) Calcd. for C22H19N3OS: 373.1249, found: 374.1309 [M+H]+.
3-Isobutyl-1-(6-methoxybenzo[d]thiazol-2-yl)indeno[1,2-c]pyrazol-4(1H)-one (4m): 

Yellow solid; yield: 56%; mp 158–160 °C; FTIR (KBr): νmax 759, 1089, 1390, 1496, 1550, 1605 (C=N stretch), 1705 (C=O stretch), 2868, 2956 (aliphatic C–H stretch), 3061 (aromatic C–H stretch) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.02 (6H, d, J = 6.6 Hz, –CH2CH(CH3)2), 2.26–2.33(1H, m, –CH2CH(CH3)2), 2.66 (2H, d, J = 7.28 Hz, –CH2CH(CH3)2), 3.91 (3H, s, OCH3), 7.12 (1H, dd, J = 2.28, 6.60 Hz, Ar-H), 7.32–7.37 (2H, m, Ar-H), 7.51(1H, t, J = 7.48Hz, Ar-H), 7.59 (1H, d, J = 7.24 Hz, Ar-H), 7.91 (1H, d, J = 8.92 Hz, Ar-H), 8.50 (1H, d, J= 7.48 Hz, 8-H); 13C NMR (100 MHz, CDCl3): δ = 22.52 (–CH2CH(CH3)2), 29.74 (–CH2CH(CH3)2), 36.41 (–CH2CH(CH3)2), 55.94 (OCH3), 104.56, 115.82, 123.45, 123.83, 124.28, 124.41, 127.13, 130.61, 132.79, 133.55, 134.79, 136.96, 140.30, 145.23, 150.48, 152.66, 158.00, 184.23 (C-4); HRMS: m/z (M+) Calcd. for C22H19N3O2S: 389.1198, found: 390.1258 [M+H]+.
1-(6-Chlorobenzo[d]thiazol-2-yl)-3-isobutylindeno[1,2-c]pyrazol-4(1H)-one (4n): 

Yellow solid; yield: 49%; mp 140–142 °C; FTIR (KBr): νmax 775, 1109, 1388, 1494, 1537, 1593 (C=N stretch), 1708 (C=O stretch), 2870, 2962 (aliphatic C–H stretch), 3088 (aromatic C–H stretch) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.02 (6H, d, J = 6.60 Hz, –CH2CH(CH3)2), 2.26–2.33 (1H, m, –CH2CH(CH3)2), 2.66 (2H, d, J = 7.32 Hz, –CH2CH(CH3)2), 7.35–7.45 (2H, m, Ar-H), 7.70 (1H, t, J = 7.40 Hz, Ar-H), 7.80–7.85 (2H, m, Ar-H), 7.95 (1H, d, J = 8.68 Hz, Ar-H), 8.49 (1H, d, J= 7.48 Hz, 8-H); 13C NMR (100 MHz, CDCl3): δ = 22.47 (–CH2CH(CH3)2), 27.82 (–CH2CH(CH3)2), 36.34 (–CH2CH(CH3)2), 120.97, 121.36, 121.49, 123.57, 124.37, 124.47, 127.28, 127.54, 130.78, 133.58, 140.09, 149.54, 151.28, 152.08, 153.03, 158.93, 185.16 (C-4); HRMS: m/z (M+) Calcd. for C21H16ClN3OS: 393.0703, found: 394.0763 [M+H]+.
1-(6-Bromobenzo[d]thiazol-2-yl)-3-isobutylindeno[1,2-c]pyrazol-4(1H)-one (4o):

Yellow solid; yield 78%; mp 168–170 °C; FTIR (KBr): νmax 763, 1097, 1392, 1494, 1537, 1593 (C=N stretch), 1707 (C=O stretch), 2870, 2960 (aliphatic C–H stretch), 3088 (aromatic C–H stretch) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.02 (6H, d, J = 6.64 Hz, –CH2CH(CH3)2), 2.25–2.35 (1H, m, –CH2CH(CH3)2), 2.66 (2H, d, J = 7.28 Hz, –CH2CH(CH3)2), 7.36–7.45 (2H, m, Ar-H), 7.50– 7.58 (2H, m, Ar-H), 7.76 (1H, d, J = 8.64 Hz, Ar-H), 7.89 (1H, d, J = 8.64 Hz, Ar-H), 8.49 (1H, d, J= 7.40 Hz, 8-H); 13C NMR (100 MHz, CDCl3): δ = 22.47 (–CH2CH(CH3)2), 27.81 (–CH2CH(CH3)2), 36.34 (–CH2CH(CH3)2), 121.49, 123.85, 123.92, 124.24, 124.37, 124.47, 130.00, 130.46, 132.55, 133.57, 134.15, 140.09, 149.88, 150.21, 153.04, 159.80, 184.05 (C-4); HRMS: m/z (M+) Calcd. for C21H16BrN3OS: 437.0197, found: 438.0257 [M+H]+.
Biological studies
Enzyme assay
In vitro α-glucosidase inhibition
McCue’s protocol was followed for evaluation of in vitro α-glucosidase inhibitory activity, with some modifications (McCue et al. 2005). The present activity was carried by using α-glucosidase enzyme (Saccharomyces cereviciae). A solution of the enzyme was obtained by adding 20 μL α-glucosidase (0.5 unit/mL) in 120 μL of 0.1 M phosphate buffer (pH 6.9). In microplate wells, the enzyme solution was mixed with 10 μL of each test samples which, in turn, were prepared by dissolving in dimethylsulphoxide (DMSO) at various concentrations i.e. 12.5, 25, 50, 100 μg/mL and incubated for 15 min at 37 °C. Thereafter, this was charged with 20 μL of substrate solution to 5 mM p-nitrophenyl-α-D-glucopyranoside in 0.1 M phosphate buffer (pH 6.9) and further incubated for 15 min. A solution of 0.2 M sodium carbonate (80 μL) was added to terminate the reaction, and absorbance was measured at λ = 405 nm on ELISA microplate reader. The reaction system without test samples (4a–o) was used as control while the system without α-glucosidase was used as a blank, and Acarbose was used as positive control. Each experiment was performed in triplicate. The enzyme inhibitory rates of samples have been expressed as percentage (%) inhibition which is determined by Eq. (1) as follows:
The IC50 values of compounds 4a–o were calculated.
In vitro α-amylase inhibition
The protocol reported by Xiao et al. and Yoshikawa et al. with slight modifications was utilized for the evaluation of in vitro α-amylase inhibition activity (Xiao et al. 2006; Yoshikawa et al. 2001). Stock solutions of compounds 4a–o were prepared by dissolving the compound (5 mg) in DMSO (5 mL) at room temperature. The α-amylase inhibitory activity was examined at different concentrations of each sample i.e., 12.5, 25, 50, and 100 μg/mL. The reagent solution without the test sample was used as the control and Acarbose was used as standard reference. Substrate solution was prepared by dissolving soluble starch (500 mg) in 0.4 M NaOH (25 mL) and heated for 5 min at 100 °C. After cooling in ice cold water, the pH of the solution was achieved to 7 by adding 2 M HCl, and water was added to make the volume to 100 mL. The sample (20 μL) and substrate (40 μL) solutions were mixed in a microplate well and the mixture in each case was preincubated at 37 °C for 3 min. Thereafter, 20 μL of α-amylase solution (50 μg/mL) was added to each well, and the microplate was incubated for 15 min. The reaction was terminated by adding 0.1 M HCl (80 μL). Then 1 mM iodine solution (200 μL) was added to the reaction mixture and absorbance was measured at λ = 650 nm with ELISA microplate reader. The enzyme inhibitory activity expressed as percentage (%) inhibition was calculated by Eq. (2) as follows:
where, Abs1 = Absorbance of incubated solution containing test sample, starch and amylase, Abs2 = Absorbance of incubated solution containing test sample and starch, Abs3 = Absorbance of incubated solution containing starch and amylase, and Abs4 = Absorbance of incubated solution containing starch.
In vitro antimicrobial assay
Test microorganism
Two Gram-positive bacteria viz. B. subtilis (NCIM 20630) and S. aureus (NCIM 5021), and two Gram-negative bacteria viz. E. coli (MTCC 723) and P. aeruginosa (MTCC 7093), and one fungal strain viz. A. niger (MTCC 9933) were used for antimicrobial assay. All the bacterial cultures were obtained from Microbial Type Culture Collection (MTCC), Institute of Microbial Technology (IMTECH), Chandigarh, India. The nutrient broth utilized for the cultivation of microorganisms was procured from HiMedia Laboratories Pvt. Ltd., Mumbai, India.
Antibacterial evaluation
All the synthesized compounds 4a‒o were screened for their antibacterial activity using agar well diffusion method (Okeke et al. 2001). The test microorganisms were enthused by inoculation in 25 mL of nutrient broth (peptone 5 g/L, sodium chloride 5 g/L, HM peptone 1.5 g/L, yeast extract 1.5 g/L, pH = 7.4 ± 0.2). The media was solidified and the test bacterial strains were cultivated by pour plate method on nutrient agar plates. Wells were bored in the seeded agar plates by using sterile cork borer of 8 mm diameter and these were loaded with a 100 μL of each synthesized compound reconstituted in DMSO. All the plates were incubated at 37 °C for 24 h. The diameter of inhibition zone of the test organisms was measured by using Digital Colony Counter (Lab and Life Instruments Pvt. Ltd., India) and reported in mm. Sterile DMSO was used as a negative control, whereas Streptomycin was used as a positive control. The experiments were performed in triplicates and the mean values are reported.
Antifungal evaluation
Antifungal activity of the title compounds 4a‒o was examined against A. niger by a quantitative microspectrophotometric assay (Broekaert et al. 1990) in 96-well microplates. The growth inhibition was observed at 595 nm. Initially, the fungus was grown on potato dextrose broth (PDB) (Potatoes, infusion form = 200 g/L, Dextrose = 20 g/L, pH = 5.1 ± 0.2 at 25 °C) at 27 °C for 7 days. The spores of the fungus were collected from culture on broth plates. The sporangial suspension concentration was measured by hemocytometer and made to 1.7 × 105 spores/mL and the fungal spore suspension was stored at −40 °C. The PDB-fungal spore suspension solution was prepared by mixing PDB (25 mL) with 0.147 ml of the fungal spore suspension solution. Experiment was performed with 100 μL of each of the compounds 4a‒o to be assayed and 100 μL of PDB-fungal spore suspension solution. DMSO was used as a negative control and Fluconazole was used as a positive control. After incubation at 27 °C for 48 h, growth was observed by measuring absorbance at 595 nm on ELISA plate reader. Growth inhibition was reported by using Eq. (3) given as follows:
where Acontrol is the absorbance of the control and Asample is the absorbance of the tested microculture.
Experimental protocol of molecular docking study
Molecular docking analysis was performed using Autodock Vina in Autodock tools (Trott and Olson 2010). For α-glucosidase (PDB ID: 5NN5), the coordinates of center of grid box were center_x = −16.224, center_y = −35.668 and center_z = 95.439, the size of grid box was x_size = 24 Å, y_size = 24 Å and z_size = 24 Å, and the exhaustiveness was equal to 40. For α-amylase (PDB ID: 2QV4), the coordinates of center of grid box were center_x = 20.074, center_y = 46.866 and center_z = 29.866, the size of grid box was x_size = 28 Å, y_size = 28 Å and z_size = 28 Å, and the exhaustiveness was equal to 40.
Results and discussion
Chemistry
Protocol for the synthesis of some new benzothiazole tethered indeno[1,2-c]pyrazole derivatives (4a–o) is shown in Scheme 1. The chemistry formerly described was utilized for the synthesis of 2-acyl-(1H)-indene-1,3(2H)-diones (1) (Mor et al. 2016). Benzo[d]thiazol-2-amine/6-substitutedbenzo[d]thiazol-2-amines and 2-hydrazinylbenzo[d]-thiazole/2-hydrazinyl-6-substitutedbenzo[d]thiazoles (2) were synthesized by the method reported earlier (Mor et al. 2017). Condensation of appropriate 1,3-diketones (1) with hydrazines (2), in equimolar quantities, in dry methanol under reflux for 15 min yielded the corresponding benzothiazolyl hydrazones (3) (Mor et al. 2019). The hydrazones (3) thus obtained were subjected to reflux in glacial acetic acid to furnish the target indeno[1,2-c]pyrazol-4-ones (4a–o) in good yields.

Synthesis of indeno[1,2-c]pyrazol-4-ones (4a–o)
Structures of the newly synthesized compounds 4a–o were confirmed by their FT-IR, NMR (1H and 13C), and mass spectra. Their FT-IR spectra exhibited strong absorption bands at 1585–1608 (C=N) and 1693–1716 (C=O) cm−1. The main characteristic feature of 1H NMR spectra of derivatives 4a–o is the resonance of a signal appeared as a doublet integrating for one proton in the range of δ 8.03–8.56 ppm, (J = 7.28–8.00 Hz), which was safely assigned to 8-H. Downfield shifting of this proton is probably due to anisotropic–diamagnetic effect of lone pair of electrons present on nitrogen/sulfur of benzothiazole moiety, which finds support from the results reported earlier (Mor et al. 2016). The significant feature of 13C NMR spectra of compounds 4a–o demonstrated the downfield shifting of signal due to C-4 (carbonyl carbon) which was observed in the region at δ 182.64‒185.16 ppm. However, the remaining protons in 1H NMR and carbons in 13C NMR spectra displayed signals in the expected regions. Further, the HRMS analysis results were found in consistent with their molecular formulae (vide experimental).
Biological studies
In search of new antidiabetic agents, we recognized primarily various pyrazole derivatives as reported in the literature (Wright et al. 1964). To the best of our knowledge, this is the first report of antidiabetic activity possessed by synthesized indeno[1,2-c]pyrazol-4-ones (4a–o).
In vitro α-glucosidase inhibitory activity
All synthesized compounds 4a–o were assessed for their α-glucosidase inhibitory activity against α-glucosidase enzyme (Saccharomyces cerevisiae) at various concentrations ranging from 12.5 to 100 μg/mL following the developed earlier procedure (McCue et al. 2005) by using Acarbose as the standard (Table 1).
It is inferred from the data presented in Table 1 that all the derivatives exhibited moderate to excellent % inhibition against α-glucosidase enzyme as compared with the standard. Compound 4i was found to be more potent analogue of this series with 67.02, 86.27, 90.47, and 94.52% inhibition when explored at the concentrations of 12.5, 25, 50, and 100 μg/mL, respectively. Similarly, compound 4e exhibited a rise in % inhibition from 76.41 to 82.76% on increasing the concentration from 12.5 to 50 μg/mL in comparison to the standard drug Acarbose. Compound 4l displayed 90.78% inhibition followed by 4a that exhibited 87.26% inhibition at concentration of 100 μg/mL. Among the synthesized indeno[1,2-c]pyrazol-4-ones, 4e and 4i were found to be more active with IC50 values 6.71 and 8.18 μg/mL, respectively (Acarbose IC50 = 9.35 μg/mL). However, the derivatives 4a and 4b exhibited good inhibitory activity with IC50 values 9.87 and 10.59 μg/mL, respectively.
In vitro α-amylase inhibitory activity
The α-amylase inhibitory activity of the synthesized compounds 4a–o was screened following Xiao’s procedure (Xiao et al. 2006; Yoshikawa et al. 2001) by using Acarbose as the standard reference (Table 2).
The results of the α-amylase inhibitory activity depicted in Table 2 revealed that all the tested derivatives 4a–o displayed moderate to high % inhibition. Compounds 4a, 4e, 4i, 4l, and 4o at concentration 12.5 μg/mL, 4e, 4f, 4i, 4l, and 4o at concentration 25 μg/mL, and 4a and 4o at concentration 50 μg/mL were found to be more potent than the standard. Whereas compound 4i displayed inhibition equivalent to the standard drug Acarbose at concentration 100 μg/mL. The analogues 4c and 4j at concentration 12.5 μg/mL, 4a and 4d at concentration 25 μg/mL, 4b, 4d, 4e, and 4i at concentration 50 μg/mL, and 4a and 4c at concentration 100 μg/mL demonstrated comparable inhibitory activity as shown by the standard drug Acarbose. Furthermore, the remaining compounds were found to display lesser inhibitory activity as compared with the standard drug screened at different concentrations. Among the synthesized derivatives, 4a (IC50 = 21.23 μg/mL), 4e (IC50 = 19.25 μg/mL), 4i (IC50 = 11.90 μg/mL), and 4o (IC50 = 12.83 μg/mL) demonstrated higher activity than the standard (Acarbose, IC50 = 22.87 μg/mL). Consequently, 4a, 4e, 4i, and 4o can be considered as a possible antidiabetic agent for further studies.
Structure activity relationship (SAR) for antidiabetic activity of indeno[1,2-c]pyrazol-4-ones (4a‒o)
According to the presented data for antidiabetic activity of indeno[1,2-c]pyrazol-4-ones (4a‒o), the following SARs have been established:
-
(1)
Results of antidiabetic activity indicated that the presence of R1 = CH3 and R2 = H, Br in the synthesized compounds 4a–o has led to increase the antidiabetic activity against α-glucosidase enzyme while the derivative 4e containing R1 = CH3 and R2 = Br has been found to exhibit improved inhibitory activity against α-amylase enzyme.
-
(2)
Compound 4i containing R1 = i-propyl and R2 = Cl has been found to enhance inhibitory activity against both α-glucosidase and α-amylase enzymes.
-
(3)
Derivative 4l containing R1 = i-butyl and R2 = CH3 has improved inhibitory activity against α-glucosidase, whereas presence of R1 = i-butyl and R2 = Br in compound 4o had increased inhibitory activity against α-amylase enzyme.
From these results, it is inferred that the compounds containing R1 = CH3 and R2 = H, CH3, OCH3, Cl, and Br, are found to display more inhibitory activity against α-glucosidase as compared with the remaining derivatives containing R1 = i-propyl and i-butyl, and R2 = H, CH3, OCH3, Cl, and Br, whereas no such trend was observed against α-amylase inhibitory activity. Overall, we may conclude that there are different structural requirements for a compound to be effective against α-glucosidase and α-amylase enzymes. However, no general trend for SAR was established for both α-glucosidase and α-amylase enzymes. The above mentioned findings are summarized in Fig. 1.

Structure activity relationship (SAR) for antidiabetic activity of synthesized indeno[1,2-c]pyrazol-4-ones (4a‒o)
In vitro antibacterial activity
All synthesized indeno[1,2-c]pyrazol-4-ones (4a–o) were tested for their in vitro antibacterial activity against Gram-positive (B. subtilis, S. aureus) and Gram-negative (E. coli, P. aeruginosa) bacteria by agar well diffusion method using Streptomycin as the reference drug (Table 3) (Okeke et al. 2001).
It is revealed from the data presented in Table 3 that compound 4l and 4g exhibited the highest activity against B. subtilis, while the derivative 4h demonstrated maximum inhibition zone against S. aureus. Moreover, the analogues 4f and 4j displayed moderate activity against E. coli, while 4g demonstrated activity against P. aeruginosa. On the other hand, some compounds were inactive against the specific bacteria under study. Overall, these results suggest that the synthesized compounds 4a–o exhibit lesser activity against the bacterial strains than standard drug Streptomycin.
In vitro antifungal activity
All synthesized compounds 4a–o were evaluated for their in vitro antifungal activity against A. niger by the quantitative microspectrophotometric assay using Fluconazole as the standard drug (Broekaert et al. 1990) (Table 4).
A perusal of accumulated data from Table 4 reveals that all synthesized compounds 4a–o were found to inhibit fungal growth with inhibition ranging from 49.21 to 72.25%, 54.71 to 76.96%, 59.42 to 80.89%, and 67.01 to 84.55% at concentration 125, 250, 500, and 1000 μg/mL, respectively. Derivative 4o was found to be more active with IC50 value of 5.68 μg/mL than the standard (IC50 = 12.73 μg/mL). Compounds 4d, 4f, 4h, and 4n demonstrated comparable activity to standard drug Fluconazole. Overall, these results suggest that the synthesized compounds 4a–o demonstrated good activity at different concentrations against the fungal strain.
SARs for antimicrobial activity of indeno[1,2-c]pyrazol-4-ones (4a‒o)
The SARs approach to the synthesized derivatives 4a–o demonstrated good to moderate inhibitory potential against all the microbial strains under study. Consequently, we may conclude that there are different structural requirements for a compound to be effective against different bacterial and fungal strains. Moreover, no general trend has been established for the SARs for antimicrobial activity.
Molecular docking analysis
Molecular docking analysis of some selected synthesized compounds with enzymes α-glucosidase and α-amylase was performed to find out the mechanism of action at the molecular level.
Molecular docking with α-glucosidase
Crystal structure used for α-glucosidase was obtained from protein data bank and the PDB ID for this structure is 5NN5 (Roig-Zamboni et al. 2017). Docking protocol was validated by docking the co-crystallized ligand. In vitro assay showed that compounds 4e and 4i exhibit best α-glucosidase inhibitory potential, hence molecular docking analysis was performed using these two compounds. Compound 4e with a docking score of −7.3 showed three pi–pi interactions with amino acid residue PHE-525, TRP-376 (T shaped), and TRP-481 (T shaped) as shown in Fig. 2 and hydrophobic interactions with TRP-481 and PHE-649. Compound 4i with docking score of −8.4 showed interactions very similar to that of 4e i.e., three pi–pi interactions with amino acid residue PHE-525, TRP-376 (T shaped), and TRP-481 (T shaped) as shown in Fig. 3 and further stabilizing interactions are provided by hydrophobic contacts with ASP-282, ALA-555, and PHE-649. Both the compounds were found to bind in similar orientation with a snug fit.

Docked pose of compound 4e (green) showing three pi-pi interactions in blue color with amino acid residue PHE-525, TRP-376 (T shaped), and TRP-481 (T shaped) of α-glucosidase

Docked pose of compound 4i (grey) inside the active site of α-glucosidase showing three pi-pi interactions in blue color with amino acid residue PHE-525, TRP-376 (T shaped), and TRP-481 (T shaped)
Molecular docking with α-amylase
In vitro results showed that most of the synthesized compounds 4a‒o were stronger inhibitors of α-amylase compared with acarbose. Compounds 4i and 4o showed superior inhibition as compared with the other synthesized compounds. Therefore, 4i and 4o compounds were used for determining the binding pose and interactions responsible for the activity against human pancreatic α-amylase (PDB ID: 2QV4) (Maurus et al. 2008). First docking protocol was validated by performing docking of co-crystallized ligand. Compound 4o with docking score of −8.8 was found to be showing two hydrogen bond interactions with amino acid residues GLN-63 and THR-163, three pi–pi interactions with TRP-59 and hydrophobic contacts with TYR-62, LEU-162, and LEU-165. The binding pose of compound 4o is shown in Fig. 4. Compound 4i with docking score of −9.3 binds in active site of α-amylase with orientation and interactions similar to that of 4o. It is showing one hydrogen bond with GLN-63, one halogen bond with ASP-197, three pi–pi interactions with TRP-59 and hydrophobic contacts with TRP-58 and TYR-62 as shown in Fig. 5.

Docked pose of compound 4o (white) showing two hydrogen bonding interactions (yellow) with GLN-63 and THR-163 and three Pi-Pi interactions (blue) with TRP-59 of α-amylase

Docked pose of compound 4i (green) showing one hydrogen bond with GLN 63 (pink), one halogen bond with ASP 197 (violet), and 3 pi-pi interactions with TRP 59 (blue) of α-amylase
Conclusion
In conclusion, the present study describe the synthesis and characterization of heterocyclic frameworks i.e., indeno[1,2-c]pyrazoles (4), and their biological evaluation as inhibitor of α-glucosidase and α-amylase related to Type II diabetes, and antimicrobial activity. The chemistry for the synthesis of indeno[1,2-c]pyrazole (4) involved the reaction of 2-acyl-(1H)-indene-1,3(2H)-diones (1) with 2-hydrazinylbenzo[d]thiazole/2-hydrazinyl-6-substitutedbenzo[d]thiazoles (2) in dry methanol to give benzothiazolyl hydrazones (3), which upon subsequent refluxing in glacial acetic acid afforded the target compounds 4 in good yields. Some of the synthesized compounds exhibited significant in vitro α-glucosidase and α-amylase inhibitory activity viz. 4e was found to be more potent with IC50 value 6.71 μg/mL against α-glucosidase enzyme and 4i showed good activity with IC50 value 11.90 μg/mL against α-amylase enzyme as compared with reference drug Acarbose (IC50 = 22.87 μg/mL). Moreover, some of the compounds exhibited convincing results for antimicrobial activity, however, with a degree of variation. The antidiabetic activity was found to be more prolific than antimicrobial activity. In vitro α-glucosidase and α-amylase inhibition was further supported by docking studies of compounds 4e, 4i, and 4o. Hopefully, these findings will prove helpful to medicinal chemists for the development of new inhibitors of enzymes related to Type II diabetes.
References
Ahsan MJ, Samy JG, Soni S, Jain N, Kumar L, Sharma LK, Prasad R (2011) Discovery of novel antitubercular 3a,4-dihydro-3H-indeno[1,2-c]pyrazole-2-carboxamide/carbothioamide analogues. Bioorg Med Chem Lett 21(18):5259–5261
Ahsan MJ, Govindasamy J, Khalilullah H, Mohan G, Stables JP, Pannecouque C, De Clercq E (2012) POMA analyses as new efficient bioinformatics platform to predict and optimise bioactivity of synthesized 3a,4-dihydro-3H-indeno[1,2-c]pyrazole-2-carboxamide/carbothioamide analogues. Bioorg Med Chem Lett 22(23):7029–7035
Ajiboye BO, Ojo OA, Adeyonu O, Imiere O, Olayide I, Fadaka A, Oyinloye BE (2016) Inhibitory effect on key enzymes relevant to acute type-2 diabetes and antioxidative activity of ethanolic extract of artocarpus heterophyllus stem bark. J Acute Dis 5(5):423–429
Amnerkar ND, Bhusari KP (2010) Synthesis, anticonvulsant activity and 3D-QSAR study of some prop-2-eneamido and 1-acetyl-pyrazolin derivatives of aminobenzothiazole. Eur J Med Chem 45(1):149–159
Bhat M, Belagali SL (2016) Guanidinyl benzothiazole derivatives: Synthesis and structure activity relationship studies of a novel series of potential antimicrobial and antioxidants. Res Chem Intermed 42(7):6195–6208
Broekaert WF, Terras FRG, Cammue BPA, Vanderleyden J (1990) An automated quantitative assay for fungal growth inhibition. FEMS Microbiol Lett 69:55–60
Carvalho VF, Barreto EO, Serra MF, Cordeiro RS, Martins MA, Fortes ZB, e Silva PM (2006) Aldose reductase inhibitor zopolrestat restores allergic hyporesponsiveness in alloxan-diabetic rats. Eur J Pharm 549(1):173–178
Chaudhry F, Naureen S, Huma R, Shaukat A, al-Rashida M, Asif N, Khan MA (2017) In search of new α-glucosidase inhibitors: Imidazolylpyrazole derivatives. Bioorg Chem 71:102–109
Chugunova E, Boga C, Sazykin I, Cino S, Micheletti G, Mazzanti A, Kostina N (2015) Synthesis and antimicrobial activity of novel structural hybrids of benzofuroxan and benzothiazole derivatives. Eur J Med Chem 93:349–359
Doddaramappa SD, Rai KL, Srikantamurthy N, Chethan J (2015) Novel 5-functionalized-pyrazoles: synthesis, characterization and pharmacological screening. Bioorg Med Chem Lett 25(17):3671–3675
Gabr MT, El-Gohary NS, El-Bendary ER, El-Kerdawy MM (2015) New series of benzothiazole and pyrimido[2,1-b]benzothiazole derivatives: synthesis, antitumor activity, EGFR tyrosine kinase inhibitory activity and molecular modeling studies. Med Chem Res 24(2):860–878
Huang G, Huang H (2019) Synthesis, antiasthmatic, and insecticidal/antifungal activities of allosamidins. J Enzym Inhib Med Chem 34(1):1226–1232
International Diabetes Foundation (2017) IDF diabetes atlas. 8th ed. International Diabetes Foundation, Brussels
Kamal A, Syed MAH, Mohammed SM (2015) Therapeutic potential of benzothiazoles: a patent review (2010–2014). Expert Opin Ther Pat 25(3):335–349
Khan I, Garikapati KR, Setti A, Shaik AB, Makani VKK, Shareef MA, Kumar CG (2019) Design, synthesis, in silico pharmacokinetics prediction and biological evaluation of 1,4-dihydroindeno[1,2-c]pyrazole chalcone as EGFR/Akt pathway inhibitors. Eur J Med Chem 163:636–648
Khan I, Shareef MA, Kumar CG (2019) An overview on the synthetic and medicinal perspectives of indenopyrazoles Eur J Med Chem 178:1–12
Keri RS, Patil MR, Patil SA, Budagumpi S (2015) A comprehensive review in current developments of benzothiazole-based molecules in medicinal chemistry. Eur J Med Chem 89:207–251
Kim MB, O’Brien TE, Moore JT, Anderson DE, Foss MH, Weibel DB, Shaw JT (2012) The synthesis and antimicrobial activity of heterocyclic derivatives of totarol. ACS Med Chem Lett 3(10):818–822
Kumar P, Duhan M, Kadyan K, Sindhu J, Kumar S, Sharma H (2017) Synthesis of novel inhibitors of α-amylase based on the thiazolidine-4-one skeleton containing a pyrazole moiety and their configurational studies. Med Chem Comm 8(7):1468–1476
Lemke TL, Abebe E, Moore PF, Carty TJ (1989) Indeno [1,2‐c] pyrazolone acetic acids as semirigid analogues of the nonsteroidal anti‐inflammatory drugs. J Pharm Sci 78(4):343–347
Lemke TL, Cramer MB, Shanmugam K (1978) Heterocyclic tricycles as potential CNS agents I: 4‐aminoalkylindeno [1,2‐c] pyrazoles. J Pharm Sci 67(10):1377–1381
Maurus R, Begum A, Williams LK, Fredriksen JR, Zhang R, Withers SG, Brayer GD (2008) Alternative catalytic anions differentially modulate human α-amylase activity and specificity. Biochemistry 47(11):3332–3344
McCue P, Kwon Y-I, Shetty K (2005) Anti-amylase, anti-glucosidase and anti-angiotensin I-converting enzyme potential of selected foods. J Food Biochem 29(3):278–294
Meltzer-Mats E, Babai-Shani G, Pasternak L, Uritsky N, Getter T, Viskind O, Gruzman A (2013) Synthesis and mechanism of hypoglycemic activity of benzothiazole derivatives. J Med Chem 56(13):5335–5350
Minegishi H, Futamura Y, Fukashiro S, Muroi M, Kawatani M, Osada H, Nakamura H (2015) Methyl 3-((6-methoxy-1,4-dihydroindeno[1,2-c]pyrazol-3-yl)amino) benzoate (GN39482) as a tubulin polymerization inhibitor identified by MorphoBase and ChemProteoBase profiling methods. J Med Chem 58(10):4230–4241
Mor S, Nagoria S, Kumar A, Monga J, Lohan S (2016) Convenient synthesis, anticancer evaluation and QSAR studies of some thiazole tethered indenopyrazoles. Med Chem Res 25(6):1096–1114
Mor S, Mohil R, Nagoria S, Kumar A, Lal K, Kumar D, Singh V (2017) Regioselective synthesis, antimicrobial evaluation and QSAR studies of some 3‐aryl‐1‐heteroarylindeno [1,2‐c]pyrazol‐4 (1H)‐ones. J Heterocycl Chem 54(2):1327–1341
Mor S, Nagoria S, Sindhu S, Khatri M, Sidhu G, Singh V (2017) Synthesis of indane‐Based 1,5‐benzothiazepines derived from 3‐Phenyl‐2,3‐dihydro‐1H‐inden‐1‐one and antimicrobial studies thereof. J Heterocycl Chem 54(6):3282–3293
Mor S, Sindhu S, Nagoria S, Khatri M, Garg P, Sandhu H, Kumar A (2019) Synthesis, biological evaluation, and molecular docking studies of some N‐thiazolyl hydrazones and indenopyrazolones. J Heterocycl Chem 56(5):1622–1633
Mor S, Sindhu S, Khatri M, Singh N, Vasudeva N, Panihar N (2019) Synthesis, type II Diabetes inhibitory activity, and antimicrobial tests of benzothiazole derivatives bridged with indenedione by methylenehydrazone. Russ J Gen Chem 89(9):1867–1873
Okeke MI, Iroegbu CU, Eze EN, Okoli AS, Esimone CO (2001) Evaluation of extracts of the root of landolphia owerrience for antibacterial activity. J Ethnopharmacol 78:119–127
Patel RV, Kumari P, Rajani DP, Chikhalia KH (2013) Synthesis of coumarin-based 1,3,4-oxadiazol-2ylthio-N-phenyl/benzothiazolyl acetamides as antimicrobial and antituberculosis agents. Med Chem Res 22(1):195–210
Patil VS, Nandre KP, Ghosh S, Rao VJ, Chopade BA, Sridhar B, Bhosale SV (2013) Synthesis, crystal structure and antidiabetic activity of substituted (E)-3-(benzo[d]thiazol-2-ylamino) phenylprop-2-en-1-one. Eur J Med Chem 59:304–309
Roig-Zamboni V, Cobucci-Ponzano B, Iacono R, Ferrara MC, Germany S, Bourne Y, Sulzenbacher G (2017) Structure of human lysosomal acid α-glucosidase–a guide for the treatment of Pompe disease. Nat Commun 8(1):1–10
Sarkar S, Dwivedi J, Chauhan R (2013) Synthesis of 1-[2(substituted phenyl)-4-oxothiazolidin-3-yl]-3-(6-fluro-7-chloro-1,3-benzothiazol-2-yl)-ureas as anthelmintic agent. J Pharm Res 7(5):439–442
Scott KA, Njardarson JT (2018) Analysis of US FDA-approved drugs containing sulfur atoms. Top Curr Chem 376(1):1–34
Sawhney KN, Lemke TL (1983) Chemistry of β-triketones. 1. Structure of Schiff base intermediates of 2-acyl-1,3-indandiones. J Org Chem 48(23):4326–4329
Shafi S, Alam MM, Mulakayala N, Mulakayala C, Vanaja G, Kalle AM, Alam MS (2012) Synthesis of novel 2-mercaptobenzothiazole and 1,2,3-triazole based bis-heterocycles: their anti-inflammatory and anti-nociceptive activities. Eur J Med Chem 49:324–333
Shareef MA, Sirisha K, Khan I, Sayeed IB, Jadav SS, Ramu G, Babu BN (2019) Design, synthesis, and antimicrobial evaluation of 1,4-dihydroindeno[1,2-c]pyrazole tethered carbohydrazide hybrids: exploring their in silico ADMET, ergosterol inhibition and ROS inducing potential. Med Chem Comm 10(5):806–813
Singh SK, Dessalew N, Bharatam PV (2006) 3D-QSAR CoMFA study on indenopyrazole derivatives as cyclin dependent kinase 4 (CDK4) and cyclin dependent kinase 2 (CDK2) inhibitors. Eur J Med Chem 41(11):1310–1319
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461
Wagman AS, Boyce RS, Brown SP, Fang E, Goff D, Jansen JM, Nuss JM (2017) Synthesis, binding mode, and antihyperglycemic activity of potent and selective (5-imidazol-2-yl-4-phenylpyrimidin-2-yl)[2-(2-pyridylamino)ethyl] amine inhibitors of glycogen synthase kinase 3. J Med Chem 60(20):8482–8514
Wright JB, Dulin WE, Markillie JH (1964) The antidiabetic activity of 3,5-dimethylpyrazoles. J Med Chem 7(1):102–105
Xiao Z, Storms R, Tsang A (2006) A quantitative starch-iodine method for measuring alpha-amylase and glucoamylase activities. Anal Biochem 351(1):146–148
Yevich JP, New JS, Smith DW, Lobeck WG, Catt JD, Minielli JL, Temple Jr. DL (1986) Synthesis and biological evaluation of 1-(1,2-benzisothiazol-3-yl) and (1,2-benzisoxazol-3-yl) piperazine derivatives as potential antipsychotic agents. J Med Chem 29(3):359–369
Yoshikawa M, Nishida N, Shimoda H, Takada M, Kawahara Y, Matsuda H (2001) Polyphenol constituents from salacia species: quantitative analysis of mangiferin with α-glucosidase and aldose reductase inhibitory activities. Yakugaku zasshi J 121(5):371–378
Zhou S, Yang S, Huang G (2017) Design, synthesis and biological activity of pyrazinamide derivatives for anti-Mycobacterium tuberculosis. J Enzym Inhib Med Chem 32(1):1183–1186
Acknowledgements
The authors are grateful to the Council of Scientific and Industrial Research, New Delhi, India for providing financial support (CSIR No. 09/752(0060)/2016-EMR-I).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
About this article

Cite this article
Mor, S., Sindhu, S. Synthesis, Type II diabetes inhibitory activity, antimicrobial evaluation and docking studies of indeno[1,2-c]pyrazol-4(1H)-ones. Med Chem Res 29, 46–62 (2020). https://doi.org/10.1007/s00044-019-02457-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-019-02457-8