
Abstract
Despite the manifold recent efforts to improve patient outcomes, trauma still is a clinical and socioeconomical issue of major relevance especially in younger people. The systemic immune reaction after severe injury is characterized by a strong pro- and anti-inflammatory response. Besides its functions as energy storage depot and organ-protective cushion, adipose tissue regulates vital processes via its secretion products. However, there is little awareness of the important role of adipose tissue in regulating the posttraumatic inflammatory response. In this review, we delineate the local and systemic role of adipose tissue in trauma and outline different aspects of adipose tissue as an immunologically active modifier of inflammation and as an immune target of injured remote organs after severe trauma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction: relevance of trauma and obesity
Severe trauma is still a major cause of disability and death. In 2017, 4.5 million people around the world died due to traumatic injuries. Trauma accounts for 8.0% of all deaths, and plays an even greater role in deaths of young people. Males face an almost twice as high death rate from injury compared to females. Additionally, there were 521 million of non-fatal injuries in 2017 [40]. Thus, despite improvements of clinical trauma management and increasing survival rates, trauma remains an important cause of adverse outcome and death.
Traumatic injuries induce a very complex systemic immune reaction, involving various parts of innate and adaptive immunity, changes in endothelial function, the coagulation system and endocrine signaling as reviewed in detail in [46]. Trauma causes systemic release of damage-associated molecular patterns (DAMPs) from damaged cells, and may also cause exposure to pathogen-associated molecular patterns (PAMPs) from the environment or the interior of the body (especially the gut). DAMPs and PAMPs activate immune cells, which increase their phagocytic capacity, secrete reactive oxygen species (ROS) and a variety of pro- and also anti-inflammatory cytokines. Interleukins (IL) like IL-6, IL-8, IL-10, tumor necrosis factor (TNF) and chemoattractants as monocyte chemoattractant factor 1 (MCP-1) are only some examples for mediators in the so-called cytokine storm provoked by severe injury. Furthermore, the complement system, involved in not only phagocytosis, but also immune modulation, is activated. The coagulation system is activated in order to stop harmful bleeding, with the risk of clot formation and hypoperfusion of tissue, aggravating systemic inflammation. Whether all those mechanisms are protective or harmful is very much dependent on the balance between pro- and anti-inflammatory mediators, and on the proportion of activators and inhibitors; an excessive activation of the mainly innate proinflammatory response may thus cause systemic inflammatory response syndrome (SIRS) and subsequent (multiple) organ failure [46].
Overweight and obesity have reached pandemic proportions. According to the World Health Organization (WHO), 1.9 billion adult people were overweight (BMI ≥ 25 kg/m2) and 650 million were obese (BMI ≥ 30 kg/m2) in 2016, corresponding to 39% or 13% of the global adult population [132]. The excessive accumulation of adipose tissue can lead to severe comorbidities, among them type 2 diabetes mellitus and cardiovascular diseases but also certain types of cancer. Of note, adipose tissue itself via its secretion products is involved in the pathogenesis [69].
In the case of a traumatic injury, adipose tissue is essential for the absorption of the inflicting trauma vector [9, 55, 103, 113]. Interestingly, an obesity paradox has been described in trauma patients: in this setting, a protective function of overweight has been described on overall survival, whereas obesity grade 2 and 3 (BMI ≥ 35 kg/m2) as well as underweight increased the risk of death after trauma [26]. Due to greater forces at lower speed, obese patients tend to suffer from more severe but more distally located fracture types caused by low-impact trauma [3, 5, 68, 97, 116]. In addition, the risk for impaired wound and fracture healing are higher in the obese population [3, 113]. This argues for an involvement of adipose tissue in the systemic response to trauma. However, its role on the immuno-pathophysiology, outcome, and treatment options after severe trauma remains rather undefined. Herein, we aim to examine and reflect the role of the adipocyte-immune-organ crosstalk in severely injured patients.
Adipose tissue: more than just an energy store
Based on morphological features, adipose tissue can be roughly classified into two basic types, white adipose tissue (WAT), where univacuolar, white adipocytes are found, and brown adipose tissue (BAT), consisting of multivacuolar, mitochondria-rich, brown adipocytes [95].
WAT covers the body in the subcutaneous depot underneath the skin, while the inner viscera are surrounded by visceral depots. As fat is a poor conductor of heat, it insulates the body and protects against cooling, but it also provides support and protection. In healthy subjects, WAT accounts for approximately 20–25% of total body mass, but in obese subjects, it can increase to up to 50% [94]. WAT was long considered a passive organ where excess energy is stored in the form of triglycerides. Usually, lipids are ingested with food, and the uptake and storage of lipids is mainly regulated by the anabolic hormone insulin. In times of negative energy balance, e.g. during a period of fasting, fatty acids can be mobilized from the lipid droplets through the process of lipolysis [13]. Besides these vital metabolic functions, WAT has been increasingly recognized as important endocrine organ releasing a plethora of secretion products to the circulation, which have been named adipokines [95]. Among them are protein factors, e.g. classical hormones such as leptin or adiponectin, growth factors, chemokines and cytokines, but also certain lipid mediators, e.g. prostaglandins, and metabolites, e.g. fatty acids, nucleotides, nucleosides or lactate [141]. Via these adipokines, WAT is in continuous crosstalk with both the local microenvironment as well as all other organs systems in the body and regulates vital processes or functions such as food intake, energy homeostasis, insulin sensitivity, hemostasis, and blood pressure [31].
While WAT is best known for storing fat, BAT has the unique ability to use it for heat production in a process called non-shivering thermogenesis. The thermogenic function is achieved by uncoupling protein-1 (UCP1), which is located at the inner mitochondrial membrane. Upon activation, UCP1 uncouples the proton gradient, which is generated in the respiratory chain, from adenosine triphosphate (ATP) synthesis to produce heat instead of ATP. BAT can be found in the interscapular and paravertebral region and also in perirenal depots. For decades, it was believed that BAT in humans is only present in neonates where it is responsible for the maintenance of body temperature, but it is now well accepted that functional BAT is also present in adults. Interestingly, an intermediate beige (brite, inducible BAT-like) adipose tissue phenotype can emerge from WAT upon chronic cold exposure in a process referred to as browning [52]. Beige adipocytes can store triglycerides, but also express UCP1 and can therefore contribute to heat production. Of note, browning of WAT has recently been linked to the hypermetabolic response after burn injury, which will be discussed in detail below [54]. In addition to white, brown, and beige adipose tissue we would also like to mention an additional adipose tissue depot with potential relevance to trauma, i.e. marrow adipose tissue.
Fat tissue as traumatized organ
As WAT is the major component of the subcutis, injury of adipose tissue is caused by nearly every physical trauma mechanism. Trauma generally induces a disruption of macro-barriers such as the skin and underlying soft tissue, as well as micro-barriers such as cell membranes, resulting in a complex immune response [46]. Damaged tissue releases various damage-associated molecular patterns (DAMPs), e.g., high-mobility group box 1 protein (HMGB1), ATP, nuclear contents such as histones, and mitochondrial DNA [137, 138, 140]. In adipose tissue, besides a necrosis-caused release of HMGB1, also an active secretion of this DAMP has been shown, especially in presence of inflammatory stimuli [38, 107, 137]. Of note, there is only limited data concerning the impact of blunt or penetrating trauma on WAT. In hematoma caused by fracture and concomitant soft-tissue injury, high concentrations of cytokines such as IL-6, IL-8 and TNF were detected [43]. Operative trauma or even minimal tissue injury caused by cannulas also increased the concentrations of inflammatory mediators [37, 79]. Burn injury in particular increased the expression of IL-6, IL-8, MCP-1, and TNF in WAT, but not of adipogenic, chondrogenic, or osteogenic factors [85, 98], and leads to morphological and functional changes, such as smaller adipocytes, collagen accumulation and tissue fibrosis, or invasion of macrophages and upregulation of UCP1. UCP1 is the key marker of brown and beige adipose tissue and characterized by its thermogenic capacity as discussed earlier. In this regard, increased expression of UCP1 was proposed to significantly contribute to the hypermetabolic state after burn trauma [1, 54, 85, 98]. Destruction of adipose tissue as well as endocrine mechanisms, e. g., β-adrenergic signaling, liberate fatty acids such as oleate and linoleate into the systemic circulation [7, 114, 126]. In case of penetrating trauma, invading bacteria with their PAMPs may exacerbate the inflammatory reaction by recruitment of further immune cells. In turn, these inflammatory cells, which represent neutrophils and monocytes/macrophages and therefore the “first cellular line of defense” can further secrete cytokines and chemokines, generate reactive oxygen species (ROS) and inflammatory mediators, with local effects as described below in detail [46, 53, 54].
Traumatized adipose tissue as a modulator of the systemic post-traumatic response
WAT in different body compartments might react differentially to traumatic insults. Compared to subcutaneous adipocytes, visceral adipocytes are metabolically more active and have a higher capacity to generate free fatty acids (FFA) [47]. Visceral WAT contains larger numbers of immune cells and directly drains to the liver via the portal vein [47]. Although visceral adiposity was not associated with increased signs of inflammation [18], larger abdominal depots increased the risk of acute kidney injury [105].
Adipose tissue is an important source of inflammatory mediators such as cytokines (e.g., IL-1β, IL-6) and chemoattractants (e.g., MCP-1). Nevertheless, most of these mediators can also originate from other tissues and the exact impact of visceral and subcutaneous adipose tissue is still rather unclear. Potential mechanisms caused or influenced by mediators secreted from adipose tissue are described in this section and also shown in Fig. 1.

Adipose tissue as a modulator of systemic post-traumatic responses. After a severe traumatic insult, a plethora of factors is released into systemic circulation, many of which are also secreted by adipose tissue. Traumatized adipose tissue releases procoagulant mediators such as tissue factor (TF) and plasminogen activator inhibitor 1 (PAI-1), facilitating thrombotic events. Furthermore, various proinflammatory mediators such as interleukin (IL-) 1β, IL-6, IL-8, tumor necrosis factor (TNF), monocyte chemoattractant protein-1 (MCP-1) and high mobility group box 1 protein (HMGB1) are liberated. Together with free fatty acids (FFA), they elicit various effects such as insulin resistance and immune cell activation, and compromise physiological functions of various remote organs. The adipokine adiponectin might exert systemic anti-inflammatory effects. Leptin was recently described as a prognostic marker of multiple organ failure in critically ill patients. ARDS acute respiratory distress syndrome, ASC adipose stromal cell
DAMPs and cytokines recruit inflammatory cells to the adipose tissue, further exacerbating release of cytokines [1, 54, 115]. These mediators exhibit various systemic effects on remote organs and are of prognostic value in patients after severe trauma, with higher concentrations associated with poorer outcome [20, 35, 63, 81]. Especially HMGB1, a DAMP passively liberated from adipose tissue and other tissues via cell death as well as actively through secretion processes, plays a pivotal role in remote organ injury by triggering the release of other cytokines such as IL-6, IL-10, and MCP-1 [38, 63, 107]. In addition, stressed adipocytes are able to enhance the protease release and oxidative burst reaction of neutrophils [23]. Inflammatory mediators, proteases, and ROS released into systemic circulation provoke remote organ injury by barrier disruption, protein degradation, cell death, and accumulation of inflammatory cells in remote organs [12, 46, 99]. The lungs are particularly susceptible to injury caused by distant organ trauma [12, 25, 90]. Severe soft (adipose) tissue trauma can be sufficient to robustly increase the numbers of inflammatory cells and especially neutrophils in the lungs and to disturb lung tissue oxygenation [39, 110]. These inflammatory mediators also contribute to renal failure [16, 76, 124] and can suppress cardiac function [67, 135] (Fig. 1). Certain adipokines such as IL-17A and leptin were recently described as potential diagnostic parameters in severely injured patients: their plasma concentrations were significantly elevated after severe multiple injury, with higher values associated with an increased risk of multiple organ failure (MOF) development [42]. For leptin, however, there is conflicting data. Some studies showed higher systemic concentrations after severe trauma [42, 80], while others detected reduced leptin levels in severely burned subjects [124] and critically ill patients after surgery [87]. Thus, further studies are required to determine the exact role of leptin in traumatized patients. Adiponectin is another important adipokine with anti-inflammatory features and protective systemic effects [8, 27, 120, 134], although there is some conflicting data in the context of critically ill patients [22]. Resistin and visfatin, two proinflammatory adipokines, were increased after burn injury and critical illness and associated with poorer outcome [4, 124]. However, to date, there is a lack of causative studies demonstrating the specific influence of these adipokines in the condition of severe trauma.
As another important aspect, higher BMI and increased fat mass alter the coagulation state after trauma towards a procoagulant phenotype and increase the risk for thrombosis and pulmonary embolism [77, 104, 128]. Alterations in blood thrombo-elastometric properties of obese subjects were shown in several studies, but the underlying mechanisms are not completely unraveled [57, 73, 128]. One possible factor is the production of plasminogen activator inhibitor 1 (PAI-1) in adipose tissue. As it possesses procoagulant features and its expression is enhanced by inflammatory mediators, PAI-1 might play an important role in posttraumatic thromboembolic events [73, 108, 121]. Moreover, WAT serves as an important site of synthesis of tissue factor (TF), which is a major activator of the coagulation cascade (Fig. 1). Therefore, liberation of TF from WAT after trauma may promote a procoagulant state [29]. IL-6 derived from WAT after trauma may furthermore induce synthesis of factor VII, factor VIII, and fibrinogen in the liver, further driving clot formation [29, 33].
Adipose tissue trauma and exposure to DAMPs and PAMPs as well as catecholamines (via β2 signaling) can liberate FFA into systemic circulation [82, 119]. FFA function as an important source of energy in a catabolic state, such as after severe injuries [126, 129]. On the other hand, FFA in mesenteric lymph and plasma have cytotoxic effects, for example on endothelial cells, thus promoting vascular leakage [15, 86, 112]. Released into systemic circulation, FFA can exhibit inflammatory features [7, 11]. Insulin resistance as a frequent condition in posttraumatic metabolism can also be promoted by FFA and other lipid degradation products such as diacylglycerols [21, 58, 101]. Recent studies showed a causal link between increased lipolysis, systemic insulin resistance and further metabolic disturbances, indicating a direct impact of metabolic changes in adipose tissue on the systemic posttraumatic reaction [54, 88]. Furthermore, elevated plasma concentrations of FFA were described as a risk factor for development of acute respiratory distress syndrome (ARDS) after trauma and sepsis [7] (Fig. 1).
Taken together, adipose tissue might act as an important player in the posttraumatic systemic response and contribute to remote organ damage and failure.
Adipose tissue as a target of the systemic response after trauma
Severe trauma activates the sympathetic nervous system and thereby increases systemic levels of catecholamines such as epinephrine and norepinephrine, which positively correlate with the injury severity. High catecholamine levels are furthermore associated with poor prognosis [51, 92, 131]. Catecholamines have various effects on different tissues, such as the endothelium, muscles, and various organs [32, 50, 61, 70]. As trauma victims, especially in the case of an additional hemorrhagic shock, often suffer from early hypotension, external catecholamine administration is often required to restore and maintain a blood pressure sufficient for organ perfusion [127]. However, the metabolic state of adipose tissue is significantly altered by catecholamines. Lipolysis is stimulated via β-adrenergic pathways, liberating FFA as energy source for remote organs [93]. Excessive catecholamine stimulation may also result in a prolonged lipolytic activity with wasting of adipose tissue [14, 49]. IL-6 secreted by various tissues throughout the body after trauma also promotes lipolysis in adipocytes and decreases adipocyte size [41, 122]. As another inflammatory cytokine, TNF is able to induce lipolysis in adipose tissue and furthermore impairs its insulin sensitivity and lipid storage capacity [10].
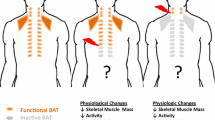
WAT is able to change its phenotype in response to trauma and turn into beige adipose tissue with thermogenic capacity. Whether this occurs through transdifferentiation of white adipocytes or de novo differentiation of specific precursor cells is still a matter of debate [44]. The expression of UCP1, the key thermogenic factor, is induced after trauma and burn injury, not only at the injured site, but also in distant areas [2, 66, 109]. Browning of WAT naturally occurs under chronic cold exposure, but it can also be induced by inflammatory mediators such as IL-6, high circulating levels of catecholamines, or their local production by alternatively activated macrophages [1, 2, 34, 54], all playing key roles in the pathophysiology after severe trauma. Thus, it is intuitive that WAT browning was shown after burn injury, major surgical trauma, and conditions associated with high catecholamine concentrations [34, 66, 109]. In turn, adipose tissue browning induces a hypermetabolic state with reduction of storage depots, lower fat content, lower weight, decreased bone mineral content and increased levels of cortisol, catecholamines, cytokines, and acute phase proteins resulting in higher heart rate and cardiac output [49].
WAT and its residing immune cells produce cytokines and chemokines that can spill over into systemic circulation, but of course, it will also receive inflammatory signals from distant organs, further aggravating the inflammatory state in a vicious cycle. The inflammatory response of WAT is furthermore determined by the general health state of the patients and their fat mass. Importantly, it can be influenced by the preoperative diet, with dietary restriction reversing the detrimental effects of diet-induced obesity on the WAT inflammatory response to trauma, opening up therapeutic options [71, 78].
To summarize, catecholamines and inflammatory mediators dysregulate homeostasis of adipose tissue, resulting not only in a hypermetabolic state with reduction of the energy storage depots, but also in aggravation of the systemic inflammatory response by the adipose tissue itself (Fig. 2).

Adipose tissue as a target of post-traumatic systemic pathophysiological conditions. Severe trauma results in organ dysfunction, blood loss and a sympathetic reaction. The liberated catecholamines induce browning of white adipose tissue by upregulating expression of uncoupling protein 1 (UCP1). Thereby, heat is produced and free fatty acids (FFA) are liberated. Catecholamines (via β-receptors, βAR) as well as cytokines, e.g. tumor necrosis factor (TNF) and interleukin (IL) 6, induce lipolysis and energy wasting. Adipocytes are activated by proinflammatory mediators and secrete cytokines, aggravating inflammatory response. Furthermore, hypoxia caused by blood loss and centralization leads to cell death in adipose tissues
Micromilieu changes in adipose tissue after trauma
Trauma alters the micromilieu in most tissues of the body and also in WAT. The so-called “lethal triad”, composed of hypothermia, acidosis and coagulopathy, is, although adapted during the past years, still of great value considering prognosis and therapeutic concepts in severely injured patients [24, 74, 106]. The lethal triad is a systemic concept, but the underlying components and mechanisms are also present in the micromilieu of adipose tissue. Due to its location at the body surface, subcutaneous WAT is particularly affected by hypothermia, as ambient temperature is usually lower than the physiological tissue temperature. Major trauma can cause substantial blood loss, resulting in hypoperfusion of tissues. Perfusion of non-vital tissues such as adipose tissue is furthermore compromised by vasoconstriction caused by endogenous catecholamines (mainly via β-adrenergic receptors) released in trauma and shock [46]. Hypoperfusion impairs removal of metabolic products and cell debris and plays a major role in the development of acidosis. With less oxygen available, anaerobic metabolic pathways leading to generation of acidic metabolites such as lactic acid become more important, further promoting adipose tissue acidosis [100, 118] (Fig. 3).

Changes of the micro-milieu at the blood-fat-tissue barrier after trauma. Severe trauma leads to barrier dysfunction, resulting in endothelial leakage, leucocyte recruitment and extravasation, and tissue inflammation. Tissue inflammation is further aggravated by adipocytes and local immune cells producing proinflammatory mediators. Some of these are again liberated into the circulation. Hypoxia, caused by blood loss, vasoconstriction and thrombus formation, promotes anaerobic metabolic pathways with generation of lactate. Thus, the posttraumatic micromilieu in adipose tissue is characterized by hypoperfusion, hypoxia, acidosis, hypothermia and a pronounced inflammatory reaction. ASC adipose stromal cell
Acute trauma-induced coagulopathy is a complex process leading to formation of microthrombi, thereby worsening tissue perfusion. Furthermore, consumption of platelets, coagulation factors and other mediators contribute to traumatic coagulopathy development. Thus, posttraumatic coagulopathic states may result in a hemorrhagic phenotype aggravating injury-induced blood loss [17, 111]. As described above, adipose tissue plays an important role in development of trauma-induced coagulopathy, thereby hampering its own blood supply and hemostatic capacity [29, 73]. Endothelial leakage after trauma occurs in the vasculature of the whole body, increasing inflammatory cell invasion into tissues. As adipose tissue secretes various inflammatory cytokines and DAMPs as described above, it recruits inflammatory cells, thus further exacerbating tissue inflammation [1, 54, 115]. Inflammatory cells liberate ROS, again aggravating endothelial leakage and tissue acidosis, resulting in a vicious cycle of micromilieu changes [46]. Overall, these changes can cause blood-organ-barrier dysfunction and failure [46] and, in case of adipose tissue, a proposed “blood-adipose tissue barrier” dysfunction (Fig. 3).
Adipocyte dysfunction after trauma
Severe injuries result in adipose tissue hypoxia, as oxygen supply is reduced due to blood loss and vasoconstriction. Adipocytes react to hypoxic conditions by changing their metabolic pathways and endocrine functions. Hypoxic adipocytes up-regulate the glycolytic pathway in response to low oxygen supply, representing a shift towards anaerobic metabolism [36, 72]. Hypoxia-inducible factor 1 (HIF-1) has been identified as a major regulator of the metabolic shift in hypoxic cells. HIF-1 is a heterodimer with the β-subunit constitutively expressed independent of oxygen supply, while the α-subunit is rapidly degraded by the proteasomal complex under normoxic, but not under hypoxic conditions [102]. HIF-1 regulates various genes involved in glucose uptake and metabolism, angiogenesis, remodeling of extracellular matrix, apoptosis and inflammation [118]. Glucose transporter 1 (GLUT1) is one of the key targets upregulated via the HIF-1 pathway, resulting in increased glucose uptake of adipocytes. Gene expression, but not protein levels, of GLUT3 and GLUT5 has also been shown to increase in response to hypoxia, while expression of the insulin-sensitive transporter GLUT4 is diminished under hypoxic conditions, impairing insulin sensitivity of adipocytes [89, 118, 130]. Glucose is degraded via anaerobic glycolysis, resulting in increased lactate levels in adipocytes [83] (Fig. 4). HIF-2, which is also stabilized and activated under hypoxic conditions, may counteract the HIF-1-induced insulin resistance to some extent, as it was shown to improve insulin sensitivity of different cell types [59, 62, 91, 117].

Posttraumatic adipocyte dysfunction. After trauma, adipocytes are exposed to pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), released locally and from neighboring damaged tissues. DAMPs, PAMPs and free fatty acids (FFA) activate receptors on the adipocyte surface, such as toll-like receptors (TLR). Thus, proinflammatory pathways as the NF-κB signaling pathway are upregulated, resulting in production and secretion of proinflammatory interleukins (IL), high mobility group box 1 protein (HMGB1), tumor necrosis factor (TNF) and procoagulant factors such as tissue factor (TF) and plasminogen activator inhibitor-1 (PAI-1). These cytokines activate immune cells, aggravating the inflammatory response. The procoagulant state facilitates thrombus formation, worsening hypoxic conditions. Thus, aerobic pathways (e.g. fatty acid (FA) oxidation) are downregulated, whereas anaerobic pathways such as glycolysis are upregulated, resulting in the generation of lactate, which is shuttled outside the cell via monocarboxylate transporter 1 (MCT-1), resulting in tissue acidosis. Hypoxia activates the hypoxia-induced 1 factor (HIF-1) pathway, upregulating glucose uptake via glucose transporter (GLUT) 1, and lactate release. Lipolysis is induced by epinephrine (E) and norepinephrine (NE) via β-receptors (βAR). After trauma, insulin sensitivity is impaired, further enhancing lipolysis and liberating FFA. By releasing complement factor D (CFD), adipocytes also activate the complement system, strengthening the posttraumatic response
To remove lactate from adipocytes, monocarboxylate transporter 1 (MCT-1) is upregulated by the transcription factor HIF-1. Lactate is thus shifted out of the cells towards the extracellular matrix and systemic circulation, promoting lactic acidosis [83]. Lactate again is an important regulator of adipocyte metabolism, as it diminishes lipolysis [65]. Independent of lactate signaling, there is also data suggesting increased lipolytic activity in hypoxic adipocytes and reduced uptake of FFA, with higher systemic FFA concentrations [136]. Insulin sensitivity of adipocytes can be altered by different mechanisms after trauma. First of all, hypoxia directly impairs insulin signaling in a HIF-1-dependent manner [89, 136]. Furthermore, inflammatory mediators such as IL-6 and TNF can further promote insulin resistance in adipocytes [45, 60, 96].
An inflammatory reaction of adipocytes can be activated by sensing of DAMPs and PAMPs, but also of saturated FFAs by classical toll-like receptors (TLRs) such as TLR2 and TLR4 [28, 56] (Fig. 4). In severe trauma, PAMPs and DAMPs can originate from distant tissues and various cell types, but also adipocytes themselves produce DAMPs as HMGB1 and release FFAs, amplifying the local inflammatory response and activating immune cells [38, 119]. These effects are at least partly mediated via the NF-κB pathway. This key pathway can be activated by inflammatory cytokines, complement activation factors (e.g. C5a), TNF, or PAMPs (e.g. lipopolysaccharides, LPS), and promotes adipocyte metabolic dysfunction and secretion of inflammatory mediators [6, 139]. HIF-1, activated by low oxygen supply, is not only a key mediator of metabolic processes but also promotes the secretion of IL-6, PAI-1, vascular endothelial growth factor (VEGF) and various other factors [125], creating an autocrine and paracrine cycle of stimulation, further aggravating the local inflammatory response [10, 28]. Inflammation is further promoted by recruited immune cells (e.g. neutrophils) which are locally activated by adipocyte-derived products [23]. This principle is also true for the fluid phase of the innate immune response mainly mounted by the complement system. For example, hypoxic adipocytes increase the generation and secretion of complement factor D (adipsin), which promotes activation of the alternative complement pathway [123, 125] (Fig. 4). As complement activation plays an important role in the pathophysiology after severe trauma, this displays another link between adipocytes and the immune response [46].
In conclusion, trauma alters metabolic functions of single adipocytes, affecting glucose utilization and both local and systemic insulin sensitivity [19, 64], causes WAT tissue browning, and results in release of FFAs and inflammatory mediators, all of which in concert lead to local (tissue) and systemic (blood) inflammation and disturbance of the metabolic balance.
Trauma and the obesity paradox
As mentioned earlier, overweight can reduce mortality in severely injured patients, whereas severe obesity (BMI ≥ 35 kg/m2) and underweight increase the mortality risk [26]. Several explanatory approaches were published in the past years. Overweight causes constant low-grade inflammation effecting the immune system as well as metabolism, but potentially increasing the systemic tolerance for the strong immune response following trauma. Furthermore, overweight causes increased systemic levels of leptin compared to lean individuals. The adipokine leptin acts as an important modulator of immune responses, as it influences T cell function and thereby protects against infections, but on the other hand reduces systemic susceptibility to the toxicity of proinflammatory stimuli [30]. In addition, overweight patients have greater energy reserve during the catabolic states after severe injury [26]. These protective factors might be outweighed by negative consequences if body mass is further increased. Obese patients suffer from pre-conditions such as decreased lung volume, impaired expiratory air flow, reduced lung compliance and reduced gas exchange, resulting in hypoventilation and hypoxia [48]. Severely obese patients required longer duration of mechanical ventilation, resulting in ventilator-associated complications as pneumonia. They were also at higher risk for ARDS and multiple organ failure. This might be due to the harmful effects of hypoxia and hypoperfusion on vital organs, and an increased inflammatory reaction associated with higher grade obesity [77]. Thus, reduced mortality in critically injured patients due to moderate overweight may be offset by the deleterious pre-existing conditions in severe obesity.
Outlook
Based on the recent advantages in understanding the impact of adipose tissue in acute diseases such as during the acute and subacute phase following trauma, novel interventional approaches and new aspects of established therapeutics have emerged to prevent the augmentation and continuance of an excessive inflammatory response. In this regard, pharmacological intervention to inhibit lipolysis and β-adrenergic signaling [75] or to improve insulin sensitivity both systemically but also locally in WAT, e.g., by administration of a PDGF receptor tyrosine kinase inhibitor [84], have demonstrated positive effects in preclinical and some clinical studies of acute injury. Future preclinical studies, e.g., in mouse models with either absence or excessive accumulation of WAT as well as large-scale multi-layered analyses in traumatized patients as recently done by the PAMPer consort [133] may provide more insights into the function of adipose tissue in regulating the host response to trauma and ensuing SIRS. Furthermore, modulating the local and systemic immune response by blocking excessive amounts of central proinflammatory mediators may circumvent the detrimental alterations of the micro-milieu and resulting dysfunction of adipocytes, thereby restoring a balanced posttraumatic response in adipose tissue.
Availability of data and materials
This review article does not report primary data or materials.
References
Abdullahi A, Auger C, Stanojcic M, Patsouris D, Parousis A, Epelman S, Jeschke MG (2019) Alternatively activated macrophages drive browning of white adipose tissue in burns. Ann Surg 269:554–563
Abdullahi A, Chen P, Stanojcic M, Sadri AR, Coburn N, Jeschke MG (2017) IL-6 signal from the bone marrow is required for the browning of white adipose tissue post burn injury. Shock 47:33–39
Abidi NA, Dhawan S, Gruen GS, Vogt MT, Conti SF (1998) Wound-healing risk factors after open reduction and internal fixation of calcaneal fractures. Foot Ankle Int 19:856–861
Al-Tarrah K, Jones SW, Moiemen N, Lord JM (2020) Potential role of adipose tissue and its hormones in burns and critically III patients. Burns 46:259–266
Bansal V, Conroy C, Lee J, Schwartz A, Tominaga G, Coimbra R (2009) Is bigger better? The effect of obesity on pelvic fractures after side impact motor vehicle crashes. J Trauma 67:709–714
Berg AH, Lin Y, Lisanti MP, Scherer PE (2004) Adipocyte differentiation induces dynamic changes in NF-kappaB expression and activity. Am J Physiol Endocrinol Metab 287:E1178–E1188
Bursten SL, Federighi DA, Parsons P, Harris WE, Abraham E, Moore EE Jr, Moore FA, Bianco JA, Singer JW, Repine JE (1996) An increase in serum C18 unsaturated free fatty acids as a predictor of the development of acute respiratory distress syndrome. Crit Care Med 24:1129–1136
Cai L, Yi F, Dai Z, Huang X, Zhao YD, Mirza MK, Xu J, Vogel SM, Zhao YY (2014) Loss of caveolin-1 and adiponectin induces severe inflammatory lung injury following LPS challenge through excessive oxidative/nitrative stress. Am J Physiol Lung Cell Mol Physiol 306:L566–L573
Carobbio S, Pellegrinelli V, Vidal-Puig A (2017) Adipose tissue function and expandability as determinants of lipotoxicity and the metabolic syndrome. Adv Exp Med Biol 960:161–196
Cawthorn WP, Sethi JK (2008) TNF-alpha and adipocyte biology. FEBS Lett 582:117–131
Chen L, Yu CX, Song B, Cai W, Liu C, Guan QB (2018) Free fatty acids mediates human umbilical vein endothelial cells inflammation through toll-like receptor-4. Eur Rev Med Pharmacol Sci 22:2421–2431
Chen L, Zhao H, Alam A, Mi E, Eguchi S, Yao S, Ma D (2019) Postoperative remote lung injury and its impact on surgical outcome. BMC Anesthesiol 19:30-019-0698-6
Choe SS, Huh JY, Hwang IJ, Kim JI, Kim JB (2016) Adipose tissue remodeling: its role in energy metabolism and metabolic disorders. Front Endocrinol (Lausanne) 7:30
Chouchani ET, Kajimura S (2019) Metabolic adaptation and maladaptation in adipose tissue. Nat Metab 1:189–200
Chung BH, Hennig B, Cho BH, Darnell BE (1998) Effect of the fat composition of a single meal on the composition and cytotoxic potencies of lipolytically-releasable free fatty acids in postprandial plasma. Atherosclerosis 141:321–332
Clark A, Neyra JA, Madni T, Imran J, Phelan H, Arnoldo B, Wolf SE (2017) Acute kidney injury after burn. Burns 43:898–908
Cohen MJ, Christie SA (2017) Coagulopathy of trauma. Crit Care Clin 33:101–118
Collier B, Dossett L, Shipman J, Day M, Lawson G, Sawyer R, May A (2010) Visceral adiposity is not associated with inflammatory markers in trauma patients. J Trauma 68:57–61
Cree MG, Wolfe RR (2008) Postburn trauma insulin resistance and fat metabolism. Am J Physiol Endocrinol Metab 294:E1–E9
Cuschieri J, Bulger E, Schaeffer V, Sakr S, Nathens AB, Hennessy L, Minei J, Moore EE, O’Keefe G, Sperry J, Remick D, Tompkins R, Maier RV, Inflammation and the Host Response to Injury Collaborative Research Program (2010) Early elevation in random plasma IL-6 after severe injury is associated with development of organ failure. Shock 34:346–351
Delarue J, Magnan C (2007) Free fatty acids and insulin resistance. Curr Opin Clin Nutr Metab Care 10:142–148
Dembinski R (2010) Adiponectin in critically ill patients: more questions than answers? Crit Care Med 38:2415–2416
Diebel LN, Liberati DM, Edelman DA, Webber JD (2013) Organ failure in the obese adipocytes prime polymorphonuclear cell inflammation under stress conditions. J Trauma Acute Care Surg 75:1047–1051 (discussion 1051–2)
Ditzel RM Jr, Anderson JL, Eisenhart WJ, Rankin CJ, DeFeo DR, Oak S, Siegler J (2020) A review of transfusion- and trauma-induced hypocalcemia: Is it time to change the lethal triad to the lethal diamond? J Trauma Acute Care Surg 88:434–439
Dushianthan A, Grocott MP, Postle AD, Cusack R (2011) Acute respiratory distress syndrome and acute lung injury. Postgrad Med J 87:612–622
Dvorak JE, Lester ELW, Maluso PJ, Tatebe L, Schlanser V, Kaminsky M, Messer T, Dennis AJ, Starr F, Bokhari F (2020) The obesity paradox in the trauma patient: normal may not be better. World J Surg 44:1817–1823
Ekmekci H, Ekmekci OB (2006) The role of adiponectin in atherosclerosis and thrombosis. Clin Appl Thromb Hemost 12:163–168
Engin AB (2017) Adipocyte-macrophage cross-talk in obesity. Adv Exp Med Biol 960:327–343
Faber DR, de Groot PG, Visseren FL (2009) Role of adipose tissue in haemostasis, coagulation and fibrinolysis. Obes Rev 10:554–563
Fantuzzi G (2005) Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol 115:911–919
Fasshauer M, Blüher M (2015) Adipokines in health and disease. Trends Pharmacol Sci 36:461–470
Fonseca RB, Mohr AM, Wang L, Clinton E, Sifri ZC, Rameshwar P, Livingston DH (2004) Adrenergic modulation of erythropoiesis following severe injury is mediated through bone marrow stroma. Surg Infect (Larchmt) 5:385–393
Fontana L, Eagon JC, Trujillo ME, Scherer PE, Klein S (2007) Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 56:1010–1013
Frontini A, Vitali A, Perugini J, Murano I, Romiti C, Ricquier D, Guerrieri M, Cinti S (2013) White-to-brown transdifferentiation of omental adipocytes in patients affected by pheochromocytoma. Biochim Biophys Acta 1831:950–959
Gaski GE, Metzger C, McCarroll T, Wessel R, Adler J, Cutshall A, Brown K, Vodovotz Y, Billiar TR, McKinley TO (2019) Early immunologic response in multiply injured patients with orthopaedic injuries is associated with organ dysfunction. J Orthop Trauma 33:220–228
Geiger K, Leiherer A, Muendlein A, Stark N, Geller-Rhomberg S, Saely CH, Wabitsch M, Fraunberger P, Drexel H (2011) Identification of hypoxia-induced genes in human SGBS adipocytes by microarray analysis. PLoS One 6:e26465
Gletsu N, Lin E, Zhu JL, Khaitan L, Ramshaw BJ, Farmer PK, Ziegler TR, Papanicolaou DA, Smith CD (2006) Increased plasma interleukin 6 concentrations and exaggerated adipose tissue interleukin 6 content in severely obese patients after operative trauma. Surgery 140:50–57
Gunasekaran MK, Viranaicken W, Girard AC, Festy F, Cesari M, Roche R, Hoareau L (2013) Inflammation triggers high mobility group box 1 (HMGB1) secretion in adipose tissue, a potential link to obesity. Cytokine 64:103–111
Gustafsson U, Suneson A, Kjellström BT (1997) Effects of peripheral high energy missile trauma on the oxygenation of lung tissue in the pig. Ann Acad Med Singap 26:22–26
Haagsma JA, James SL, Castle CD, Dingels ZV, Fox JT, Hamilton EB, Liu Z et al (2020) Burden of injury along the development spectrum: associations between the Socio-demographic Index and disability-adjusted life year estimates from the Global Burden of Disease Study 2017. Inj Prev 26:i12–i26
Han J, Meng Q, Shen L, Wu G (2018) Interleukin-6 induces fat loss in cancer cachexia by promoting white adipose tissue lipolysis and browning. Lipids Health Dis 17:14-018-0657-0
Haupt J, Krysiak N, Unger M, Bogner-Flatz V, Biberthaler P, Hanschen M, van Griensven M, Haug AT (2021) The potential of adipokines in identifying multiple trauma patients at risk of developing multiple organ dysfunction syndrome. Eur J Med Res 26:38-021-00511-z
Hauser CJ, Zhou X, Joshi P, Cuchens MA, Kregor P, Devidas M, Kennedy RJ, Poole GV, Hughes JL (1997) The immune microenvironment of human fracture/soft-tissue hematomas and its relationship to systemic immunity. J Trauma 42:895–903
Herz CT, Kiefer FW (2019) Adipose tissue browning in mice and humans. J Endocrinol 241:R97–R109
Hotamisligil GS (1999) Mechanisms of TNF-alpha-induced insulin resistance. Exp Clin Endocrinol Diabetes 107:119–125
Huber-Lang M, Lambris JD, Ward PA (2018) Innate immune responses to trauma. Nat Immunol 19:327–341
Ibrahim MM (2010) Subcutaneous and visceral adipose tissue: structural and functional differences. Obes Rev 11:11–18
Imber DA, Pirrone M, Zhang C, Fisher DF, Kacmarek RM, Berra L (2016) Respiratory management of perioperative obese patients. Respir Care 61:1681–1692
Jeschke MG, Gauglitz GG, Kulp GA, Finnerty CC, Williams FN, Kraft R, Suman OE, Mlcak RP, Herndon DN (2011) Long-term persistance of the pathophysiologic response to severe burn injury. PLoS One 6:e21245
Johansson PI, Ostrowski SR (2010) Acute coagulopathy of trauma: balancing progressive catecholamine induced endothelial activation and damage by fluid phase anticoagulation. Med Hypotheses 75:564–567
Johansson PI, Stensballe J, Rasmussen LS, Ostrowski SR (2012) High circulating adrenaline levels at admission predict increased mortality after trauma. J Trauma Acute Care Surg 72:428–436
Kajimura S, Spiegelman BM, Seale P (2015) Brown and beige fat: physiological roles beyond heat generation. Cell Metab 22:546–559
Kaminski DA, Randall TD (2010) Adaptive immunity and adipose tissue biology. Trends Immunol 31:384–390
Kaur S, Auger C, Jeschke MG (2020) Adipose tissue metabolic function and dysfunction: impact of burn injury. Front Cell Dev Biol 8:599576
Kershaw EE, Flier JS (2004) Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 89:2548–2556
Könner AC, Brüning JC (2011) Toll-like receptors: linking inflammation to metabolism. Trends Endocrinol Metab 22:16–23
Kornblith LZ, Howard B, Kunitake R, Redick B, Nelson M, Cohen MJ, Callcut R (2015) Obesity and clotting: body mass index independently contributes to hypercoagulability after injury. J Trauma Acute Care Surg 78:30–36
Kraegen EW, Cooney GJ (2008) Free fatty acids and skeletal muscle insulin resistance. Curr Opin Lipidol 19:235–241
Kunkemoeller B, Chen K, Lockhart SM, Wang X, Rask-Madsen C (2021) The transcriptional coregulator CITED2 suppresses expression of IRS-2 and impairs insulin signaling in endothelial cells. Am J Physiol Endocrinol Metab 321:E252–E259
Lagathu C, Bastard JP, Auclair M, Maachi M, Capeau J, Caron M (2003) Chronic interleukin-6 (IL-6) treatment increased IL-6 secretion and induced insulin resistance in adipocyte: prevention by rosiglitazone. Biochem Biophys Res Commun 311:372–379
Laverty R (1978) Catecholamines: role in health and disease. Drugs 16:418–440
Lee YS, Kim JW, Osborne O, Oh DY, Sasik R, Schenk S, Chen A, Chung H, Murphy A, Watkins SM, Quehenberger O, Johnson RS, Olefsky JM (2014) Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell 157:1339–1352
Levy RM, Mollen KP, Prince JM, Kaczorowski DJ, Vallabhaneni R, Liu S, Tracey KJ, Lotze MT, Hackam DJ, Fink MP, Vodovotz Y, Billiar TR (2007) Systemic inflammation and remote organ injury following trauma require HMGB1. Am J Physiol Regul Integr Comp Physiol 293:R1538–R1544
Li L, Messina JL (2009) Acute insulin resistance following injury. Trends Endocrinol Metab 20:429–435
Liu C, Wu J, Zhu J, Kuei C, Yu J, Shelton J, Sutton SW, Li X, Yun SJ, Mirzadegan T, Mazur C, Kamme F, Lovenberg TW (2009) Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J Biol Chem 284:2811–2822
Longchamp A, Tao M, Bartelt A, Ding K, Lynch L, Hine C, Corpataux JM, Kristal BS, Mitchell JR, Ozaki CK (2015) Surgical injury induces local and distant adipose tissue browning. Adipocyte 5:163–174
Maass DL, White J, Horton JW (2002) IL-1beta and IL-6 act synergistically with TNF-alpha to alter cardiac contractile function after burn trauma. Shock 18:360–366
Maheshwari R, Mack CD, Kaufman RP, Francis DO, Bulger EM, Nork SE, Henley MB (2009) Severity of injury and outcomes among obese trauma patients with fractures of the femur and tibia: a crash injury research and engineering network study. J Orthop Trauma 23:634–639
Manna P, Jain SK (2015) Obesity, oxidative stress, adipose tissue dysfunction, and the associated health risks: causes and therapeutic strategies. Metab Syndr Relat Disord 13:423–444
Mashaly HA, Provencio JJ (2008) Inflammation as a link between brain injury and heart damage: the model of subarachnoid hemorrhage. Cleve Clin J Med 75(Suppl 2):S26-30
Mauro CR, Nguyen BT, Yu P, Tao M, Gao I, Seidman MA, Nguyen LL, Ozaki CK (2013) Inflammatory “adiposopathy” in major amputation patients. Ann Vasc Surg 27:346–352
Mazzatti D, Lim FL, O’Hara A, Wood IS, Trayhurn P (2012) A microarray analysis of the hypoxia-induced modulation of gene expression in human adipocytes. Arch Physiol Biochem 118:112–120
McCully BH, Dean RK, McCully SP, Schreiber MA (2014) Diet-induced obesity prevents the development of acute traumatic coagulopathy. J Trauma Acute Care Surg 77:873–877
Mitra B, Tullio F, Cameron PA, Fitzgerald M (2012) Trauma patients with the ‘triad of death.’ Emerg Med J 29:622–625
Mohseni S, Joseph B, Peden CJ (2021) Mitigating the stress response to improve outcomes for older patients undergoing emergency surgery with the addition of beta-adrenergic blockade. Eur J Trauma Emerg Surg
Nechemia-Arbely Y, Barkan D, Pizov G, Shriki A, Rose-John S, Galun E, Axelrod JH (2008) IL-6/IL-6R axis plays a critical role in acute kidney injury. J Am Soc Nephrol 19:1106–1115
Newell MA, Bard MR, Goettler CE, Toschlog EA, Schenarts PJ, Sagraves SG, Holbert D, Pories WJ, Rotondo MF (2007) Body mass index and outcomes in critically injured blunt trauma patients: weighing the impact. J Am Coll Surg 204:1056–1061
Nguyen B, Tao M, Yu P, Mauro C, Seidman MA, Wang YE, Mitchell J, Ozaki CK (2013) Preoperative diet impacts the adipose tissue response to surgical trauma. Surgery 153:584–593
Pachler C, Ikeoka D, Plank J, Weinhandl H, Suppan M, Mader JK, Bodenlenz M, Regittnig W, Mangge H, Pieber TR, Ellmerer M (2007) Subcutaneous adipose tissue exerts proinflammatory cytokines after minimal trauma in humans. Am J Physiol Endocrinol Metab 293:E690–E696
Papathanassoglou ED, Moynihan JA, Ackerman MH, Mantzoros CS (2001) Serum leptin levels are higher but are not independently associated with severity or mortality in the multiple organ dysfunction/systemic inflammatory response syndrome: a matched case control and a longitudinal study. Clin Endocrinol (Oxf) 54:225–233
Partrick DA, Moore FA, Moore EE, Biffl WL, Sauaia A, Barnett CC Jr (1996) Barney Resident Research Award winner. The inflammatory profile of interleukin-6, interleukin-8, and soluble intercellular adhesion molecule-1 in postinjury multiple organ failure. Am J Surg 172:425–429
Penn AH, Schmid-Schönbein GW (2008) The intestine as source of cytotoxic mediators in shock: free fatty acids and degradation of lipid-binding proteins. Am J Physiol Heart Circ Physiol 294:H1779–H1792
Pérez de Heredia F, Wood IS, Trayhurn P (2010) Hypoxia stimulates lactate release and modulates monocarboxylate transporter (MCT1, MCT2, and MCT4) expression in human adipocytes. Pflug Arch 459:509–518
Pichavaram P, Shawky NM, Hartney TJ, Jun JY, Segar L (2021) Imatinib improves insulin resistance and inhibits injury-induced neointimal hyperplasia in high fat diet-fed mice. Eur J Pharmacol 890:173666
Prasai A, El Ayadi A, Mifflin RC, Wetzel MD, Andersen CR, Redl H, Herndon DN, Finnerty CC (2017) Characterization of adipose-derived stem cells following burn injury. Stem Cell Rev Rep 13:781–792
Qin X, Dong W, Sharpe SM, Sheth SU, Palange DC, Rider T, Jandacek R, Tso P, Deitch EA (2012) Role of lipase-generated free fatty acids in converting mesenteric lymph from a noncytotoxic to a cytotoxic fluid. Am J Physiol Gastrointest Liver Physiol 303:G969–G978
Quasim T, McMillan DC, Wallace AM, Kinsella J (2004) The relationship between leptin concentrations, the systemic inflammatory response and illness severity in surgical patients admitted to ITU. Clin Nutr 23:233–238
Raje V, Ahern KW, Martinez BA, Howell NL, Oenarto V, Granade ME, Kim JW, Tundup S, Bottermann K, Gödecke A, Keller SR, Kadl A, Bland ML, Harris TE (2020) Adipocyte lipolysis drives acute stress-induced insulin resistance. Sci Rep 10:18166-020-75321-0
Regazzetti C, Peraldi P, Grémeaux T, Najem-Lendom R, Ben-Sahra I, Cormont M, Bost F, Le Marchand-Brustel Y, Tanti JF, Giorgetti-Peraldi S (2009) Hypoxia decreases insulin signaling pathways in adipocytes. Diabetes 58:95–103
Reino DC, Pisarenko V, Palange D, Doucet D, Bonitz RP, Lu Q, Colorado I, Sheth SU, Chandler B, Kannan KB, Ramanathan M, Xu DZ, Deitch EA, Feinman R (2011) Trauma hemorrhagic shock-induced lung injury involves a gut-lymph-induced TLR4 pathway in mice. PLoS ONE 6:e14829
Riopel M, Moon JS, Bandyopadhyay GK, You S, Lam K, Liu X, Kisseleva T, Brenner D, Lee YS (2020) Inhibition of prolyl hydroxylases increases hepatic insulin and decreases glucagon sensitivity by an HIF-2α-dependent mechanism. Mol Metab 41:101039
Rizoli SB, Jaja BNR, Di Battista AP, Rhind SG, Neto AC, da Costa L, Inaba K, da Luz LT, Nascimento B, Perez A, Baker AJ, de Oliveira Manoel AL (2017) Catecholamines as outcome markers in isolated traumatic brain injury: the COMA-TBI study. Crit Care 21:37
Robidoux J, Martin TL, Collins S (2004) Beta-adrenergic receptors and regulation of energy expenditure: a family affair. Annu Rev Pharmacol Toxicol 44:297–323
Rogal J, Binder C, Kromidas E, Roosz J, Probst C, Schneider S, Schenke-Layland K, Loskill P (2020) WAT-on-a-chip integrating human mature white adipocytes for mechanistic research and pharmaceutical applications. Sci Rep 10:6666-020-63710-4
Rosen ED, Spiegelman BM (2014) What we talk about when we talk about fat. Cell 156:20–44
Rotter V, Nagaev I, Smith U (2003) Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem 278:45777–45784
Sabharwal S, Root MZ (2012) Impact of obesity on orthopaedics. J Bone Jt Surg Am 94:1045–1052
Saraf MK, Herndon DN, Porter C, Toliver-Kinsky T, Radhakrishnan R, Chao T, Chondronikola M, Sidossis LS (2016) Morphological changes in subcutaneous white adipose tissue after severe burn injury. J Burn Care Res 37:e96-103
Scholz M, Cinatl J, Schädel-Höpfner M, Windolf J (2007) Neutrophils and the blood-brain barrier dysfunction after trauma. Med Res Rev 27:401–416
Seheult J, Fitzpatrick G, Boran G (2017) Lactic acidosis: an update. Clin Chem Lab Med 55:322–333
Sell H, Dietze-Schroeder D, Kaiser U, Eckel J (2006) Monocyte chemotactic protein-1 is a potential player in the negative cross-talk between adipose tissue and skeletal muscle. Endocrinology 147:2458–2467
Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3:721–732
Sethi JK, Vidal-Puig AJ (2007) Thematic review series: adipocyte biology. Adipose tissue function and plasticity orchestrate nutritional adaptation. J Lipid Res 48:1253–1262
Sharma OP, Oswanski MF, Joseph RJ, Tonui P, Westrick L, Raj SS, Tatchell T, Waite PJ, Gandaio A (2007) Venous thromboembolism in trauma patients. Am Surg 73:1173–1180
Shashaty MG, Kalkan E, Bellamy SL, Reilly JP, Holena DN, Cummins K, Lanken PN, Feldman HI, Reilly MP, Udupa JK, Christie JD (2014) Computed tomography-defined abdominal adiposity is associated with acute kidney injury in critically ill trauma patients*. Crit Care Med 42:1619–1628
Sherren PB, Hussey J, Martin R, Kundishora T, Parker M, Emerson B (2014) Lethal triad in severe burns. Burns 40:1492–1496
Shimizu T, Yamakuchi M, Biswas KK, Aryal B, Yamada S, Hashiguchi T, Maruyama I (2016) HMGB1 is secreted by 3T3-L1 adipocytes through JNK signaling and the secretion is partially inhibited by adiponectin. Obesity (Silver Spring) 24:1913–1921
Shimomura I, Funahashi T, Takahashi M, Maeda K, Kotani K, Nakamura T, Yamashita S, Miura M, Fukuda Y, Takemura K, Tokunaga K, Matsuzawa Y (1996) Enhanced expression of PAI-1 in visceral fat: possible contributor to vascular disease in obesity. Nat Med 2:800–803
Sidossis LS, Porter C, Saraf MK, Borsheim E, Radhakrishnan RS, Chao T, Ali A, Chondronikola M, Mlcak R, Finnerty CC, Hawkins HK, Toliver-Kinsky T, Herndon DN (2015) Browning of subcutaneous white adipose tissue in humans after severe adrenergic stress. Cell Metab 22:219–227
Silberschmid M, Lund C, Szczepanski K, Lyager S (1978) Lung function and morphology after hemorrhagic shock, soft tissue trauma and regional ischemia in dogs. Eur Surg Res 10:404–414
Simmons JW, Powell MF (2016) Acute traumatic coagulopathy: pathophysiology and resuscitation. Br J Anaesth 117:iii31–iii43
Speidel MT, Booyse FM, Abrams A, Moore MA, Chung BH (1990) Lipolyzed hypertriglyceridemic serum and triglyceride-rich lipoprotein cause lipid accumulation in and are cytotoxic to cultured human endothelial cells. High density lipoproteins inhibit this cytotoxicity. Thromb Res 58:251–264
Spitler CA, Hulick RM, Graves ML, Russell GV, Bergin PF (2018) Obesity in the polytrauma patient. Orthop Clin N Am 49:307–315
Stanojcic M, Abdullahi A, Rehou S, Parousis A, Jeschke MG (2018) Pathophysiological response to burn injury in adults. Ann Surg 267:576–584
Stanojcic M, Chen P, Harrison RA, Wang V, Antonyshyn J, Zúñiga-Pflücker JC, Jeschke MG (2014) Leukocyte infiltration and activation of the NLRP3 inflammasome in white adipose tissue following thermal injury. Crit Care Med 42:1357–1364
Strauss EJ, Frank JB, Walsh M, Koval KJ, Egol KA (2007) Does obesity influence the outcome after the operative treatment of ankle fractures? J Bone Jt Surg Br 89:794–798
Taniguchi CM, Finger EC, Krieg AJ, Wu C, Diep AN, LaGory EL, Wei K, McGinnis LM, Yuan J, Kuo CJ, Giaccia AJ (2013) Cross-talk between hypoxia and insulin signaling through Phd3 regulates hepatic glucose and lipid metabolism and ameliorates diabetes. Nat Med 19:1325–1330
Trayhurn P (2013) Hypoxia and adipose tissue function and dysfunction in obesity. Physiol Rev 93:1–21
Tuzson J, Kertai P (1968) The action of endotoxin in mobilizing free fatty acid. J Pharm Pharmacol 20:226–227
Vachharajani V, Cunningham C, Yoza B, Carson J Jr, Vachharajani TJ, McCall C (2012) Adiponectin-deficiency exaggerates sepsis-induced microvascular dysfunction in the mouse brain. Obesity (Silver Spring) 20:498–504
Van Gaal LF, Mertens IL, De Block CE (2006) Mechanisms linking obesity with cardiovascular disease. Nature 444:875–880
van Hall G, Steensberg A, Sacchetti M, Fischer C, Keller C, Schjerling P, Hiscock N, Møller K, Saltin B, Febbraio MA, Pedersen BK (2003) Interleukin-6 stimulates lipolysis and fat oxidation in humans. J Clin Endocrinol Metab 88:3005–3010
Volanakis JE, Narayana SV (1996) Complement factor D, a novel serine protease. Protein Sci 5:553–564
Wade CE, Mora AG, Shields BA, Pidcoke HF, Baer LA, Chung KK, Wolf SE (2013) Signals from fat after injury: plasma adipokines and ghrelin concentrations in the severely burned. Cytokine 61:78–83
Wang B, Wood IS, Trayhurn P (2007) Dysregulation of the expression and secretion of inflammation-related adipokines by hypoxia in human adipocytes. Pflug Arch 455:479–492
Warner WA (1969) Release of free fatty acids following trauma. J Trauma 9:692–699
Wiles MD (2013) Blood pressure management in trauma: from feast to famine? Anaesthesia 68:445–449
Winfield RD, Mellnick VM, Chamieh J, Nohra E, Tan WH, Ramirez R, Raptis C, Turnbull IR, Bochicchio K, Reese S, Spinella PC, Bochicchio GV (2016) Adipose tissue location and contribution to postinjury hypercoagulability. J Trauma Acute Care Surg 81:79–85
Wolfe RR (1997) Substrate utilization/insulin resistance in sepsis/trauma. Baillieres Clin Endocrinol Metab 11:645–657
Wood IS, Wang B, Lorente-Cebrián S, Trayhurn P (2007) Hypoxia increases expression of selective facilitative glucose transporters (GLUT) and 2-deoxy-d-glucose uptake in human adipocytes. Biochem Biophys Res Commun 361:468–473
Woolf PD, McDonald JV, Feliciano DV, Kelly MM, Nichols D, Cox C (1992) The catecholamine response to multisystem trauma. Arch Surg 127:899–903
World Health Organisation (WHO) (2021) Fact sheets—obesity and overweight. Accessed 10 2021
Wu J, Vodovotz Y, Abdelhamid S, Guyette F, Yaffe M, Gruen D, Cyr A, Okonkwo D, Kar UK (2021) Multi-omic analysis in injured humans: patterns align with outcomes and treatment responses. Cell Rep Med 2(12):100478
Yadav A, Kataria MA, Saini V, Yadav A (2013) Role of leptin and adiponectin in insulin resistance. Clin Chim Acta 417:80–84
Yang S, Hu S, Hsieh YC, Choudhry MA, Rue LW III, Bland KI, Chaudry IH (2006) Mechanism of IL-6-mediated cardiac dysfunction following trauma-hemorrhage. J Mol Cell Cardiol 40:570–579
Yin J, Gao Z, He Q, Zhou D, Guo Z, Ye J (2009) Role of hypoxia in obesity-induced disorders of glucose and lipid metabolism in adipose tissue. Am J Physiol Endocrinol Metab 296:E333–E342
Zhang J, Zhang L, Zhang S, Yu Q, Xiong F, Huang K, Wang CY, Yang P (2017) HMGB1, an innate alarmin, plays a critical role in chronic inflammation of adipose tissue in obesity. Mol Cell Endocrinol 454:103–111
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464:104–107
Zhang XY, Liu Y, He T, Yang TT, Wu J, Cianflone K, Lu HL (2018) Anaphylatoxin C5a induces inflammation and reduces insulin sensitivity by activating TLR4/NF-kB/PI3K signaling pathway in 3T3-L1 adipocytes. Biomed Pharmacother 103:955–964
Zhao H, Kilgas S, Alam A, Eguchi S, Ma D (2016) The role of extracellular adenosine triphosphate in ischemic organ injury. Crit Care Med 44:1000–1012
Zinngrebe J, Debatin KM, Fischer-Posovszky P (2020) Adipocytes in hematopoiesis and acute leukemia: friends, enemies, or innocent bystanders? Leukemia 34:2305–2316
Acknowledgements
MHL was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Projektnummer 251293561—SFB 1149. PFP was supported by the Deutsche Forschungsgemeinschaft—Projektnummer 398707781—Heisenberg professorship.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Projektnummer 251293561-SFB 1149, Projektnummer 398707781.
Author information
Authors and Affiliations
Contributions
All authors contributed to this review’s conception and design. LW wrote the first draft of the manuscript. LW and MHL provided figures. All authors performed comprehensive literature research, worked on preliminary versions of manuscript and agreed to its final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Ethics approval
This review article summarizes published data and does not require ethical approval.
Consent to publish
All authors gave their consent for publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wrba, L., Halbgebauer, R., Roos, J. et al. Adipose tissue: a neglected organ in the response to severe trauma?. Cell. Mol. Life Sci. 79, 207 (2022). https://doi.org/10.1007/s00018-022-04234-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-022-04234-0