
Abstract
The recent severe acute respiratory syndrome, known as Coronavirus Disease 2019 (COVID-19) has spread so much rapidly and severely to induce World Health Organization (WHO) to declare a state of emergency over the new coronavirus SARS-CoV-2 pandemic. While several countries have chosen the almost complete lock-down for slowing down SARS-CoV-2 spread, the scientific community is called to respond to the devastating outbreak by identifying new tools for diagnosis and treatment of the dangerous COVID-19. With this aim, we performed an in silico comparative modeling analysis, which allows gaining new insights into the main conformational changes occurring in the SARS-CoV-2 spike protein, at the level of the receptor-binding domain (RBD), along interactions with human cells angiotensin-converting enzyme 2 (ACE2) receptor, that favor human cell invasion. Furthermore, our analysis provides (1) an ideal pipeline to identify already characterized antibodies that might target SARS-CoV-2 spike RBD, aiming to prevent interactions with the human ACE2, and (2) instructions for building new possible neutralizing antibodies, according to chemical/physical space restraints and complementary determining regions (CDR) mutagenesis of the identified existing antibodies. The proposed antibodies show in silico high affinity for SARS-CoV-2 spike RBD and can be used as reference antibodies also for building new high-affinity antibodies against present and future coronaviruses able to invade human cells through interactions of their spike proteins with the human ACE2. More in general, our analysis provides indications for the set-up of the right biological molecular context for investigating spike RBD–ACE2 interactions for the development of new vaccines, diagnostic kits, and other treatments based on the targeting of SARS-CoV-2 spike protein.
Similar content being viewed by others

Introduction
The scientific community is called to respond to a pandemic of respiratory disease that has spread with impressive rate among people of all the world. The new coronavirus has been called SARS-CoV-2 and the related disease indicated as COVID-19. WHO reports that positive patients in the world increased from 1,353,361, with 79,235 ascertained deaths (9 April 2020) to 10,021,401, with 499,913 confirmed deaths (29 June 2020) in less than 3 months, due to COVID-19 complications. It also appears that these numbers might be a smaller number of real cases due to our inability in quantifying rescued or asymptomatic people.
To limit death rate and SARS-CoV-2 spread, it needs to develop a vaccine and to identify new small molecules able to prevent or treat COVID-19 complications, as well as to prepare new quick diagnosis kits, able to quantify the real number of people exposed to SARS-CoV-2. Among the main actors of SARS-CoV-2 infection, SARS-CoV-2 spike proteins, RNA-dependent RNA polymerases, and proteases deserve to be mentioned. Indeed, RNA-dependent RNA polymerase has become one of the main targets of a nucleoside analog antiviral drug, the remdesivir, already used for reducing complications due to Ebola, Dengue, and MERS-CoV infections [1,2,3,4,5,6,7]. At the same time, viral protease inhibitors [8,9,10,11,12] are under investigation for their ability in preventing virus protein cleavage (with specific reference to spike protein cleavage) [13] leading to the fusion of virus proteins with host cell membranes. Also, anti-inflammatory antibodies/drugs in combination with anticoagulant molecules are under investigation for limiting coagulopathies [14,15,16,17,18] and cytokine signaling impressively triggered by SARS-CoV-2 infection [19,20,21,22,23]. Finally, the same SARS-CoV-2 spike protein has become the most investigated target due to its ability in forming interactions with the human ACE2 receptor, causing fusion events that make possible for the virus to penetrate host human cells [24,25,26,27].
The crucial role played by the spike protein is also due to the possibility to use the recombinant SARS-CoV-2 spike protein for triggering an immune response, working as a vaccine, that may help in preventing and treating COVID-19, similarly to what recently proposed [28,29,30,31,32,33,34].
For clarifying SARS-CoV-2 infection mechanisms, several research groups have recently solved the structure of the entire SARS-CoV-2 spike protein (6vsb.pdb [24]; 6vxx.pdb and 6vyb.pdb [25]), in pre-fusion conformation, and/or SARS-CoV-2 spike RBD domain in complex with the human ACE2 (6vw1.pdb; 6lzg.pdb).
In light of the available cited crystallized/cryo-em solved structures, here we propose a strategy for identifying/drawing new SARS-CoV-2 therapeutic antibodies directed against the RBD of SARS-CoV-2 spike protein that could be used for contrasting SARS-CoV-2 infection, aiming to prevent pre-/post-fusion spike conformation interconversion, responsible for virus invasion, and to provide a molecular structural context for studying new diagnosis kits based on the interactions between our engineered antibodies and the human SARS-CoV-2 spike RBD.
Materials and methods
Crystal structure sampling via folding recognition and multiple sequence alignments (MSA)
CoV-Spike and ACE2 homologous protein-crystallized structures were searched using the folding recognition methods implemented in pGenThreader and i-Tasser. With this aim, the amino acid sequence of the SARS-CoV-2 spike protein monomer (fasta sequence of the monomer taken from 6vsb.pdb, chain A) and of the ACE2 (fasta sequence of the monomer taken from 6m18.pdb, chain B) were used as query sequences for running pGenThreader (http://bioinf.cs.ucl.ac.uk/psipred/) and i-Tasser (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) to screen the PDB, searching for the most similar deposited crystallized structures [35,36,37,38].
The sequences of the retrieved 48 crystallized structures (with reference to those crystallized structures indicated with “Certain” or “High” confidence level in the pGenThreader output) were aligned using ClustalW [39] implemented in the Jalview package [40]. The 3D coordinates from the 48 crystallized structures were superimposed using the “super” command available in PyMOL [41]. The “super” command allows aligning the selected proteins under investigation for performing a comparative structural analysis, due to its ability in providing a sequence-independent structure-based pairwise alignment. Notably, the “super” command is more robust than the “align” command because it successfully performs also superimposition of proteins with a lower sequence similarity [37, 42].
Protein–protein and protein–ligand binding regions were highlighted by selecting residues within 4 Å at the protein–protein interface or from the investigated ligands, in the superimposed structures.
All the generated 3D all-atom models were energetically minimized using the Yasara Minimization server [43] or the Rosetta “relax” application within the Rosetta “scoring and prep” tools (https://www.rosettacommons.org/demos/latest/tutorials/scoring_and_prep/scoring_and_prep) [57,58,59], independently.
3D atomic models preparation of SARS-CoV-2 spike protein in post-fusion conformation and SARS-CoV-2 spike-ACE2 interactions in pre-fusion conformations
The 3D comparative model of SARS-CoV-2 spike trimer in post-fusion conformation was built by multi-template modeling using Modeller [44]. More in detail, the human SARS-CoV-2 spike protein sequence was aligned to the sequences of the available entire post-fusion conformation of the murine coronavirus spike protein (6b3o.pdb, [45]) and the remaining available crystallized subdomains of other coronavirus spike proteins in post-fusion conformations (5yl9.pdb [46]; 1wyy.pdb [47] and 1wdf.pdb [48]). Sequences of the cited crystallized structure fragments were used as query sequences for sampling the corresponding entire spike monomer sequences, by reciprocal-blastp, to be aligned with sequences of the investigated structures for comparative purposes. The obtained MSA was used for driving the multi-template modeling.
Then, a complex 3D model representing the pre-fusion spike trimer interacting with three ACE2 functional receptor units was built by superimposing the recently solved cryo-EM prefusion structure of SARS-CoV-2 spike trimer complex (6vsb.pdb, [24]; 6vyb.pdb and 6vxx.pdb [25]), the SARS-CoV-2 spike RBD crystallized in complex with the human ACE2 (6vw1.pdb; 6lzg.pdb) the SARS-CoV-1 spike trimer interacting with one ACE2 functional receptor (conformations 1–3, 6acg.pdb, 6acj.pdb, 6ack.pdb, [49] and 6cs2.pdb, [50]), the SARS-CoV-1 spike-RBD crystallized in complex with the human ACE2 (2ajf.pdb, [51]).
For investigating pre-/post-fusion conformation interconversion, we superimposed the pre-fusion available crystallized structures of SARS-CoV2 spike proteins and the generated 3D models about the pre-fusion conformation of the spike trimer in complex with three ACE2 units, to the obtained 3D model of the post-fusion conformation. All the generated 3D all-atom models were energetically minimized using the Yasara Minimization server [43] or the Rosetta “relax” application within the Rosetta “scoring and prep” tools (https://www.rosettacommons.org/demos/latest/tutorials/scoring_and_prep/scoring_and_prep) [57,58,59], independently.
Antibody 3D modeling and mutagenesis
Starting from the 3D atomic coordinates of the crystallized neutralizing antibodies m396 (2dd8.pdb [52]) and S230 (6nb7.pdb, [53]), both complexed with the SARS-CoV-1 spike RBD domain, we modelled the interactions of m396 and S230 (6nb7.pdb, [53]) with SARS-CoV-2 spike RBD domain, by superimposing the fragment antigen-based (FAB) portions of m396 (2dd8.pdb [52]) and S230 (6nb7.pdb, [53]) with the SARS-CoV-2 spike RBD domain, complexed with ACE2 (6vw1.pdb), using PyMOL.
For creating a more specific antibody directed against SARS-CoV-2 spike RBD, we replaced residues of the CDR regions of the m396 crystallized FAB portion with residues that may complement and fulfill better the SARS-CoV-2 RBD surface, also based on interactions observed in the crystallized structures of SARS-CoV-2-RBD-ACE2 protein complex. Mutagenesis analyses and modeling of the incomplete residues within the crystallized structures were performed using SPDBV [54] and/or PyMOL [55].
The proposed complete IgG chimeric antibodies were obtained by superimposing the above-cited m396 and the resulting engineered FAB portions, in complex with SARS-CoV-1/2 RBD, to the 3D atomic model of a crystallized IgG, available on the PDB (1igt.pdb, [56]) using SPDBV and PyMOL. More in detail, m396, and the engineered FAB portions were superimposed, through the PyMOL “super” command, to the FAB portions of the crystallized structure of an IgG (1igt.pdb) starting from the structural alignment of their backbones. Then, it was possible to model missing residues located at the FAB–Fc interface, solving clashes and breaks in the backbone [37, 38, 42] by using SPDBV.
Each glycosylation ladder coming from the crystal structures here investigated (1igt.pdb; 2dd8.pdb; 6nb7.pdb) was alternatively retained within the generated structural models.
After superimposition operations, allowing backbone connections, we renumbered all the atoms and the residues present in the resulting final pdb file, using an in-house developed Perl script. All the generated 3D all-atom models were energetically minimized using the Yasara Minimization server [43] or the Rosetta “relax” application within the Rosetta “scoring and prep” tools (https://www.rosettacommons.org/demos/latest/tutorials/scoring_and_prep/scoring_and_prep) [57,58,59], independently. The obtained final models were examined in VMD, PyMOL, and SPDBV by visual inspection searching for putative unsolved clashes [38, 42].
FoldX energy calculations
The FoldX AnalyseComplex assay was performed to determine the interaction energy between the four generated antibodies and the RBD domains of SARS-CoV-1/2 spike proteins, but also for determining the interaction energy between ACE2 and the interacting spike RBDs for comparative purposes.
The way the FoldX AnalyseComplex operates is by unfolding the selected targets and determining the stability of the remaining molecules and then subtracting the sum of the individual energies from global energy. More negative energies indicate a better binding. Positive energies indicate no binding [60, 61]. The energy calculated for the crystallized m396-SARS-CoV-1 RBD protein complex was used as a reference value.
Rosetta energy calculations
Rosetta applications combine several tools to model and analyze macromolecular structures (https://www.rosettacommons.org/docs/latest/Home). We chose to use the “residue_energy_breakdown” and the “InterfaceAnalyzer” applications (https://www.rosettacommons.org/demos/latest/tutorials/scoring_and_prep/scoring_and_prep) from Rosetta “scoring and prep” tools for analyzing SARS-CoV-2 spike RBD–ACE2 interface interactions. The Rosetta “residue_energy_breakdown” application was used to examine the contribution of each protein residue to the score for the lowest energy backbone relaxed structures, whereas the Rosetta “InterfaceAnalyzer” application was used for evaluating interactions across the antibody–receptor interface [57, 62,63,64].
Results
Modelling of the SARS-CoV-2 spike protein in post-fusion conformation
The main event that allows virus envelop fusion with the host human cell plasma membrane concerns a conformational change occurring at the SARS-CoV-2 spike protein that converts from pre-fusion conformation to post-fusion conformation after interactions with ACE2 and spike protein cleavage. While SARS-CoV-2 spike protein trimer has been resolved by cryo-em (6vsb.pdb [24]; 6vxx.pdb and 6vyb.pdb [25]), the post-fusion conformation is not available yet. According to [13] Coutard et al., protein cleavage at site S1/S2 and S2′ produces the division of the spike protein in two subdomains, i.e. the N-ter S-I ectodomain (containing the RBD interacting with ACE2) and the C-ter S-II membrane-anchored subdomain, that will form the SARS-CoV-2 spike protein in post-fusion conformation, able to trigger the fusion of the viral envelope with host cell plasma membrane determining host cell invasion.
For modelling 3D post-fusion conformation of SARS-CoV-2 spike protein, we searched for SARS-CoV-2 spike protein homologous structures and found that 48 crystallized structures that included poses of the whole SARS-CoV-2 spike proteins or about protein domains of SARS-CoV-2 spike proteins in complex with protein interactors (i.e. ACE2), several pre-fusion conformations of other coronavirus spike proteins, one spike protein in post-fusion conformation and three further protein subdomains about spike proteins in post-fusion conformation (Supplementary Table 1).
Thus, we built an MSA by aligning the sequence of the human SARS-CoV-2 spike protein, the sequence of the available whole post-fusion conformation of a spike protein (6b3o.pdb, [45]), sequences of the remaining crystallized subdomains of other virus spike proteins in post-fusion conformations (5yl9.pdb [46]; 1wyy.pdb [47] and 1wdf.pdb [48]), together with their complete counterpart sequences sampled by reciprocal-blastp (Fig. 1).

Extract of the MSA of SARS-CoV-2 spike protein monomer with the sequences of the crystallized structures of the spike whole protein or protein fragments observed in the post-fusion conformation from other coronaviruses, resulting from sequence cleavage. a Grey boxes indicate protein regions involved in cleavage events, whereas black boxes indicate the position of the proposed cleavage sites. a, b Cyan boxes indicate the cluster of residues conserved in the sampled sequences and maintained in the post-fusion conformation, considered as anchor points for preparing the 6b3o.pdb based SARS-CoV-2 spike sequence-structure alignment, used for building the 3D comparative model of SARS-CoV-2 spike protein in post-fusion conformation (amino acids S704-A771, see a; amino acids L922-S1147, see b; YP_009724390.1 residues numbering)
In the provided MSA (Fig. 1), it is possible to observe the conserved S1/S2 and S2′ cleavage sites, according to [13] and the sequence of the C-terminal domain resulting from the cleavage. Furthermore, it is also possible to observe several clusters of conserved residues (Fig. 1) that were used as anchor points of the proposed SARS-CoV-2 spike sequence/multi-template-structure alignment for building the 3D model of a monomer of SARS-CoV-2 spike protein in post-fusion conformation (Fig. 2). Among those conserved protein regions, six clusters of residues host conserved cysteine residues (Fig. 2) forming disulfide bridges crucial for stabilizing both pre-fusion and post-fusion SARS-CoV-2 spike protein conformation [45]. The modelled SARS-CoV-2 spike post-fusion conformation consists of residues 704–771 and 922–1147, YP_009724390.1 residues numbering, resulting from protein cleavage [13]. Those protein fragments are the main constituents of the post-fusion conformation, and also the only fragments which can be modelled using as a protein template the unique/greatest existing solved post-fusion conformation structure of a sequence/structure related spike protein (6b3o.pdb, amino acids 741–807 and 972–1248, NP_045300.1/6b3o.pdb residues numbering) [45].

SARS-CoV-2 spike protein (S-II domain) 3D model in post-fusion conformation. Lateral view (a), top view (b) and bottom view (c) of the SARS-CoV-2 spike protein trimer 3D comparative model, reported in cartoon colored representation
The trimer of the SARS-CoV-2 spike protein in post-fusion conformation was obtained by duplicating two times the obtained monomer and superimposing the resulting three SARS-CoV-2 spike protein monomers on the three spike protein monomers reported in 6b3o.pdb (Fig. 2). The releaxed 3D comparative model of SARS-CoV-2 spike protein trimer built by multi-template comparative modeling showed an RMSD lower than 0.5 Å with the available spike protein in post-fusion conformation (6b3o.pdb). The resulting model (Fig. 2) appeared elongated and narrow (with respect to the pre-fusion conformation), according to what observed in fragments of the spike proteins crystallized in post-fusion conformations, whose sequences are reported in Fig. 1 and whose PDB_IDs are listed in Supplementary Table 1.
The resulting SARS-CoV-2 spike post-fusion spike conformation was obtained following the loss of the cited N-terminal domain (residues 1–703, black cartoon, YP_009724390.1 residues numbering Fig. 3) and of a middle protein portion (residues 772–921, black cartoon). The resulting post-fusion structure shows regions that underwent few conformational changes, with respect to the pre-fusion structure [i.e. see residues 715–771 (yellow cartoon), 983–1028 (red cartoon), 1029–1126 (magenta cartoon), YP_009724390.1 residues numbering, Fig. 3], whereas other regions show important conformational changes [i.e. see residues 704–715 (yellow cartoon), 922–979 (green cartoon), 1127–1146 (orange cartoon), YP_009724390.1 residues numbering, Fig. 3]. Notably, regions showing few conformational changes are those hosting conserved cysteine residues involved in disulfide bridges in both pre-fusion and post-fusion structures (Figs. 1, 3).

SARS-CoV-2 spike protein regions involved in the pre/post-fusion conformational transitions. Topology panel: schematic of the SARS-CoV-2 spike protein organization according to the below reported 3D model protein regions. Colors and residues numbering reflect the localization in the SARS-CoV-2 spike protein sequence of the below reported protein domain 3D structures (YP_009724390.1 residues numbering). UH upstream helix; FP the region hosting the fusion peptide; HR1 heptad repeat 1; CH central helix; BH β-hairpin region; CD connector domain; HR2 heptad repeat 2, according to [45]. a, f Lateral views of the SARS-CoV-2 spike protein trimer in pre-/post-fusion conformation, respectively, are reported in colored cartoon representation. b, g Lateral views of the SARS-CoV-2 spike monomer in pre-/post-fusion conformation, respectively, are reported in colored cartoons. c, h Indicate the zoomed views of the Q926-K1028 protein region (green/red cartoon representation). d, i Indicate the zoomed views of the M1029-D1146 protein region (magenta/orange cartoon representation). e, j Indicate the zoomed views of the S704-I771 protein region (yellow cartoon representation). Yellow sticks in d, e, i, j indicate disulfide bridges. Residue labels indicate residues to be used as a reference for identifying quickly the cited protein region terminal portions or cysteine residues involved in disulfide bridges. Notably, yellow, magenta and red cartoon indicate the monomer regions involved in few conformational changes, whereas green and orange cartoon indicate regions involved in large conformational changes. Black cartoon portions in b indicate protein regions lost after cleavage events and/or not available in the crystallized structures. Cyan cartoon portion in g indicates the 1146–1197 protein region and was obtained by comparative modeling using as a protein template the only available SARS-CoV-1 spike protein with a solved structure for the corresponding protein region in the post-fusion conformation, as observed in 6b3o.pdb. Notably, the corresponding protein region was solved in none of the investigated crystallized spike proteins in pre-fusion conformations (Supplementary Table 1)
On the other hand, the important conformational changes observed at regions 704–715 and 1127–1146 appears to be related to the loss of the N-terminal portion (residues 1–703, black cartoon), which causes the reorientation of the 704–715 peptide, involved in the perturbation of the 1127–1146 protein region that can re-orient itself in the free space, due to the N-terminal missing residues. Similarly, the important conformational changes observed at the 922–982 protein region (Fig. 3) appears to be due to the loss of the middle protein region (residues 772–921, black cartoon Fig. 3), whose removal causes the reorientation and the relaxation of the 922–982 amino acid protein region (green cartoon, Fig. 3), that may occupy the space previously occupied by the cleaved N-terminal region (1–703 residues, black cartoon).
Notably, a further stabilization of the post-fusion conformation depends on the re-orientation of the C-terminal 1121–1196 protein region (orange and cyan cartoon, Fig. 4). Indeed, the C-terminal portion (with specific reference to residues 1179–1196) of each monomer is deeply involved in intermonomer hydrophilic and ionic interactions with residues of the 922–982 re-oriented amino acid protein region (with specific reference to residues 926–949), of the close monomers. Notably, the 1147–1196 protein region was not solved in the pre-fusion structure (Fig. 4), but the previous protein segment, i.e. residues 1121–1146, showed a completely different orientation in the available pre-fusion structures.

SARS-CoV-2 spike protein regions involved in the pre/post-fusion molecular packing. a, g Lateral views of the spike protein trimer in post/pre-fusion conformation, respectively. Colored regions indicate the main protein portions responsible for the different molecular packing of the spike protein in the post/pre-fusion conformations. b, h Lateral zoomed views of the spike protein trimer core (red–green–orange cartoon) in the post/pre-fusion conformations. c, e Top and bottom views of the spike protein trimer in post-fusion conformation reported in a. d, f Top and bottom views of the spike protein trimer colored regions in post-fusion conformation reported in b. i, k Top and bottom views of the spike protein trimer in pre-fusion conformation reported in g. j, l Top and bottom views of the spike protein trimer colored regions in post-fusion conformation reported in h. The reported colors indicate the same regions described in Fig. 3
Modelling of the interactions between the SARS-CoV-2 spike protein and the human ACE2 along pre-/post-fusion conformation interconversion
Among the sampled crystallized structures, it was possible to observe three PDB_IDs about the entire SARS-CoV-2 spike proteins and two about SARS-CoV-2 spike RBD protein interacting with the human ACE2 (Supplementary Table 1). Furthermore, it was possible to highlight several crystallized structures about SARS-CoV-1 and MERS-CoV spike proteins as single proteins or in complex with their receptors or dedicated antibodies (Supplementary Table 1). Notably, among the sampled structures, also the four entries used for building the 3D comparative model of the post-fusion conformation, were considered (Supplementary Table 1).
For modeling main interactions occurring between SARS-CoV-2 spike proteins and ACE2, thanks to the high percentage of identical residues shared by spike RBD from several CoV strains (Fig. 5), it was possible to structurally align three objects consisting of the human ACE2-SARS-CoV-1 spike-RBD protein complex (2ajf.pdb) to the human ACE2-SARS-CoV-2 spike-RBD protein complex (6vw1.pdb, 6lzg.pdb) and the SARS-CoV-2 spike protein trimer (6vsb.pdb; 6vxx.pdb; 6vyb.pdb). More in detail, the superimposition performed using PyMol was led by the structural alignment of the RBD of ACE2-SARS-CoV1 (2ajf.pdb) and ACE-2-SARS-CoV-2 (6vw1.pdb, 6lzg.pdb) spike proteins (Fig. 6), followed by the structure alignment with SARS-CoV-2 spike protein trimer (6vsb.pdb; 6vxx.pdb; 6vyb.pdb). Notably, we obtained an efficient superimposition of the two RBD domains (RMSD lower than 0.5 Å) of the human SARS-CoV-1 and SARS-CoV-2 spike proteins also due to their high percentage of identical residues (> 75%).

Multiple sequence alignment of RBDs from 11 SARS-CoV and 3 MERS-CoV strains. The reported residues numbering refers to the indicated sequences sampled by blastp or to the indicated crystallized structure sequences

Side view (a–f) and top view (g–l) of the human SARS-CoV-2 spike protein interacting with 3 units of the human ACE2 N-terminal domain (a–e; g–k). SARS-CoV-2 spike protein trimer (6vsb.pdb) is reported in white cartoon representation with the 3 spike RBDs reported in red (in the closed pre-fusion state) or green (in the open pre-fusion state) cartoon. The open pre-fusion state allows establishing pre-invasion interactions with the ACE2 N-terminal domain. SARS-CoV-2 spike protein trimer C-terminal domain, resulting from protein cleavage that triggers the post-fusion conformation, is reported in black cartoon representation in f (lateral view) and l (top view)
It was possible to superimpose the crystallized SARS-CoV-2 spike protein in pre-fusion conformation and the modelled SARS-CoV-2 spike protein trimers in post-fusion conformation for showing the deep conformational changes occurring along conformation interconversion (Fig. 6). Indeed, it is known that RBD subunits are further mobile being able to exhibit multiple conformational states that modulate the accessibility of RBDs, triggering interactions with receptor proteins of the host cell plasma membranes [53]. In the proposed conformational changes reported in the scheme of Fig. 6, the SARS-CoV-2 spike protein goes from a closed state in which RBD residues interacting with ACE2 are not accessible (Fig. 6a, g), to a partially open state with one (Fig. 6b, h) or two (Fig. 6c, i) RBD accessible subunits, till a final open state in which RBD residues interacting with ACE2 at each RBD subunits are accessible and ready to establish interactions with ACE2 (Fig. 6d, j). The established RBD–ACE2 interactions are the first step of the fusion of plasma membrane with the viral protein envelop, followed by cleavage events and conformational changes that determine the set-up of the post-fusion conformation (Fig. 6e, f, k, l). The resulting post-fusion conformation appears to be elongated (about 180 Å long) and narrower (Figs. 3, 4, 6f, l) than the pre-fusion conformation (about 88 Å long, a structural comparison is viewable in Figs. 3, 4, 6e, k). The top portion of the post-fusion conformation extends beyond ACE2 receptors (Fig. 6e, k), known for being anchored to the plasma membrane and involved in internalization events [8, 49, 52, 53, 65].
SARS-CoV-1 and SARS-CoV-2 spike RBD residues involved in direct interactions with ACE2
From the available crystallized structures and from the obtained 3D structure models, it was possible to highlight SARS-CoV-1 spike RBD (2ajf.pdb) and SARS-CoV-2 spike RBD residues (6) involved in the binding of the human ACE2 (Fig. 7 and Supplementary Table 1). Notably, ion-pair interactions observed between SARS-CoV-1 spike RBD and the human ACE2 are also observed between SARS-CoV-2 spike RBD and the human ACE2. The reported data represent an updated/integrated analysis of a similar one reported in [66], in light of the recently deposited SARS-CoV-2 spike RBD in complex with the human ACE2 (6vw1.pdb).

SARS-CoV-1 and SARS-CoV-2 RBD residues involved in direct interactions with ACE2. H. sapiens ACE2 is reported in white cartoon representation. SARS-CoV-1 RBD is reported in magenta cartoon representation, whereas SARS-CoV-2 RBD is reported in yellow cartoon representation. a, c Residues involved in polar interactions between SARS-CoV-1 RBD (magenta sticks) and ACE2 (white sticks). b, d Residues involved in polar interactions between SARS-CoV-2 RBD (yellow sticks) and ACE2 (black sticks). Polar interactions are represented by black dashed lines in the exploded views reported in c and d
Comparative analysis of existing SARS-CoV-1 spike RBD directed neutralizing antibodies and predicted interactions with SARS-CoV-2 spike RBD
RBD from SARS-CoV-1 was crystallized in complex with the FAB domain of two different antibodies, namely m396 (2dd8.pdb, [52]) and S230 (6nb7.pdb, [53]). Both of them show high affinity for SARS-CoV-1 spike RBD [52, 53]. Nevertheless, they show different peculiarities in their mechanism of action.
Indeed, S230 after binding RBD, similarly to ACE2, can trigger the SARS-CoV spike transition to the post-fusion conformation and it is not clarified yet, if virus–cell fusion may be triggered by S230 also when S230-RBD interactions occur close to the surface of the host cell plasma membrane protein targets of the SARS-CoV-1 [53]. At variance with S230, the m396 antibody appears to be able to prevent SARS-CoV-1 spike-ACE2 interactions and SARS-CoV-1 spike pre-/post-fusion conformation transition, neutralizing virus attack [52].
Thanks to the high percentage of identical residues (> 75%) between SARS-CoV-1 and SARS-CoV-2 spike RBD domains and to their highly similar tertiary structure, as observed from the RMSD of 0.5 Å between the coordinates of RBDs from SARS-CoV-1 (6nb7.pdb, [53] and 2dd8.pdb, [52]) and SARS-CoV-2 (6vw1.pdb [67] and 6vsb.pdb, [24]) spike proteins, it was possible to evaluate interactions between m396 and SARS-CoV-2 spike RBD and to propose a sequence/structure of an ideal FAB m396-based chimeric antibody for targeting SARS-CoV-2 spike RBD domain, preventing fusion events with ACE2 and thus the following infection.
With this aim, we first highlighted the different RBD portions bound to the known antibodies. Then, we superimposed SARS-CoV-1 RBD to SARS-CoV-2 RBD for highlighting differences in residues involved in direct interactions with m396 CDR regions and with S230 CDR regions (Fig. 8 and Table 1).

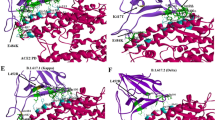
SARS-CoV-1 spike and SARS-CoV-2 spike monomers in pre-fusion conformation interacting with SARS-CoV-1 spike RBD selective antibodies S230 (6nb7.pdb) and m396 (2dd8.pdb). a Superimposition of the tertiary structure of SARS-CoV-1 (6nb7.pdb) and SARS-CoV-2 (6vsb.pdb) spike protein monomers reported in pink cartoon representation. SARS-CoV-1 and SARS-CoV-2 RBDs are reported in grey cartoon representation. S230 FAB ab portion (6nb7.pdb) is reported in yellow (light chain) and pink (heavy chain) cartoon representation. m396 FAB ab portion (2dd8.pdb) is reported in orange (light chain) and blue (heavy chain) cartoon representation. b Zoomed view of the superimposition of SARS-CoV-1 Spike and SARS-CoV-2 Spike RBD domains interacting with S230 and m396 FAB antibodies (see a for colors). c, d Super zoomed and rotated views of the crystallized SARS-CoV-1 Spike RBD residues interacting with S230 ab. e, f Super zoomed and rotated views of SARS-CoV-2 Spike RBD predicted residues interacting with S230 ab. g, h Super zoomed and rotated views of the crystallized SARS-CoV-1 Spike RBD residues interacting with m396 ab. i, j Super zoomed and rotated views of SARS-CoV-2 Spike RBD predicted residues interacting with m396 ab. c–j Residues at the RBD–ab interface in the 3.5–4 Å distance range are reported in sticks representation. White sticks indicate RBD residues; orange and blue sticks indicate m396 ab residues, yellow and pink sticks indicate S230 ab residues
SARS-CoV-2 spike RBD-directed neutralizing antibody engineering
Due to the uncertain data concerning fusion events and mechanism of action of S230 antibody, we built a new SARS-CoV-1/2 RBD-directed antibody focusing and starting from the analysis of monomer–monomer interface interactions observed between the m396 antibody crystallized in complex with SARS-CoV-1 RBD [52], superimposed to SARS-CoV-1 spike RBD/ACE2 complex (2ajf.pdb), and by comparing them with monomer–monomer interface interactions observed between the modelled m396 antibody in complex with SARS-CoV-2 RBD, superimposed to SARS-CoV-2 RBD/ACE2 (6vw1.pdb; 6lzg.pdb) protein complex (Fig. 9).

Molecular framework of the investigated proteins hosting SARS-CoV-spike RBDs, light and heavy chain of the m396 antibody and the human ACE2, simultaneously. The shown spike RBD, ACE2, and m396 protein portions are those in a reciprocal distance range of 4 Å. Upper panel: superimposition of the crystallized SARS-CoV-1 spike RBD (white cartoon representation) in complex with m396 antibody (2d88.pdb, orange, and blue cartoon) and ACE2 (2ajf.pdb, cyan cartoon). Bottom panel: superimposition of SARS-CoV-2 spike RBDs (from 6vw1.pdb, white cartoon representation), ACE2 from 6vw1.pdb (cyan cartoon) and m396 from 2d88.pdb (orange and blue cartoon)
Then, we highlighted m396 CDR residues (Table 2) for replacing them aiming to increase m392 affinity versus SARS-CoV-1/2 spike RBDs. Residues to be mutated/replaced were chosen according to space-restraints and chemical needs for better complementing SARS-CoV-1/2 spike RBD surface, based on the available SARS-CoV-1/2 RBD structures in complex with ACE2, aiming to produce something that resembled ACE2 surface (Tables 1, 2). Some of the proposed mutated residues (Table 3) are surely allowed because already observed at the corresponding sites of other known antibodies, according to Chotia/Kabat rules (http://www.bioinf.org.uk/abs/chothia.html; [68]).
Residue replacement was directly performed in the newly generated 3D model hosting the interacting m396-SARS-CoV-2 spike RBD. Similarly, a complex of the modified m396 antibody interacting with SARS-CoV-1 RBD was also created. All m396 CDR mutated residues are reported in Table 3. Furthermore, mutated residues within m396 CDR interacting with SARS-CoV-2 spike RBD residues can be observed in Fig. 10.

m396 neutralizing antibody, native and engineered, in complex with SARS-CoV-2 spike RBD. a, b Exploded view and perspective view of native m396 neutralizing antibody (in orange blue cartoon) in complex with SARS-CoV-2 spike RBD (in white cartoon representation). Residues at the m396/RBD interface in a distance range within 4 Å are indicated by white sticks (RBD), orange sticks (m396 CDR-H residues), and blue sticks (m396 CDR-L residues). c, d Exploded view and perspective view of the engineered m396 predicted neutralizing antibody (in orange blue cartoon) in complex with SARS-CoV-2 spike RBD (in white cartoon representation). Residues at the engineered m396/RBD interface in a distance range within 4 Å are indicated by white sticks (RBD), orange sticks (engineered m396 CDR-H residues), and blue sticks (engineered m396 CDR-L residues)
The engineered FAB portions were, thus, aligned and superimposed on the FAB portion of a crystallized IgG (1igt.pdb, [56]). The sequence of the chimeric antibodies can be observed in Supplementary Fig. 1, whereas their complete structure can be observed in Supplementary Fig. 2.
FoldX-free energy calculation
The interaction energies calculated between the SARS-CoV-2 spike RBD domain and m396 native antibody FAB portion give a negative value (Table 4), confirming that there might be a binding interaction between m396 native antibody FAB portion and SARS-CoV-2 spike RBD. This result is encouraging, also due to the indirect validation obtained by getting similar interaction energies for the crystallized SARS-CoV-1 RBD in complex with m396 (2d88.pdb) and for SARS-CoV-1 and SARS-CoV-2 spike RBD domains crystallized in complex with ACE2 (2ajf.pdb and 6vw1.pdb, respectively) (Table 4). Furthermore, a strong interaction (in terms of interaction energies calculated by FoldX Analyse complex assay) is also predicted between SARS-CoV-2 spike RBD (but also SARS-CoV-1 spike RBD) and the modified m396 antibody (see Table 4), suggesting that the engineered m396 might be more efficient than the native m396 in binding the SARS-CoV-2 (more than SARS-CoV-1) spike RBD.
Rosetta energy calculations
The Rosetta “relax” application was used for relaxing energetically the investigated crystallized structures and 3D models in the Rosetta force field and for each relaxation run the script provided a new PDB file, whose structural energy features are reported in Table 5. The “residue_energy_breakdown” tool confirmed that the m396/SARS-CoV-2 spike RBD complex might have a number of interchain interactions comparable to those observed at the interface of the crystallized m396/SARS-CoV-1 spike RBD complex (Supplementary Tables 2 and 3). Furthermore, the proposed mutations favor new interactions at the interface of the m396-based mutated antibody and SARS-CoV-1/2 spike RBDs, at the level of the proposed ten mutations and in their neighborhoods (Supplementary Tables 2–5).
Then, the investigated crystallized structures and 3D models were analysed with the Rosetta “InterfaceAnalyzer” application, for estimating interchain binding energies. The analyses provided negative binding energies (see dG_separated parameter, Table 5) for all the investigated structures, indicating a binding interaction between m396 native antibody FAB portion and SARS-CoV-2 spike RBD, comparable in terms of binding energy, to the ones calculated for the crystallized SARS-CoV-1 spike RBD/m396 protein complex. Notably, the resulting binding energies obtained for the 3D models hosting the investigated m396 and m396-based modified antibodies in complex with SARS-CoV-1/2 spike RBD domains were comparable (see dG_separated parameter, Table 5) to the binding energies calculated for the 3D models hosting the crystallized SARS-CoV-1/2 spike RBD domains in complex with ACE2 (2ajf.pdb and 6vw1.pdb, respectively) (Table 5; Supplementary Tables 2–7).
Discussion
The indicated pipeline has allowed to set up a molecular framework hosting SARS-CoV-2 spike protein, ACE2 receptor, and different antibodies in the same pdb session that could be handled with different molecular visualizers. In this molecular framework, it is possible to study and predict, at molecular level, interactions between the different “pieces” of the framework that may help in understanding virus invasion mechanisms, developing new vaccines or antibodies, identifying small molecules with high affinity for viral proteins and establishing quick/safe diagnosis selective/specific kits. Indeed, the scientific community is now focused in the development of new weapons for containing SARS-CoV-2 spread and COVID-19 complications as it could be observed in the enormous effort in developing new vaccines based on a virus protein/nucleic acid portion able to induce an efficient and specific immunogenic response [69,70,71,72,73], or in developing a neutralizing therapeutic antibody highly specific for SARS-CoV-2 spike RBD [29, 30, 74,75,76,77], maybe starting from antibodies isolated from hyperimmune plasma from convalescent patients [78], or in identifying chemicals with high affinity for SARS-CoV-2 crucial proteins [1,2,3, 79,80,81,82], maybe chosen also among repositories of inhibitors of proteases or RNA-dependent polymerases [83,84,85]. Notably, it needs to develop trustable titer serologic assay kits, for establishing the level of the antibodies in different biological samples. Indeed, the most important question, i.e. the ones about the real number of people exposed to the virus, has not a clear answer, yet [77, 86,87,88].
Within the presented molecular framework, we have highlighted a set of possible efficient interactions between the crystallized m396 antibody and SARS-CoV-2 spike RBD, raising the question about the possibility to test directly m396 on cultured cells exposed to the virus and then, hopefully, on patients.
Starting from that observation, we have also proposed a set of modifications of m396 CDR residues resulting in a higher specific antibody, to be expressed and tested on cultured cells. Along the development of our antibody engineering modeling session, an important paper was published and another is under revision in support of the hypothesis that m396 may be able to bind SARS-CoV-2 spike protein [74, 75].
FoldX and Rosetta analyses employed independently for monitoring binding energies at the antibody receptor interface, provided coherent results, strengthening the hypothesis that the original m396 antibody might have high affinity also against SARS-CoV-2 spike protein, as indicated by the calculated negative binding energy (see the Foldx interaction energy parameter or the Rosetta dG_separated parameter). Furthermore, the employed tools indicate a local break down in structural energy in correspondence of the proposed mutated sites at the antibody–receptor complex interfaces, reflecting an increased affinity (in terms of newly established interactions) among the investigated m396-based mutated antibodies and SARS-CoV-1/2 spike RBDs.
Although the calculated binding energies cannot be considered an absolute estimation of the real affinity between the investigated proteins, the binding energies obtained for the analysed crystallized structures and 3D engineered models, were of the same order of magnitude suggesting that the original m396 and the m396-based modified antibody might be very efficient in binding the SARS-CoV-2 (as well as SARS-CoV-1) spike RBD.
It was also possible to pose in the proposed molecular framework the recent proposed SARS-CoV-2 spike RBD directed CR3022 FAB antibody (6yla.pdb; 6w41.pdb, [75]), showing that it binds a different site of RBD that protrudes towards the central cavity of the spike protein trimer (data not shown). It appears that the RBD-antibody interaction is possible only if at least two RBDs on the trimeric spike protein are in the “open” state of the prefusion conformation and slightly rotated, in a site distant from ACE2 receptor-binding region, according to what proposed by the authors [75]. Dedicated studies are necessary for understanding if steric hindering problems might arise using the whole antibody, and deepening the comprehension of the not competitive mechanism that would be observed between CR3022 and RBD in presence of ACE2 receptor.
Studying all the cited interactions in the same pdb-molecular session has allowed highlighting the most crucial ACE2 portions involved in direct interactions with SARS-CoV-2 RBD, suggesting that the administration of the recombinant RBD, a spike monomer or the entire spike trimer, if correctly folded, might result in the efficient triggering of antibody production from our plasma b-cells, reducing COVID-19 complications (supporting what has been recently proposed [69,70,71,72,73]).
At the same time, the ACE2–RBD interaction estimated in our molecular framework has strengthened the hypothesis to use the recombinant ACE2 for limiting COVID-19 infection complications (according to what recently proposed [89, 90]).
A molecular framework like the ones here proposed will also help in studying the putative role of ACE inhibitors in perturbing ACE2–RBD interactions. Indeed, it was recently proposed that patients treated with ACE inhibitors might be more exposed to SARS-CoV-2 infection [91]. Although ACE1 (refseq accession number: NP_068576.1, representing the main target of ACE inhibitors) and ACE2 (NP_690043.1, testis isoform or NP_000780, somatic isoform, among the most studied isoforms) share the 40% of identical residues, few uncertain data about ACE inhibitors and a possible greater selectivity for ACE1 versus ACE2, or on their effect on ACE1/2 expression regulation are available in the literature [91, 92]. From a structural comparison, it is observed that the RMSD of the crystallized native ACE2 coordinates (1r42.pdb, [93]) and ACE1 coordinates (1o8a.pdb, [94]) is lower than 2.5 Å.
Notably, the presence of ACE inhibitors captopril and enalaprilat (1uze.pdb [95], 4c2p.pdb, [96]) and lisinopril (1o86.pdb, [94]) produces an RMSD lower than 0.3 Å in the atomic coordinates of the cited crystallized structures with reference to the native ACE2 (1r42.pdb, [93]).
Conversely, we cannot establish if the slightly higher RMSD observed between the native ACE2 (1r42.pdb) and ACE2 complexed with SARS-CoV-1 spike RBD (0.41 Å, 2ajf.pdb) and SARS-CoV-2 spike RBD (1.2 Å, 6vw1.pdb) can be attributed exclusively to interactions with SARS-CoV-1/2 spike RBDs because the observed RMSDs are of the same order of magnitude of the experimental resolution of the investigated crystallized structures.
However, also admitting that ACE1 inhibitors at the employed dosage would target ACE2, with the same efficiency observed versus ACE1, the presence of those inhibitors in ACE2 binding cavity should not be able to induce an important conformational change in ACE2, which might favor a greater affinity of ACE2 versus SARS-CoV-2 spike RBD.
Thus, the only mechanism for which, patients treated with ACE inhibitors would be more exposed to SARS-CoV-2, would rely on a positive feedback induced by ACE inhibitors in ACE2 expression. Nevertheless, pieces of evidence in support of this hypothesis need to be deepened [92, 97]. Along with manuscript revisions, a couple of interesting papers have been published on this concern showing that patients treated with ACE inhibitors show a slightly less severe set of complications due to COVID-19 than patients that do not use ACE inhibitors [98, 99].
In conclusion, the presented analysis highlights the importance to use fold recognition tools along the approach to a drug design problem according to a rational protocol (similar to what previously reported [37, 38, 42]), like the ones presented. Indeed, in this case, fold recognition tools have helped us in identifying crystallized structures of ACE2 and SARS-CoV spike proteins similar to those under investigation in just 2 days. Furthermore, performing structural comparative analysis has allowed us to identify a possible good starting point, like the ones represented by m396, already crystallized in complex with SARS-CoV-1 spike RBD, for building the proposed antibodies in just few weeks. The same strategy might be promptly applied also for future infections by those researchers involved in drawing new antibodies and/or developing new vaccines, i.e. for dealing with future coronaviruses outbreaks.
To the best of our knowledge, the reported SARS-CoV-2 spike protein trimer 3D model is the first model describing a possible conformational change leading to a reliable SARS-CoV-2 spike protein in post-fusion conformation. The proposed model, based on the only available spike protein (6b3o.pdb) crystallized in post-fusion conformation, will help in understanding the mechanism allowing the virus envelop fusion with host cell plasma membranes, through and following interactions with ACE2. Notably, it was possible to propose a 3D model for the main states reached by the SARS-CoV-2 spike protein through the cited conformational changes, leading to the post-fusion conformation, because a closed state in which RBD residues interacting with ACE2 are not accessible, a partially open state with one or two RBD accessible subunits, a final open state, in which RBD residues interacting with ACE2 at each RBD subunits are accessible and ready to establish interactions with ACE2, and a post-fusion conformation, were already described for MERS-CoV (6nb3.pdb; 6nb4.pdb) and SARS-CoV-1 (6nb6.pdb; 6nb7.pdb; 6b3o.pdb) spike proteins [45, 53]. Considering that ACE2 works as a dimer and the spike protein consists of a trimer, it might be even speculated that a fusion complete mechanism might involve three ACE2 dimers and two spike trimers, simultaneously.
Notably, since 27 May 2020, three crystallized structures of SARS-CoV-2 spike protein in the closed conformation (with not accessible RBD subunits) or in the partially open conformations (with one or two accessible RBD subunits, without ACE2 interacting subunits) have been deposited (6x2a.pdb, 6x2b.pdb, and 6x2c.pdb) and are coherent with our proposed corresponding models. Indeed, the RMSD between the coordinates of the crystallized (6x2c.pdb) closed state and our starting crystallized structures (6vsb.pdb; 6vxx.pdb) depicting a possible closed state of SARS-CoV-2 spike protein is lower than 1.3 Å. Furthermore, the RMSD between the coordinates of the crystallized partially open states of SARS-CoV-2 spike protein (6x2b.pdb, showing two accessible RBDs and 6x2a.pdb, showing one accessible RBD) and our corresponding 3D models, showing two or one accessible RBDs, is lower than 1.4 Å, in both cases.
Our analysis through the provided 3D model in post-fusion conformation, coherently to the crystallized 3D structure of SARS-CoV-2 spike protein in pre-fusion conformation, further confirms the presence and stability of a sort of channel at the interface of the three monomers that could represent a good target site for a virtual screening of a chemical/drug library, aiming to identify a small molecule/peptide with high affinity for the central region of the spike protein trimer, which should be able to block conformational changes leading to post-fusion conformation. Indeed, it was recently observed that a peptide named EK1 (5zvm.pdb, [100]), showing high affinity for the 920–935 protein region (YP_009724390.1 residues numbering), was able to prevent conformational changes and post-fusion conformation stabilization. The screening of a drug/peptide library would help in identifying an already approved drug/peptide, maybe structurally related to EK1, with high affinity for the spike trimer central channel, that might be immediately translated to the patient bed-side, after successful in silico analyses, in the context of the drug-repositioning approaches [101, 102].
Notably, the provided molecular framework for investigating/drawing new antibodies, based on space-restraints needs, would be used for the set-up of new antibodies based on the available tissue-specific immunoglobulin structures, as the proposed IgG2A (1igt.pdb, [56]) or other specialized antibodies, already optimized for targeting specific cells or receptors (i.e.1hzh.pdb, [103]), also among those that may successfully target the respiratory tract (1r70.pdb, [104] or 2qtj.pdb [105] or 6ue7.pdb [106]), that might be administered even by aerosol [107, 108].
At the same time, already at the preclinical level, the administered vaccines based on the administration of the entire SARS-CoV-2 spike protein ([69,70,71] or on the administration of the single SARS-CoV-2 spike RBD, will induce the production of specific antibodies that might be sequenced and modelled in silico. On this concern, the provided molecular network will help in quantifying interactions between SARS-CoV-2 spike RBD (also in cases of different RBD variants [109]) and the newly investigated antibodies, i.e. lower the calculated binding energy in the modelled complex, higher the likelihood to obtain strong RBD-antibody interactions, that would result in more efficient treatments.
The discovered antibodies with the highest affinity for the spike RBD might also be implemented in an antibody titer serologic kit for diagnosis, aiming to the early identification and quantification of SARS-CoV-2 in sera, also in asymptomatic people.
Conversely, a new diagnosis kit could also be based on the native RBD or a modified synthetic RBD, with greater affinity for the detected human antibodies directed against SARS-CoV-2 spike RBD protein, for determining the real number of healthy people already exposed to the virus in the population, by quantifying anti-SARS-CoV-2 antibodies in sera/plasma of rescued patients and/or asymptomatic peoples.
The lacking knowledge about the real number of people exposed to the virus (including asymptomatic people, people with mild symptoms and rescued people that never needed hospitalization or quarantine) is the only important data that we still miss. Without data about the real number of people, exposed to the virus, in the population, coming back to normal life will be extremely slow.
Abbreviations
- SARS:
-
Severe acute respiratory syndrome
- CoV:
-
Coronavirus
- RBD:
-
Receptor binding domain
- ACE2:
-
Angiotensin-converting enzyme 2
- FAB:
-
Fragment antigen binding
- WHO:
-
Word Health Organization
- CDR:
-
Complementary-determining regions
- SPDBV:
-
SwissPDBViewer
References
Gordon CJ, Tchesnokov EP, Feng JY et al (2020) The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J Biol Chem. https://doi.org/10.1074/jbc.AC120.013056
Elfiky AA (2020) Ribavirin, remdesivir, sofosbuvir, galidesivir, and tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): a molecular docking study. Life Sci. https://doi.org/10.1016/j.lfs.2020.117592
Wang M, Cao R, Zhang L et al (2020) Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res 30(3):269–271
Wu C, Liu Y, Yang Y et al (2020) Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. https://doi.org/10.1016/j.apsb.2020.02.008
Kirchdoerfer RN, Ward AB (2019) Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat Commun. https://doi.org/10.1038/s41467-019-10280-3
Shiraki K, Daikoku T (2020) Favipiravir, an anti-influenza drug against life-threatening RNA virus infections. Pharmacol Ther 209:107512. https://doi.org/10.1016/j.pharmthera.2020.107512
Andersen PI, Krpina K, Ianevski A et al (2019) Novel antiviral activities of obatoclax, emetine, niclosamide, brequinar, and homoharringtonine. Viruses. https://doi.org/10.3390/v11100964
Hoffmann M, Kleine-Weber H, Schroeder S et al (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. https://doi.org/10.1016/j.cell.2020.02.052
Ton A-T, Gentile F, Hsing M et al (2020) Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 billion compounds. Mol Inform. https://doi.org/10.1002/minf.202000028
Zhang L, Lin D, Sun X et al (2020) Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. https://doi.org/10.1126/science.abb3405
Jin Z, Du X, Xu Y et al (2020) Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582(7811):289–293. https://doi.org/10.1038/s41586-020-2223-y
Palese LL (2020) The structural landscape of SARS-CoV-2 main protease: hints for inhibitor search 1–13. https://doi.org/10.26434/chemrxiv.12209744.v1
Coutard B, Valle C, de Lamballerie X et al (2020) The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir Res. https://doi.org/10.1016/j.antiviral.2020.104742
Tang N, Bai H, Chen X et al (2020) Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost. https://doi.org/10.1111/jth.14817
Han H, Yang L, Liu R et al (2020) Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin Chem Lab Med. https://doi.org/10.1515/cclm-2020-0188
Tang N, Li D, Wang X, Sun Z (2020) Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. https://doi.org/10.1111/jth.14768
Yin S, Huang M, Li D, Tang N (2020) Difference of coagulation features between severe pneumonia induced by SARS-CoV2 and non-SARS-CoV2. J Thromb Thrombolysis. https://doi.org/10.1007/s11239-020-02105-8
Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP (2020) The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol 20(6):363–374. https://doi.org/10.1038/s41577-020-0311-8
Mehta P, Mcauley DF, Brown M et al (2020) Correspondence COVID-19: consider cytokine storm syndromes and. Lancet 6736:19–20. https://doi.org/10.1016/S0140-6736(20)30628-0
Clerkin KJ, Fried JA, Raikhelkar J et al (2020) Coronavirus disease 2019 (COVID-19) and cardiovascular disease. Circulation. https://doi.org/10.1161/CIRCULATIONAHA.120.046941
Quagliariello V, Passariello M, Coppola C et al (2019) Cardiotoxicity and pro-inflammatory effects of the immune checkpoint inhibitor Pembrolizumab associated to Trastuzumab. Int J Cardiol 292:171–179. https://doi.org/10.1016/j.ijcard.2019.05.028
Colson P, Rolain JM, Lagier JC et al (2020) Chloroquine and hydroxychloroquine as available weapons to fight COVID-19. Int J Antimicrob Agents 55(4):105932
Sodhi M, Etminan M (2020) Safety of ibuprofen in patients with COVID-19; causal or confounded? Chest. https://doi.org/10.1016/j.chest.2020.03.040
Wrapp D, Wang N, Corbett KS et al (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. https://doi.org/10.1126/science.abb2507
Walls AC, Park Y-J, Tortorici MA et al (2020) Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. https://doi.org/10.1016/j.cell.2020.02.058
Yan R, Zhang Y, Li Y et al (2020) Structural basis for the recognition of the SARS-CoV-2 by full-length human ACE2. Science. https://doi.org/10.1126/science.abb2762
Wang Q, Zhang Y, Wu L et al (2020) Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. https://doi.org/10.1016/j.cell.2020.03.045
Lei C, Fu W, Qian K et al (2020) Potent neutralization of 2019 novel coronavirus by recombinant ACE2-Ig. bioRxiv:2020.02.01.929976. https://doi.org/10.1101/2020.02.01.929976
Wang C, Li W, Drabek D et al (2020) A human monoclonal antibody blocking SARS-CoV-2 infection. Nat Commun 11:2511. https://doi.org/10.1038/s41467-020-16256-y
Park T, Lee S-Y, Kim S et al (2020) Spike protein binding prediction with neutralizing antibodies of SARS-CoV-2. bioRxiv:2020.02.22.951178. https://doi.org/10.1101/2020.02.22.951178
Sui J, Li W, Murakami A et al (2004) Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.0307140101
Gao Q, Bao L, Mao H et al (2020) Development of an inactivated vaccine candidate for SARS-CoV-2. Science (80-). https://doi.org/10.1126/science.abc1932
Koyama T, Weeraratne D, Snowdon JL, Parida L (2020) Emergence of drift variants that may affect covid-19 vaccine development and antibody treatment. Pathogens. https://doi.org/10.3390/pathogens9050324
Tian X, Li C, Huang A et al (2020) Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg Microbes Infect 9(1):382–385. https://doi.org/10.1080/22221751.2020.1729069
Lobley A, Sadowski MI, Jones DT (2009) pGenTHREADER and pDomTHREADER: new methods for improved protein fold recognition and superfamily discrimination. Bioinformatics 25:1761–1767
Yang J, Zhang Y (2015) Protein structure and function prediction using I-TASSER. Curr Protoc Bioinform 52:5.8.1–5.8.15. https://doi.org/10.1002/0471250953.bi0508s52
Trisolini L, Gambacorta N, Gorgoglione R et al (2019) FAD/NADH dependent oxidoreductases: from different amino acid sequences to similar protein shapes for playing an ancient function. J Clin Med 8:2117. https://doi.org/10.3390/jcm8122117
Pierri CL, Parisi G, Porcelli V (2010) Computational approaches for protein function prediction: a combined strategy from multiple sequence alignment to molecular docking-based virtual screening. Biochim Biophys Acta 1804(9):1695–1712. https://doi.org/10.1016/j.bbapap.2010.04.008
Persson B (2000) Bioinformatics in protein analysis. EXS 88:215–231
Waterhouse AM, Procter JB, Martin DM et al (2009) Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191
Ordog R (2008) PyDeT, a PyMOL plug-in for visualizing geometric concepts around proteins. Bioinformation 2:346–347
Pierri CL, Bossis F, Punzi G et al (2016) Molecular modeling of antibodies for the treatment of TNFα-related immunological diseases. Pharmacol Res Perspect 4:e00197. https://doi.org/10.1002/prp2.197
Krieger E, Joo K, Lee J et al (2009) Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins 77 Suppl 9(Suppl 9):114–122. https://doi.org/10.1002/prot.22570
Sanchez R, Sali A, Sánchez R, Sali A (2000) Comparative protein structure modeling. Introduction and practical examples with modeller. Methods Mol Biol 143:97–129
Walls AC, Tortorici MA, Snijder J et al (2017) Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1708727114
Yan L, Meng B, Xiang J et al (2018) Crystal structure of the post-fusion core of the Human coronavirus 229E spike protein at 1.86 Å resolution. Acta Crystallogr Sect D Struct Biol. https://doi.org/10.1107/S2059798318008318
Duquerroy S, Vigouroux A, Rottier PJM et al (2005) Central ions and lateral asparagine/glutamine zippers stabilize the post-fusion hairpin conformation of the SARS coronavirus spike glycoprotein. Virology. https://doi.org/10.1016/j.virol.2005.02.022
Xu Y, Liu Y, Lou Z et al (2004) Structural basis for coronavirus-mediated membrane fusion: crystal structure of mouse hepatitis virus spike protein fusion core. J Biol Chem. https://doi.org/10.1074/jbc.M403760200
Song W, Gui M, Wang X, Xiang Y (2018) Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1007236
Kirchdoerfer RN, Wang N, Pallesen J et al (2018) Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Sci Rep. https://doi.org/10.1038/s41598-018-34171-7
Li F, Li W, Farzan M, Harrison SC (2005) Structural biology: structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science (80-). https://doi.org/10.1126/science.1116480
Prabakaran P, Gan J, Feng Y et al (2006) Structure of severe acute respiratory syndrome coronavirus receptor-binding domain complexed with neutralizing antibody. J Biol Chem. https://doi.org/10.1074/jbc.M600697200
Walls AC, Xiong X, Park YJ et al (2019) Unexpected receptor functional mimicry elucidates activation of coronavirus fusion. Cell. https://doi.org/10.1016/j.cell.2018.12.028
Guex N, Schwede T, Peitsch MC (2001) Protein tertiary structure modeling. Curr Protoc Protein Sci 23(1):2.8.1–2.8.17. https://doi.org/10.1002/0471140864.ps0208s23
DeLano WL (2002) The PyMOL molecular graphics system, version 1.1. Schrödinger LLC, New York. https://doi.org/10.1038/hr.2014.17
Harris LJ, Larson SB, Hasel KW, McPherson A (1997) Refined structure of an intact IgG2a monoclonal antibody. Biochemistry 36:1581–1597. https://doi.org/10.1021/bi962514+
Alford RF, Leaver-Fay A, Jeliazkov JR et al (2017) The Rosetta all-atom energy function for macromolecular modeling and design. J Chem Theory Comput. https://doi.org/10.1021/acs.jctc.7b00125
Nivón LG, Moretti R, Baker D (2013) A Pareto-optimal refinement method for protein design scaffolds. PLoS ONE. https://doi.org/10.1371/journal.pone.0059004
Conway P, Tyka MD, DiMaio F et al (2014) Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Sci. https://doi.org/10.1002/pro.2389
Van Durme J, Delgado J, Stricher F et al (2011) A graphical interface for the FoldX forcefield. Bioinformatics 27:1711–1712. https://doi.org/10.1093/bioinformatics/btr254
Schymkowitz J, Borg J, Stricher F et al (2005) The FoldX web server: an online force field. Nucleic Acids Res 33:W382–W388. https://doi.org/10.1093/nar/gki387
Benjamin Stranges P, Kuhlman B (2013) A comparison of successful and failed protein interface designs highlights the challenges of designing buried hydrogen bonds. Protein Sci. https://doi.org/10.1002/pro.2187
Bazzoli A, Vance DJ, Rudolph MJ et al (2017) Using homology modeling to interrogate binding affinity in neutralization of ricin toxin by a family of single domain antibodies. Proteins Struct Funct Bioinform. https://doi.org/10.1002/prot.25353
Lewis SM, Kuhlman BA (2011) Anchored design of protein–protein interfaces. PLoS ONE. https://doi.org/10.1371/journal.pone.0020872
Deshotels MR, Xia H, Sriramula S et al (2014) Angiotensin II mediates angiotensin converting enzyme type 2 internalization and degradation through an Angiotensin II type I receptor-dependent mechanism. Hypertension. https://doi.org/10.1161/HYPERTENSIONAHA.114.03743
Wan Y, Shang J, Graham R et al (2020) Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol. https://doi.org/10.1128/jvi.00127-20
Shang J, Ye G, Shi K et al (2020) Structural basis of receptor recognition by SARS-CoV-2. Nature. https://doi.org/10.1038/s41586-020-2179-y
Martin ACR, Thornton JM (1996) Structural families in loops of homologous proteins: automatic classification, modelling and application to antibodies. J Mol Biol. https://doi.org/10.1006/jmbi.1996.0617
Chen WH, Strych U, Hotez PJ, Bottazzi ME (2020) The SARS-CoV-2 vaccine pipeline: an overview. Curr Trop Med Rep. https://doi.org/10.1007/s40475-020-00201-6
Kim E, Erdos G, Huang S et al (2020) Microneedle array delivered recombinant coronavirus vaccines: immunogenicity and rapid translational development. EBioMedicine 55:102743. https://doi.org/10.1016/j.ebiom.2020.102743
Ahmed SF, Quadeer AA, McKay MR (2020) Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses. https://doi.org/10.3390/v12030254
Thevarajan I, Nguyen THO, Koutsakos M et al (2020) Breadth of concomitant immune responses prior to patient recovery: a case report of non-severe COVID-19. Nat Med 26(4):453–455. https://doi.org/10.1038/s41591-020-0819-2
Lurie N, Saville M, Hatchett R, Halton J (2020) Developing Covid-19 vaccines at pandemic speed. N Engl J Med 382(21):1969–1973. https://doi.org/10.1056/NEJMp2005630
Joyce MG, Sankhala RS, Chen W-H, Choe M, Bai H, Hajduczki A, Yan L, Sterling SL, Peterson CE, Green EC, Smith C, de Val N, Amare M, Scott P, Laing ED, Christopher KM (2020) A cryptic site of vulnerability on the receptor binding domain of the SARS-CoV-2 spike glycoprotein. bioRxiv. https://doi.org/10.1101/2020.03.15.992883v1
Yuan M, Wu NC, Zhu X et al (2020) A highly conserved cryptic epitope in the receptor-binding domains of SARS-CoV-2 and SARS-CoV. Science (80-) 7269:eabb7269. https://doi.org/10.1126/science.abb7269
Bhattacharya M, Sharma AR, Patra P et al (2020) Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): immunoinformatics approach. J Med Virol. https://doi.org/10.1002/jmv.25736
Amanat F, Stadlbauer D, Strohmeier S et al (2020) A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat Med. https://doi.org/10.1038/s41591-020-0913-5
Chen L, Xiong J, Bao L, Shi Y (2020) Convalescent plasma as a potential therapy for COVID-19. Lancet Infect Dis 20(4):398–400. https://doi.org/10.1016/S1473-3099(20)30141-9
Caly L, Druce JD, Catton MG et al (2020) The FDA-approved DRUG Ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir Res. https://doi.org/10.1016/j.antiviral.2020.104787
Zhou Y, Hou Y, Shen J et al (2020) Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. https://doi.org/10.1038/s41421-020-0153-3
Gordon DE, Jang GM, Bouhaddou M et al (2020) A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. https://doi.org/10.1038/s41586-020-2286-9
Romano M, Ruggiero A, Squeglia F, Maga G, Berisio R (2020) A structural view of SARS-CoV-2 RNA replication machinery: RNA synthesis, proofreading and final capping. Cells 9:1267
van der Linden L, Vives-Adrián L, Selisko B et al (2015) The RNA template channel of the RNA-dependent RNA polymerase as a target for development of antiviral therapy of multiple genera within a virus family. PLoS Pathog 11(3):e1004733. https://doi.org/10.1371/journal.ppat.1004733
Consiglio A, Grillo G, Licciulli F et al (2011) PlantPIs–an interactive web resource on plant protease inhibitors. Curr Protein Pept Sci 12(5):448–454. https://doi.org/10.2174/138920311796391052
Savarino A (2005) Expanding the frontiers of existing antiviral drugs: possible effects of HIV-1 protease inhibitors against SARS and avian influenza. J Clin Virol 34(3):170–178. https://doi.org/10.1016/j.jcv.2005.03.005
Bendavid E, Mulaney B, Sood N et al (2020) COVID-19 antibody seroprevalence in Santa Clara County, California. medRxiv:2020.04.14.20062463. https://doi.org/10.1101/2020.04.14.20062463
Sethuraman N, Jeremiah SS, Ryo A (2020) Interpreting diagnostic tests for SARS-CoV-2. JAMA. https://doi.org/10.1001/jama.2020.8259
Bryan A, Pepper G, Wener MH et al (2020) Performance characteristics of the Abbott Architect SARS-CoV-2 IgG assay and seroprevalence in Boise, Idaho. J Clin Microbiol. https://doi.org/10.1128/JCM.00941-20
Zhang H, Penninger JM, Li Y et al (2020) Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. https://doi.org/10.1007/s00134-020-05985-9
Batlle D, Wysocki J, Satchell K (2020) Soluble angiotensin-converting enzyme 2: a potential approach for coronavirus infection therapy? Clin Sci (Lond). https://doi.org/10.1042/CS20200163
Zheng YY, Ma YT, Zhang JY, Xie X (2020) COVID-19 and the cardiovascular system. Nat Rev Cardiol 17(5):259–260. https://doi.org/10.1038/s41569-020-0360-5
Kuster GM, Pfister O, Burkard T et al (2020) SARS-CoV2: should inhibitors of the renin–angiotensin system be withdrawn in patients with COVID-19? Eur Heart J. https://doi.org/10.1093/eurheartj/ehaa235
Towler P, Staker B, Prasad SG et al (2004) ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J Biol Chem. https://doi.org/10.1074/jbc.M311191200
Natesh R, Schwager SLU, Sturrock ED, Acharya KR (2003) Crystal structure of the human angiotensin-converting enzyme–lisinopril complex. Nature. https://doi.org/10.1038/nature01370
Natesh R, Schwager SLU, Evans HR et al (2004) Structural details on the binding of antihypertensive drugs captopril and enalaprilat to human testicular angiotensin I-converting enzyme. Biochemistry. https://doi.org/10.1021/bi049480n
Yates CJ, Masuyer G, Schwager SLU et al (2014) Molecular and thermodynamic mechanisms of the chloride-dependent human angiotensin-I-converting enzyme (ACE). J Biol Chem. https://doi.org/10.1074/jbc.M113.512335
Ferrario CM, Jessup J, Chappell MC et al (2005) Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. https://doi.org/10.1161/CIRCULATIONAHA.104.510461
Alifano M, Alifano P, Forgez P, Iannelli A (2020) Renin-angiotensin system at the heart of COVID-19 pandemic. Biochimie. https://doi.org/10.1016/j.biochi.2020.04.008
Zhang P, Zhu L, Cai J et al (2020) Association of inpatient use of angiotensin converting enzyme inhibitors and angiotensin II receptor blockers with mortality among patients with hypertension hospitalized with COVID-19. Circ Res. https://doi.org/10.1161/circresaha.120.317134
Xia S, Yan L, Xu W et al (2019) A pan-coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Sci Adv. https://doi.org/10.1126/sciadv.aav4580
Law GL, Tisoncik-Go J, Korth MJ, Katze MG (2013) Drug repurposing: a better approach for infectious disease drug discovery? Curr Opin Immunol 25(5):588–592. https://doi.org/10.1016/j.coi.2013.08.004
Cheng F, Desai RJ, Handy DE et al (2018) Network-based approach to prediction and population-based validation of in silico drug repurposing. Nat Commun. https://doi.org/10.1038/s41467-018-05116-5
Saphire EO, Parren PWHI, Pantophlet R et al (2001) Crystal structure of a neutralizing human IgG against HIV-1: a template for vaccine design. Science (80-) 293:1155–1159. https://doi.org/10.1126/science.1061692
Furtado PB, Whitty PW, Robertson A et al (2004) Solution structure determination of monomeric human IgA2 by X-ray and neutron scattering, analytical ultracentrifugation and constrained modelling: a comparison with monomeric human IgA1. J Mol Biol. https://doi.org/10.1016/j.jmb.2004.03.007
Bonner A, Almogren A, Furtado PB et al (2009) The nonplanar secretory IgA2 and near planar secretory IgA1 solution structures rationalize their different mucosal immune responses. J Biol Chem. https://doi.org/10.1074/jbc.M807529200
Kumar N, Arthur CP, Ciferri C, Matsumoto ML (2020) Structure of the secretory immunoglobulin A core. Science (80-). https://doi.org/10.1126/science.aaz5807
Zeitlin L, Cone RA, Whaley KJ (1999) Using monoclonal antibodies to prevent mucosal transmission of epidemic infectious diseases. Emerg Infect Dis 5(1):54–64. https://doi.org/10.3201/eid0501.990107
Cerutti A, Chen K, Chorny A (2011) Immunoglobulin responses at the mucosal interface. Annu Rev Immunol. https://doi.org/10.1146/annurev-immunol-031210-101317
Chiara M, Horner DS, Pesole G (2020) Comparative genomics suggests limited variability and similar evolutionary patterns between major clades of SARS-Cov-2. bioRxiv:2020.03.30.016790. https://doi.org/10.1101/2020.03.30.016790
Acknowledgements
Authors would like to thank the Italian Association for Mitochondrial Research (http://www.mitoairm.it), IT resources made available by ReCaS, a project funded by the MIUR (Italian Ministry for Education, University and Research) in the “PON Ricerca e Competitività 2007–2013-Azione I-Interventi di rafforzamento strutturale” PONa3_00052, Avviso 254/Ric, University of Bari (“Fondi Ateneo ex-60%”2016”; “ProgettoCompetitivo 2018” and “FFABR 2017–2018”). Authors would like to thank MIUR for having funded the project “Salute, alimentazione, qualità della vita”: individuazione di un set di biomarker dell’apoptosi” for an innovative industrial Ph.D. course—PON RI 2014–2020, CUP H92H18000160006. Authors would also like to thank Angelo Onofrio (Biotechnologist), Luna Laera (Biotechnologist), and Daniela Tortorella (Biotechnologist) for critical reading and/or stimulating discussions.
About technical questions.
Detailed instructions for the set-up of the shown molecular framework have been provided in the manuscript. Nevertheless, we can also provide free assistance for academic analyses, upon request. We can also provide dedicated technical support for analyses requested by private companies through our BROWSer s.r.l. spin-off (in this case, please, write to info@browser-bioinf.com and CLP in Cc).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
18_2020_3580_MOESM8_ESM.tif
Supplementary Figure 1 MSA of the antigen-binding fragment (Fab) of m396 ab (native and modified) with 1IGT.pdb sequence. (A) Pairwise alignment of the light chains from 1IGT (murine IgG) and m396 antibody (native and modified). (B) Pairwise alignment of the heavy chains of the Fab portions from 1IGT and m396 antibody (native and modified. (TIFF 5482 kb)
18_2020_3580_MOESM9_ESM.jpg
Supplementary Figure 2 The overall structure of the 1IGT antibody used as a protein template for building our chimeric mAb models based on m396 ab. The heavy chains are reported in orange cartoons, and the light chains are reported in blue cartoons, for the native m396 (panel a) and for the modified m396 (panel b). SARS-CoV-2 spike proteins are reported in cyan/magenta/green cartoon representation with the exclusion of RBD reported in black cartoon representation. Breaks in the tertiary structures of the antibody backbones (portion in orange cartoon representation), at the interface of the FAB/Fc portion, indicate sites hosting residues that were ligated after superimposition of the different portions (1igt.pdb and native/mutated m396) for obtaining the final complete 3D model of the proposed antibodies. (JPEG 6731 kb)
Rights and permissions
About this article

{kind=link}
Cite this article
Mercurio, I., Tragni, V., Busto, F. et al. Protein structure analysis of the interactions between SARS-CoV-2 spike protein and the human ACE2 receptor: from conformational changes to novel neutralizing antibodies. Cell. Mol. Life Sci. 78, 1501–1522 (2021). https://doi.org/10.1007/s00018-020-03580-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-020-03580-1