
Abstract
Understanding the mechanisms that underlie age-related macular degeneration (AMD) has led to the identification of key molecules. Hypoxia-inducible transcription factors (HIFs) have been associated with choroidal neovascularization and the progression of AMD into the neovascular clinical phenotype (nAMD). HIFs regulate the expression of multiple growth factors and cytokines involved in angiogenesis and inflammation, hallmarks of nAMD. This knowledge has propelled the development of a new group of therapeutic strategies focused on gene therapy. The present review provides an update on current gene therapies in ocular angiogenesis, particularly nAMD, from both basic and clinical perspectives.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Age-related macular degeneration (AMD) is the most common cause of visual loss in developed countries, with particular incidence in the geriatric population [1]. AMD is the third global cause of blindness (8.7% of patients’ blindness), preceded by cataract and glaucoma [2]. By the year 2020, AMD patients are predicted at 196 million, rising to 288 million in 2040, in association with the global aging of human population [3]. Patients with AMD suffer from impaired fine and color vision, with particular clinical relevance when the fovea (center of the macula, containing the highest density of cone photoreceptors) is affected and the central field of vision is compromised [4, 5].
Cellular events in AMD involve disruption of photoreceptors, retinal pigment epithelial (RPE) cells, Bruch’s membrane (BM), and the choroid. The observed cellular events are a result of destabilized homeostasis of reactive oxygen species (ROS) response [6], phagocytosis [7, 8], extracellular matrix remodeling [9], and alternative complement-related inflammation [10].
Etiologically, AMD is a multifactorial disease. Genetic variants associated with AMD include complement factor (CF)H [11] and CFH-related genes 1 to 5 [12], complement protein (C)3 [13], C9 [14], age-related maculopathy susceptibility (ARMS)2 gene [15], and the vascular endothelial growth factor (VEGF) and VEGF receptor (VEGFR) axis [16, 17]. In addition, other genetic variants have shown a causal link to AMD, such as tissue inhibitor metalloproteinase (TIMP) 3 [18], fibrillin [19], collagen 4A3, and metalloproteinase (MMP) 19 and − 9 [20]. Moreover, environmental factors, e.g., advanced age, female gender, white race, smoking, increased body mass index, hypertension, and hyperopia, have been suggested to predispose to AMD [21]. In addition, certain biomarkers as carboxyethylpyrrole [22] and homocysteine [23, 24] have been shown to correlate with AMD. Despite this knowledge, AMD etiology is still evasive, urging a need for further research to deepen the understanding of AMD initiation and progression.
Here, the role of hypoxia and the hypoxia-inducible factors (HIFs) are revised in a perspective of AMD initiation and progression, as well as putative therapeutic targets, as they are the predominant molecular pathways of neovascular AMD (nAMD) [25,26,27,28,29].
Types of AMD
In general terms, AMD is designated according to disease progression and symptoms into early, intermediate, and advanced AMD [30]. Advanced cases (3% of the population [31]) are categorized into two types: “dry” or geographic atrophic form, and “wet” or neovascular form [30]. Atrophic AMD (aAMD) is more common but spanning only 20% of greater central visual loss. nAMD comprehends fewer cases, but has been related with 80% of greater central vision loss [32,33,34]. Albeit not a clinical constant, some cases of aAMD progress into nAMD, illustrating a possible clinical evolution or a certain redundancy between the two advanced types of AMD [4, 35, 36].
Drusen maculopathy
A common characteristic of AMD is pigment alteration in the RPE and hard or soft extracellular deposits between the RPE and the BM, referred to as drusen deposits [5, 37,38,39]. Soft drusen have been associated with higher risk of developing advanced AMD [40] and involved in the progression into geographic atrophy and causally associated with the initiation of choroidal neovascularization (CNV). CNV results in the formation of immature and fragile blood vessels that cause exudates into the subretinal spaces [41]. In rare cases, large soft drusen assemble and form pigment epithelial detachments (PEDs), which lead to an elevation of the RPE under the retina and impair the oxygenation of RPE cells, physiologically dependent on the intricate contact with the choroidal vascular bed [42].
The Beaver Dam Eye Study estimates that the prevalence of drusen is 2% in persons 43–54 years of age and 24% in persons over 75 years of age [43, 44]. Drusen are accepted as one of the causes of AMD, but the molecular pathway of drusen formation is still elusive [5, 37,38,39].
Drusen deposits are understood as the result of an incomplete processing of photoreceptor outer segments (POS) by the RPE [45], which cannot transmigrate through the BM for removal by the choriocapillaries. Such deposits create a barrier that decreases the diffusion of oxygen and nutrients from the choroid to the photoreceptors and the RPE in one direction, and decreases the removal of debris by the choriocapillaries into the other direction [4, 5, 37, 46]. Drusen contain lipids [47, 48], glycoconjugates [49, 50], glycosaminoglycan [51], and pigments, such as lipofuscin [52]. Concomitantly, the presence of advanced glycation end-products in drusen and on the BM, a product of aging, seems to contribute to AMD pathogenesis [53]. Finally, when cholesterol accumulates, soft drusen are formed [54, 55] and a higher risk association for advanced AMD is denoted. Drusen proteomic analysis has demonstrated the presence of multiple proteins, among which TIMP3 [18], clusterin, vitronectin, and serum albumin were the most common proteins in normal donor drusen, whereas crystallins were more frequently present in AMD donor drusen [38, 56, 57]. Drusen inflammatory and complement proteins, such as beta amyloid [58], immunoglobulin light chains, factor X, and C5 and − 5b, have received special attention [59], specifically since CFH [11] and -B [60] gene mutations have been associated with AMD [61].
Lipofuscin [52], an aging pigment containing the autofluorescent oxidatively modified lipid N-retinylidene-N-retinylethanolamine (A2E) [62, 63] together with minimal amounts of oxidatively modified proteins [64], is the product of impaired phagocytosis of the POS due to decreased RPE lysosomal activity [65]. Physiologically, lipofuscin is degraded completely or exocytosed by the RPE cells basolaterally and removed by the choroidal blood stream [66]. In AMD, lipofuscin accumulates intracellularly in the RPE and on the BM and is highly present in drusen deposits [67]. In addition, age-dependent reduction in enzyme and mitochondria functions results in ROS accumulation, which contributes to the pathogenesis of AMD [6, 8].
In sum, drusen maculopathy results from the combination of accumulated lipofuscin, ROS, and complement alternative-related inflammation, and the decreased extracellular matrix remodeling and phagocytosis. These culminate in the thickening of Bruch’s membrane, creating a barrier that reduces the diffusion of oxygen and nutrients from the choroid to the photoreceptors and the RPE, and decreases the removal of debris by the choriocapillaries [4, 5, 37, 46]. Ultimately, macular drusenopathy leads to a status of relative hypoxia within the subretinal compartments and contributes to the pathogenesis of AMD (Fig. 1) [25, 68, 69]. In response, the RPE is triggered to secrete proangiogenic factors, such as VEGF into the choroidal space, in an attempt to minimize hypoxia-induced events. Hypoxia stimulates secretion of not only VEGF but several vasculogenic and inflammatory cytokines by the RPE, thus contributing to the development of CNV [70,71,72,73].

Schematic representation of nAMD pathogenesis. a In physiologic conditions, a flow of oxygen and nutrients raises from the choroid to the outer retina. The RPE produces physiologic levels of VEGF to sustain the proximity of choroidal vessels and maintenance of a normoxic status. b In early AMD, the disruption of several cellular mechanisms damages RPE cells and results in the formation of sub-RPE drusen deposits and thickening of Bruch’s membrane. These events create a relative hypoxic condition in the outer retina, due to decreased oxygen and nutrients flow from the distanced choroid. In response, RPE cells upregulate production of VEGF, which leads to choroidal neovascularization and ultimately a dystrophic RPE layer
Treatment of AMD
To date, there is neither an effective preventive treatment [74,75,76] nor a cure for AMD. Yet if untreated, AMD will evolve to a severe and irreversible disciform scar, resulting in extremely compromised vision or even blindness [4, 5].
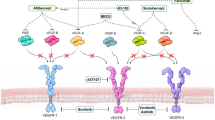
Nowadays, nAMD is treated by routine intraocular injections of anti-VEGF agents [77,78,79,80,81,82,83], and to some extent by photodynamic therapy and thermal laser [84, 85]. The use of intravitreal anti-VEGF agents has revolutionized the treatment of nAMD with pegaptanib being the first approved anti-VEGF agent. Since pegaptanib binds to a single isoform of VEGF, it showed less efficacy. Later bevacizumab and ranibizumab have been introduced as anti-VEGF agents in the treatment of nAMD. Both these molecules bind all isoforms of VEGF. In comparison with bevacizumab, ranibizumab immunogenicity demonstrates 5- to 20-fold greater potency due to higher affinity to VEGF molecules. The latest approved drug in the treatment of nAMD, aflibercept, consists of the VEGF-binding domains of human VEGFR1 and VEGFR2 fused to the Fc domain of human immunoglobulin-G1. Aflibercept acts as a decoy receptor binding not only VEGF but also placental growth factor (PLGF) [86,87,88].
In regard to aAMD, only antioxidant therapy in multiple forms is palliatively applied, which delays progression in 20–25% of eyes [75, 76, 89].
The Hypoxia-inducible Factors
HIFs, the main regulators of hypoxia, are members of the basic helix-loop-helix Per-Arnt-Sim (bHLH-PAS) family. HIFs are composed of α and β subunits, where the latter is the constitutively expressed hydrocarbon receptor nuclear translocator (ARNT). Three isoforms have been identified for α subunits: HIF-1α, -2α and -3α [26, 27, 90, 91] with HIF-1α and HIF-2α being more prone for active responses to hypoxia. HIF-1α is widely expressed in all human normoxic and hypoxic tissues [92], while HIF-2α expression has been associated with physiologically hypoxic tissues [93] and spans 48% structure homology to HIF-1α [94]. The less characterized HIF-3α is expressed in adult thymus, lung, brain, heart, and kidney [95], and different splice variants of HIF-3α have opposite functions varying from activation to repression of hypoxia-inducible genes [91, 96].
Structurally, HIF-α subunits span at the N-terminus a basic helix-loop-helix (bHLH) and a Period-ARNT-Sim (PAS) domains responsible for DNA binding and dimerization to ARNT [97]. HIF-α transcription factors at the C-terminus contain two transactivation domains (TAD) which recruit coactivators [98, 99], and an inhibitory domain (ID) between the two TADs [100]. Oxygen-dependent protein regulation has been mapped to an internal oxygen-dependent degradation domain (ODD) [101]. In addition, two nuclear localization signals (C-terminal NLS and N-terminal NLS) have been identified, determining a near exclusive nuclear subcellular localization of HIF-α subunits [99, 102].
Regulation of HIF expression
Interestingly, HIF-α subunits are constitutively expressed, yet protein levels and transcription activity are regulated by oxygen-dependent posttranslational modification (PTM), followed by epigenetic regulation of HIF-mediated transcription [103].
In normal oxygen conditions (Fig. 2), HIF-α is hydroxylated in two conserved prolines by a family of three oxygen-dependent orthologues of prolyl hydroxylase domain (PHD1-3) [104, 105]. Subsequent to hydroxylation of prolines 402 and 564 of HIF-1α (P405 and P531 in HIF-2α), the von Hippel–Lindau (pVHL), acting as the substrate-recognizing component of an E3 ubiquitin ligase complex, recognizes and polyubiquitinates HIF-α, resulting in 26S proteasome-mediated degradation of HIF-α proteins [106,107,108,109]. An additional oxygen-dependent asparaginyl hydroxylase factor inhibiting HIF-1α (FIH-1) regulates HIF transcriptional activation in normoxia. Hydroxylation of asparagine 803 of HIF-1α C-terminal TAD (N851 of HIF-2α) inhibits the binding of coactivators (CBP/p300) and renders HIF-α subunits transcriptionally inactive in normoxia [98, 99, 110, 111].

Regulatory mechanisms of HIF. a In normoxia, two prolines residues within the ODD of HIF-α are hydroxylated. Besides, a lysine acetylation by ARD1 ensues. These molecular reactions allow recognition of HIF-α by VHL ubiquitin ligase, which results in polyubiquitination of HIF-α and proteasome-mediated degradation. Simultaneously, an asparagine hydroxylation by FIH-1 blocks the interaction with CPB/p300 coactivators and critically impairs HIF-α transactivation. b In hypoxic conditions, PHD1-3, FIH-1 and ARD1 are rendered non-functional. HIF-α translocates to the nucleus and heterodimerizes with ARNT, forming the transcriptionally active HIF. The dimer is able to bind to HRE and initiate transcription of hypoxia-inducible genes. Moreover, MAPK phosphorylation of HIF-α enhances HIF transcriptional activity and recruitment of CPB/p300 coactivators
In addition, non-oxygen-dependent regulation of HIF-α has been attributed to lysine acetylation within the ODD by the ADP-ribosylation factor domain protein 1 (ARD1). Acetylation of lysine 532 promotes interaction between VHL and HIF-1α and stimulates HIF protein degradation. Despite oxygen-independent, PTM of HIF-α by acetylation is regulated by hypoxia-induced downregulation of ARD1 expression [112, 113].
In hypoxic conditions (Fig. 2), molecular oxygen is limited, the oxygen-dependent PHDs activity is compromised, and HIF remains unhydroxylated bypassing ubiquitin–proteasome degradation. Once stabilized and translocated to the nucleus, HIF-α heterodimerizes with ARNT, and the dimer binds to hypoxia-response elements (HRE; 5′RA/GCGTG3′) [114, 115], thus mediating upregulation of hypoxic genes [116]. Finally, phosphorylation of HIF-1α by p42/44 [117] and p38 [118] mitogen-activated protein kinases (MAPK) enhances heterodimerization and transcriptional activity of HIF, leading to the upregulation of hypoxic genes.
Canonical HIF pathway
In hypoxia, the nuclear and transcriptionally active HIF heterodimer binds HRE, and the hypoxia-inducible genes are expressed [26, 119, 120]. Classically, the hypoxia-inducible genes are principally involved in cell differentiation, proliferation, and survival or apoptosis, cellular energy and metabolism, extracellular matrix (ECM) degradation and chemotaxis, and angiogenesis. HIF-mediated genes are involved also in a myriad of pathologies from stroke and ischemia, neovascular diseases, and several tumors and metastatic disease [26, 120].
In regard to tumor metastasis, HIFs are directly enrolled in tumor cellular growth by upregulating survival and proliferation genes, such as twist-related protein 1 (TWIST1) [121], integrins αvβ3, αvβ5 and αvβ6 [122], and several cadherins [123]. Concomitantly, HIF genes regulate ECM degradation and chemotaxis, modulating tumor metastasis directly. Matrixmetalloproteinases such as MMP2 [124] and MMP9 [125] together with urokinase-like plasminogen activator and its receptor (uPA; uPAR) and plasminogen activator inhibitor (PAI)-1 [126,127,128] are upregulated by HIFs in metastatic tumors. Moreover, the HIF-driven genes C–X–C motif chemokine receptor 4 (CXCR4) [129, 130] and C–C motif chemokine receptor 7 (CCR7) [131] contribute to metastasis guidance and dissemination of certain cancers.
Angiogenesis is regulated by HIFs, with VEGF and VEGFR major family as the principal intermediary [132,133,134]. In addition, calcitonin receptor-like receptor (CRLR) [135], stem cell factor (SCF) [136], and angiopoietin 2 (ANGPT2) [137] are overexpressed by HIFs and contribute to VEGF-independent angiogenesis.
Regarding cellular energy and metabolism, HIFs directly regulate the glycolytic shift from Krebs cycle to anaerobic pathways in hypoxia by upregulation of glucose transporters GLUT1 and GLUT3 [138, 139], glycolytic enzymes 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) [140], phosphoglycerate kinase 1 (PGK1) [141, 142], pyruvate kinase M2 (PKM2) [143], pyruvate dehydrogenase kinase 1 (PDK1) [144], lactate dehydrogenase A (LDHA) [114], c-Myc [145], and monocarboxylate transporter 4 (MCT4) [146].
On cellular differentiation processes, HIFs maintain cells undifferentiated by promoting the expression of octamer-binding transcription factor 4 (OCT-4), and stage-specific embryonic antigen-1 (SSEA-1) and SSEA-4 [147], therefore contributing the stemness of specific progenitor niches in adult tissues. In addition, HIFs contribute to de-differentiation of cancer cells by modulating the Notch-dependent epithelial-mesenchymal transition signal involved in the maintenance of undifferentiated cells [148, 149]. On the other hand, HIFs through erythropoietin and its receptor (EPO; EPOR) [150, 151] produce proliferation and differentiation of hematopoietic stem cells into red blood cells. In addition, HIF-1α upregulates the transcription factor SOX-9 expression, which participates in chondrogenesis [152].
Finally, HIFs have been reported to induce apoptosis via the p53 tumor-suppressor gene [153] and the B-cell lymphoma (Bcl-2) gene family—BNip3 [154] and Noxa [155]. On the other hand, HIF can contribute to prevent apoptosis by the upregulation of nucleophosmin (NPM) [156] and human urocortin 2 (hUcn2) [157].
The role of HIF pathway in nAMD
Some tissues in the eye, including the cornea and the macula, are physiologically avascular and prone to hypoxia. In the eye, HIFs play specific physiologic roles in contributing to the avascularity: in the cornea, a splice variant of HIF-3α is responsible for blocking hypoxia-inducible neoangiogenesis during sleep [158], while in the foveal pit, HIFs contribute to the expression of VEGFR2 to sequester VEGF and impair vascularization into the macula [159]. In the particular case of the RPE layer, HIFs have been suggested to contribute to the apical secretion of pigment epithelium-derived factor (PEDF; antiangiogenic factor) and baso-lateral VEGF (angiogenic factor), thus simultaneously ensuring avascularity of the POS layer and vascular proximity from the choriocapillaries [27].
With regard to nAMD, genes encoding the canonical angiogenic factors VEGF [16, 132,133,134, 160, 161] and VEGFR [162, 163] have been widely associated with the development and progression of the disease. In addition, multiple molecular effectors have been associated with progression of nAMD [26] by contributing to: endothelial cell proliferation and recruitment of inflammatory cells—placental growth factor (PLGF) [164] and its receptor VEGFR1 [162], platelet-derived growth factor B (PDGF-B) [165] and its receptor PDGFRB [166], ANGPT-1, ANGPT-2 and their receptor Tie2 [161, 167], angiopoietin-like 4 (ANGPTL4) [168], stromal-derived growth factor-1 (SDF-1) [169] and its receptor CXCR4 [130]; ECM degradation—PAI-1 [126], and MMP2 and MMP9 [125]; vascular permeability and vasodilation—VEGF (particularly VEGF-A), VEGFR1, VEGFR2 [162, 163]; endothelial sprouting—ANGPT-2 [170]; and fibroplasia—transforming growth factor beta (TGF-β) [171].
Interestingly, all of the aforementioned factors, which directly contribute to nAMD, are HIF target genes [26, 27, 29, 69, 172, 173]. Drusen maculopathy and the age-related thickening of the BM together contribute to the increased hypoxic status of the RPE and thus lead to increased levels of HIF transcription factors. The expression of HIFs has been detected in choroidal neovascular membranes from clinical samples of nAMD patients [28, 29] and further corroborated to contribute to nAMD in mouse models of CNV [174, 175]. Moreover, HIFs also increase RPE apoptosis and autophagy, similarly to the choriocapillaries, which stimulates the endothelial proliferation [176,177,178] observed in nAMD.
Addressing unmet needs in nAMD treatment
Nowadays, nAMD practitioners widely recur to anti-VEGF agents [79,80,81,82] for treatment, albeit with limitations or side effects: high probability of ocular infection, increased ocular pressure, cataract, vitreous hemorrhaging and retinal detachment; elevated price; transient (demands repeated intravitreal injections); limited recovery of patients’ visual acuity in a long period [179,180,181,182]. Some of these issues evolve from the use of mono-factor therapies, such as anti-VEGF drugs, to address a multifactorial, multi-genetic disease, as is the case of nAMD. In order to improve effectiveness, combination of treatment with other proangiogenic inhibitors as blocking PDGF ([183, 184], clinicaltrials.gov/NCT01940900), steroids, e.g., triamcinolone [185], or AINEs, such as ketorolac [186], and integrin antagonists [187] has been suggested as adjuvants to anti-VEGF regiments. Collectively, these novel therapeutic approaches indicate the need to simultaneously address a multitude of nAMD-associated factors. Considering its pivotal role in nAMD and master regulators of many factors associated with the disease, therapeutic targeting of HIFs reveals an attractive possibility.
Pharmacological inhibition of HIF in nAMD
With HIFs as a central pathway in the pathogenesis of AMD [188,189,190,191], pharmacological inhibition of HIF would delimitate the production and secretion of many angiogenic factors and cytokines, in contrast to anti-VEGF drugs.
The inhibition of HIFs can be achieved indirectly by impairing pathways that mediate hypoxic responses, such as phosphoinositide 3-kinases/mammalian target of rapamycin (PI3K/mTOR) [192], topoisomerase I [193]/II [194], microtubules [195], Hsp90 [196], farnesyltransferase [197, 198], histone deacetylase [199], or thioredoxin 1 (TRX1) [200]. In animal models of ocular neovascular diseases, pharmacological inhibition of hypoxia-related pathways has displayed some success. Some of these drugs include cardiac glycosides as digoxin (inhibits p53 protein synthesis and target nuclear factor (NF)-κB; nuclear competitors of HIF) [201], anthracyclines as doxorubicin and daunorubicin (inhibits topoisomerase II, hence blocking the binding of HIF to DNA) [202], YC-1 (inactivates the upregulated PI3K/mTOR pathway, thus inducing degradation of HIF-1α) [203], and honokiol—which inactivates multiple pathways including NF-κB, mTOR, epidermal growth factor receptor (EGFR), signal transducer and activator of transcription 3 (STAT3), and caspase-mediated pathways, while also blocking the binding of HIFs to VEGF promoter and simultaneously decreasing VEGF secretion in RPE cells by HIF-independent effects on EGFR and STAT3 [204]. Expectedly, many of the indirect HIF inhibitors have shown considerable side effects both in vitro and in vivo studies, which implies a preferable direct inhibition approach. A multi-kinase inhibitor of uPAR co-receptor dimerization, UPARANT, has been demonstrated to indirectly inhibit HIF [205,206,207,208] with promising effects on systemic administration and unobserved side effects in animal models of ocular diseases, including nAMD [206].
Direct inhibition can affect different stages of HIFs, by reducing protein levels, interfering with dimerization, or binding to HREs. The first specific direct HIF inhibitor, echinomycin, has been reported to interfere with the HIF transcription factor binding to DNA [209], although reports of the effects of echinomycin on ocular cells or models are currently missing. Alternative direct inhibition of HIF has been achieved by overexpressing specific HIF modulators, such as PHD2 [210] or microRNAs [211]. Due to the intracellular nature, HIF modulator molecules must undergo gene therapy strategies.
Gene therapy for the treatment of nAMD
Nowadays, a few inherited mono-genetic diseases can be treated by gene replacement therapies. Interestingly, gene therapy for a rare form of Leber congenital amaurosis (LCA) by the replacement of Rpe65 gene—Luxturna—has been approved by the FDA [212].
The eye has been considered as an ideal organ for gene therapy-based transduction: it is compartmentalized and on the fringes of the immune system actions; the adult eye cell proliferation is limited and thus non-integrating vectors are feasible; easy access for direct treatment and evaluating effects; and need for small quantities of drug which correlate to neglectable-to-none systematic side effects. For AMD, gene therapy should be delivered in the macular subretinal space since RPE cells and photoreceptors are the principal targets of treatment [213]. Subretinal injections are becoming more standard with the currently approved ocular gene therapy treatment.
Generally, gene transfer can be achieved by viral and non-viral vectors [214], and the target gene can be replaced or inactivated. Viral vectors are more sustainable and effective than non-viral vectors, and thus preferable. Commonly used viral vectors in gene therapy models include recombinant adeno-associated virus (rAAV) vectors, adenovirus (Ad), and integrating-deficient lentivirus (IDLV) [215]. rAAV vectors have been the most effective for retinal gene therapy due to sustainable transduction of photoreceptors and RPE. Nonetheless, due to their small size, DNA capacity is limited to genes smaller than 4.7 kb [216]. In addition, rAAV vectors are non-integrating to the mammalian genome, thus displaying good safety profile as it is reported in multiple animal models, including large-eyed animals, such as dogs and primates [217]. It is noteworthy that minimal immune response to the rAAV vectors can occur [218, 219], although the use of specific viral serotypes, such as rAAV2, has demonstrated good tolerability in patients [220].
Ongoing clinical trials for gene therapies for the treatment of nAMD exploit protein-based and RNA interference (RNAi) antiangiogenic strategies. Considerable effort has been applied on protein-based gene therapy trial for nAMD, where the transduced cells overexpress angiostatic proteins in order to arrest CNV. As an example, subretinal rAAV-mediated gene transfer of the VEGF inhibitor—sVEGFR1—decreases choroidal vascularization in animal models by sequestration of VEGF and forming inactive heterodimers incapable of activating the VEGF receptors [221]. AAV vectors with other angiostatic factors, such as PEDF [222] and angiostatin [223], also have successfully arrested CNV in animal models. At present, clinical trials include Ad transducing PEDF protein [224], rAAV2 transducing sVEGFR1 (clinicaltrials.gov NCT01494805, NCT01024998, [221]), and the first lentiviral vector clinical trial, RetinoStat (clinicaltrials.gov NCT01301443, [225]), transducing two antiangiogenic genes: endostatin and angiostatin.
Anti-HIF gene therapy in nAMD
Specifically targeting HIF transcription factors have been an attractive strategy for the treatment of the multifactorial nAMD [226,227,228]. Currently, only animal models of CNV-associated with nAMD have been addressed with anti-HIF gene therapies. Assessing HIFs directly appears to mitigate angiogenesis and inflammatory responses, both associated with nAMD disease initiation and progression.
Inhibition of HIFs by gene therapy constructs has most commonly been achieved by anti-HIF microRNAs (miRNA). The expression of miRNA-20b modulates VEGF by targeting HIF-1α and STAT3 in MCF-7 breast cancer cells [229, 230] and has been suggested as a putative candidate for nAMD gene therapy [26, 27]. Nevertheless, miRNAs have been suggested to display considerable unspecificity by targeting multiple pathways; therefore, the use of HIF-specific RNAi has been reported beneficial in AMD models [211].
Use of protein-based gene therapy strategies for the treatment of CNV-associated with nAMD has been less explored, partly due to the lack of RPE- and HIF-specific modulators of the hypoxia pathway. Gene transfer of PHD2 in vivo resulted in the mitigation of HIF-mediated angiogenesis in a mouse model of nAMD [210] by reducing several nAMD-associated angiogenic factors and cytokines. PHD2 has been suggested as the ideal candidate for targeting HIF in RPE cells and has considerable HIF-selectivity in hypoxia [231] rendering it a putative candidate for anti-HIF gene therapy treatments of nAMD.
Future perspectives
Treatment of multifactorial diseases, such as nAMD, presents an immense clinical challenge. Despite prevalent use and significant success, current treatments for patients afflicted with nAMD by the administration of anti-VEGF drugs are far from optimal. Treatments must be administered on routine basis with substantial cost for health care systems and associated risks for the patients. Development of sustainable treatments could considerably improve nAMD therapies.
Gene therapy presents a possibility of sustainability when compared to pharmacological or surgical treatments, since the treatment is localized to the target cells, sustained with one dose, and regulated through gene-construct engineering. The regulation of gene expression is desirable to minimize side effects [226, 232]. The use of tissue- and cell-specific promoters has increased targeted expression of therapeutic genes in gene therapy. In nAMD, the involvement of RPE cells is pivotal, and RPE-specific promotors, such as pRpe65 (encoding RPE65 protein) and pVMD2 (promoter for the vitelliform macular dystrophy 2, encoding bestrophin-1 protein), grant expression of therapeutic genes specifically in RPE cells. Albeit non-integrative viral vectors are used for gene therapy in ophthalmology, the post-mitotic character of adult RPE and photoreceptors results in long-term expression of transduced genes. Despite the attractiveness of sustainable treatments for nAMD, in opposition to the need for routine injections currently applied with anti-VEGF drugs, the risk of long-term secondary events of the therapeutic gene should be considered. Engineering of inducible cell-specific promoters to the administration of exogenous genes for the treatment of nAMD has been proposed [233]. Combining tissue-specific promoters (pRpe65) with HRE regulatory elements, hypoxia-mediated spatial and temporal regulation of angiostatic proteins can be achieved to mitigate CNV in mouse models of nAMD [234].
The subretinal injection routinely used to deliver gene therapy to RPE cells requires a vitrectomy to hamper retinal detachments, which could result in surgical complications. Present intravitreal transduction of gene therapy vectors most commonly is limited to the inner layers of the retina [235, 236], yet modified adeno-associated virus (AAV) capsids have been reported to transduce both photoreceptor and RPE cells in non-human primates [237]. Development of novel mutant virus packaging capsids of different serotype will certainly improve gene expression kinetics and cellular tropism for the future of gene therapies [238,239,240].
Conclusion
Anti-HIF therapies have demonstrated considerable improvement in models of nAMD when compared to anti-VEGF drugs. Targeting a transcription factor can present its own challenges, yet advances on gene therapy strategies have paved bright conceptual avenues for future anti-HIF sustainable and long-term treatments for nAMD patients.
References
Leibowitz HM, Krueger DE, Maunder LR et al (1980) The Framingham Eye Study monograph: an ophthalmological and epidemiological study of cataract, glaucoma, diabetic retinopathy, macular degeneration, and visual acuity in a general population of 2631 adults, 1973–1975. Surv Ophthalmol 24:335–610
Flaxman SR, Bourne RRA, Resnikoff S et al (2017) Global causes of blindness and distance vision impairment 1990–2020: a systematic review and meta-analysis. Lancet Glob Health 5:e1221–e1234. https://doi.org/10.1016/S2214-109X(17)30393-5
Wong WL, Su X, Li X et al (2014) Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health 2:e106–e116. https://doi.org/10.1016/S2214-109X(13)70145-1
Gehrs KM, Anderson DH, Johnson LV, Hageman GS (2009) Age-related macular degeneration—emerging pathogenetic and therapeutic concepts. Ann Med 38:450–471. https://doi.org/10.1080/07853890600946724
Algvere PV, Kvanta A, Seregard S (2016) Drusen maculopathy: a risk factor for visual deterioration. Acta Ophthalmol 94:427–433. https://doi.org/10.1111/aos.13011
Winkler BS, Boulton ME, Gottsch JD, Sternberg P (1999) Oxidative damage and age-related macular degeneration. Mol Vis 5:32
Mitter SK, Song C, Qi X et al (2014) Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 10:1989–2005. https://doi.org/10.4161/auto.36184
Golestaneh N, Chu Y, Xiao Y-Y et al (2017) Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis 8:e2537–e2537. https://doi.org/10.1038/cddis.2016.453
Fernandez-Godino R, Pierce EA, Garland DL (2016) Extracellular matrix alterations and deposit formation in AMD. Adv Exp Med Biol 854:53–58. https://doi.org/10.1007/978-3-319-17121-0_8
Gemenetzi M, Lotery AJ (2016) Complement pathway biomarkers and age-related macular degeneration. Eye 30:1–14. https://doi.org/10.1038/eye.2015.203
Mattapallil MJ, Caspi RR (2017) Compliments of factor H: what's in it for AMD? Immunity 46:167–169. https://doi.org/10.1016/j.immuni.2017.02.008
Skerka C, Chen Q, Fremeaux-Bacchi V, Roumenina LT (2013) Complement factor H related proteins (CFHRs). Mol Immunol 56:170–180. https://doi.org/10.1016/j.molimm.2013.06.001
Natoli R, Fernando N, Jiao H et al (2017) Retinal macrophages synthesize C3 and activate complement in AMD and in models of focal retinal degeneration. Invest Ophthalmol Vis Sci 58:2977–2990. https://doi.org/10.1167/iovs.17-21672
Nishiguchi KM, Yasuma TR, Tomida D et al (2012) C9–R95X polymorphism in patients with neovascular age-related macular degeneration. Invest Ophthalmol Vis Sci 53:508–512. https://doi.org/10.1167/iovs.11-8425
Grassmann F, Heid IM, Weber BHF, International AMD Genomics Consortium (IAMDGC) (2017) Recombinant haplotypes narrow the ARMS2/HTRA1 association signal for age-related macular degeneration. Genetics 205:919–924. https://doi.org/10.1534/genetics.116.195966
Barchitta M, Maugeri A (2016) Association between vascular endothelial growth factor polymorphisms and age-related macular degeneration: an updated meta-analysis. Dis Markers 2016:8486406–8486409. https://doi.org/10.1155/2016/8486406
Lazzeri S, Orlandi P, Figus M et al (2012) The rs2071559 AA VEGFR-2 genotype frequency is significantly lower in neovascular age-related macular degeneration patients. Sci World J 2012:420190–420196. https://doi.org/10.1100/2012/420190
Kamei M, Hollyfield JG (1999) TIMP-3 in Bruch's membrane: changes during aging and in age-related macular degeneration. Invest Ophthalmol Vis Sci 40:2367–2375
Ratnapriya R, Zhan X, Fariss RN et al (2014) Rare and common variants in extracellular matrix gene Fibrillin 2 (FBN2) are associated with macular degeneration. Hum Mol Genet 23:5827–5837. https://doi.org/10.1093/hmg/ddu276
Fritsche LG, Igl W, Bailey JNC et al (2016) A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet 48:134–143. https://doi.org/10.1038/ng.3448
Age-Related Eye Disease Study Research Group (2000) Risk factors associated with age-related macular degeneration. A case-control study in the age-related eye disease study: age-related eye disease study report number 3. Ophthalmology 107:2224–2232
Lu L, Gu X, Hong L et al (2009) Synthesis and structural characterization of carboxyethylpyrrole-modified proteins: mediators of age-related macular degeneration. Bioorg Med Chem 17:7548–7561. https://doi.org/10.1016/j.bmc.2009.09.009
Kamburoglu G, Gumus K, Kadayifcilar S, Eldem B (2006) Plasma homocysteine, vitamin B12 and folate levels in age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol 244:565–569. https://doi.org/10.1007/s00417-005-0108-2
Rochtchina E, Wang JJ, Flood VM, Mitchell P (2007) Elevated serum homocysteine, low serum vitamin B12, folate, and age-related macular degeneration: the Blue Mountains Eye Study. Am J Ophthalmol 143:344–346. https://doi.org/10.1016/j.ajo.2006.08.032
Arjamaa O, Aaltonen V, Piippo N et al (2017) Hypoxia and inflammation in the release of VEGF and interleukins from human retinal pigment epithelial cells. Graefes Arch Clin Exp Ophthalmol. https://doi.org/10.1007/s00417-017-3711-0
Vadlapatla RK, Vadlapudi AD, Mitra AK (2013) Hypoxia-inducible factor-1 (HIF-1): a potential target for intervention in ocular neovascular diseases. Curr Drug Targets 14:919–935
Peet DJ, Kittipassorn T, Wood JP et al (2017) HIF signalling: the eyes have it. Exp Cell Res 356:136–140. https://doi.org/10.1016/j.yexcr.2017.03.030
Sheridan CM, Pate S, Hiscott P et al (2009) Expression of hypoxia-inducible factor−1α and −2α in human choroidal neovascular membranes. Graefes Arch Clin Exp Ophthalmol 247:1361–1367. https://doi.org/10.1007/s00417-009-1133-3
Inoue Y, Yanagi Y, Matsuura K et al (2007) Expression of hypoxia-inducible factor 1alpha and 2alpha in choroidal neovascular membranes associated with age-related macular degeneration. Br J Ophthalmol 91:1720–1721. https://doi.org/10.1136/bjo.2006.111583
Ferris FL, Wilkinson CP, Bird A et al (2013) Clinical classification of age-related macular degeneration. Ophthalmology 120:844–851. https://doi.org/10.1016/j.ophtha.2012.10.036
Colijn JM, Buitendijk GHS, Prokofyeva E et al (2017) Prevalence of age-related macular degeneration in Europe: the past and the future. Ophthalmology 124:1753–1763. https://doi.org/10.1016/j.ophtha.2017.05.035
Murphy RP (1986) Age-related macular degeneration. Ophthalmology 93:969–971
Sunness JS (1999) The natural history of geographic atrophy, the advanced atrophic form of age-related macular degeneration. Mol Vis 5:25
Ferris FL, Fine SL, Hyman L (1984) Age-related macular degeneration and blindness due to neovascular maculopathy. Arch Ophthalmol 102:1640–1642. https://doi.org/10.1001/archopht.1984.01040031330019
Kvanta A (2008) Neovascular age-related macular degeneration: too many theories, too little knowledge? Acta Ophthalmol 86:468–469. https://doi.org/10.1111/j.1755-3768.2008.01283.x
Grunwald JE, Pistilli M, Ying G-S et al (2015) Growth of geographic atrophy in the comparison of age-related macular degeneration treatments trials. Ophthalmology 122:809–816. https://doi.org/10.1016/j.ophtha.2014.11.007
Khan KN, Mahroo OA, Khan RS et al (2016) Differentiating drusen: Drusen and drusen-like appearances associated with ageing, age-related macular degeneration, inherited eye disease and other pathological processes. Prog Retin and Eye Res 53:70–106. https://doi.org/10.1016/j.preteyeres.2016.04.008
Crabb JW, Miyagi M, Gu X et al (2002) Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci USA 99:14682–14687. https://doi.org/10.1073/pnas.222551899
Johnson LV, Forest DL, Banna CD et al (2011) Cell culture model that mimics drusen formation and triggers complement activation associated with age-related macular degeneration. Proc Natl Acad Sci USA 108:18277–18282. https://doi.org/10.1073/pnas.1109703108
Sarks JP, Sarks SH, Killingsworth MC (1994) Evolution of soft drusen in age-related macular degeneration. Eye (Lond) 8(Pt 3):269–283. https://doi.org/10.1038/eye.1994.57
Ichiyama Y, Sawada T, Ito Y et al (2017) Optical coherence tomography angiography reveals blood flow in choroidal neovascular membrane in remission phase of neovascular age-related macular degeneration. Retina 37:724–730. https://doi.org/10.1097/IAE.0000000000001576
Balaratnasingam C, Yannuzzi LA, Curcio CA et al (2016) Associations between retinal pigment epithelium and drusen volume changes during the lifecycle of large drusenoid pigment epithelial detachments. Invest Ophthalmol Vis Sci 57:5479–5489. https://doi.org/10.1167/iovs.16-19816
Klein R, Klein BE, Linton KL (1992) Prevalence of age-related maculopathy. The Beaver Dam eye study. Ophthalmology 99:933–943
Klein R, Klein BEK, Tomany SC, Moss SE (2002) Ten-year incidence of age-related maculopathy and smoking and drinking: the Beaver Dam Eye Study. Am J Epidemiol 156:589–598. https://doi.org/10.1093/aje/kwf092
Sarks SH (1976) Ageing and degeneration in the macular region: a clinico-pathological study. Br J Ophthalmol 60:324–341. https://doi.org/10.1136/bjo.60.5.324
Ko A, Cao S, Pakzad-Vaezi K et al (2013) Optical coherence tomography-based correlation between choroidal thickness and drusen load in dry age-related macular degeneration. Retina 33:1005–1010. https://doi.org/10.1097/IAE.0b013e31827d266e
Curcio CA, Presley JB, Millican CL, Medeiros NE (2005) Basal deposits and drusen in eyes with age-related maculopathy: evidence for solid lipid particles. Exp Eye Res 80:761–775. https://doi.org/10.1016/j.exer.2004.09.017
Curcio CA, Presley JB, Malek G et al (2005) Esterified and unesterified cholesterol in drusen and basal deposits of eyes with age-related maculopathy. Exp Eye Res 81:731–741. https://doi.org/10.1016/j.exer.2005.04.012
D’souza YB, Jones CJP, Short CD, Bonshek RE (2010) Basal laminar drusen and soft drusen have similar glycan composition. Can J Ophthalmol 45:297–299. https://doi.org/10.3129/i09-232
Mullins RF, Johnson LV, Anderson DH, Hageman GS (1997) Characterization of drusen-associated glycoconjugates. Ophthalmology 104:288–294
Kliffen M, Mooy CM, Luider TM et al (1996) Identification of glycosaminoglycans in age-related macular deposits. Arch Ophthalmol 114:1009–1014. https://doi.org/10.1001/archopht.1996.01100140217021
Sparrow JR, Boulton M (2005) RPE lipofuscin and its role in retinal pathobiology. Exp Eye Res 80:595–606. https://doi.org/10.1016/j.exer.2005.01.007
Lin T, Walker GB, Kurji K et al (2013) Parainflammation associated with advanced glycation endproduct stimulation of RPE in vitro: implications for age-related degenerative diseases of the eye. Cytokine 62:369–381. https://doi.org/10.1016/j.cyto.2013.03.027
Delcourt C, Michel F, Colvez A et al (2001) Associations of cardiovascular disease and its risk factors with age-related macular degeneration: the POLA study. Ophthalmic Epidemiol 8:237–249
Cougnard-Gregoire A, Delyfer M-N, Korobelnik J-F et al (2014) Elevated high-density lipoprotein cholesterol and age-related macular degeneration: the Alienor study. PLoS ONE 9:e90973. https://doi.org/10.1371/journal.pone.0090973
Nakata K, Crabb JW, Hollyfield JG (2005) Crystallin distribution in Bruch's membrane-choroid complex from AMD and age-matched donor eyes. Exp Eye Res 80:821–826. https://doi.org/10.1016/j.exer.2004.12.011
Yuan X, Gu X, Crabb JS et al (2010) Quantitative proteomics: comparison of the macular Bruch membrane/choroid complex from age-related macular degeneration and normal eyes. Mol Cell Proteomics 9:1031–1046. https://doi.org/10.1074/mcp.M900523-MCP200
Anderson DH, Talaga KC, Rivest AJ et al (2004) Characterization of beta amyloid assemblies in drusen: the deposits associated with aging and age-related macular degeneration. Exp Eye Res 78:243–256
Mullins RF, Russell SR, Anderson DH, Hageman GS (2000) Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J 14:835–846
Mantel I, Ambresin A, Moetteli L et al (2014) Complement factor B polymorphism and the phenotype of early age-related macular degeneration. Ophthalmic Genet 35:12–17. https://doi.org/10.3109/13816810.2013.766217
Johnson LV, Leitner WP, Staples MK, Anderson DH (2001) Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res 73:887–896. https://doi.org/10.1006/exer.2001.1094
Adler L, Boyer NP, Anderson DM et al (2015) Determination of N-retinylidene-N-retinylethanolamine (A2E) levels in central and peripheral areas of human retinal pigment epithelium. Photochem Photobiol Sci 14:1983–1990. https://doi.org/10.1039/c5pp00156k
Eldred GE, Lasky MR (1993) Retinal age pigments generated by self-assembling lysosomotropic detergents. Nature 361:724–726. https://doi.org/10.1038/361724a0
Ng K-P, Gugiu B, Renganathan K et al (2008) Retinal pigment epithelium lipofuscin proteomics. Mol Cell Proteomics 7:1397–1405. https://doi.org/10.1074/mcp.M700525-MCP200
Guha S, Liu J, Baltazar G et al (2014) Rescue of compromised lysosomes enhances degradation of photoreceptor outer segments and reduces lipofuscin-like autofluorescence in retinal pigmented epithelial cells. Adv Exp Med Biol 801:105–111. https://doi.org/10.1007/978-1-4614-3209-8_14
Terman A, Brunk UT (1998) Lipofuscin: mechanisms of formation and increase with age. APMIS 106:265–276
Booij JC, Baas DC, Beisekeeva J et al (2010) The dynamic nature of Bruch's membrane. Prog Retin Eye Res 29:1–18. https://doi.org/10.1016/j.preteyeres.2009.08.003
Schlingemann RO (2004) Role of growth factors and the wound healing response in age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol 242:91–101. https://doi.org/10.1007/s00417-003-0828-0
Kvanta A (2006) Ocular angiogenesis: the role of growth factors. Acta Ophthalmol 84:282–288. https://doi.org/10.1111/j.1600-0420.2006.00659.x
Bhutto I, Lutty G (2012) Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch's membrane/choriocapillaris complex. Mol Aspects Med 33:295–317. https://doi.org/10.1016/j.mam.2012.04.005
Mammadzada P, Gudmundsson J, Kvanta A, André H (2016) Differential hypoxic response of human choroidal and retinal endothelial cells proposes tissue heterogeneity of ocular angiogenesis. Acta Ophthalmol 94:805–814. https://doi.org/10.1111/aos.13119
Campochiaro PA (2015) Molecular pathogenesis of retinal and choroidal vascular diseases. Prog Retin Eye Res 49:67–81. https://doi.org/10.1016/j.preteyeres.2015.06.002
Alizadeh E, Mammadzada P, André H (2018) The different facades of retinal and choroidal endothelial cells in response to hypoxia. Int J Mol Sci 19:3846. https://doi.org/10.3390/ijms19123846
Chew EY, Clemons TE, Agrón E et al (2013) Long-term effects of vitamins C and E, β-carotene, and zinc on age-related macular degeneration: AREDS report no. 35. Ophthalmology 120:1604–11.e4. https://doi.org/10.1016/j.ophtha.2013.01.021
Ho L, van Leeuwen R, Witteman JCM et al (2011) Reducing the genetic risk of age-related macular degeneration with dietary antioxidants, zinc, and ω-3 fatty acids: the Rotterdam study. Arch Ophthalmol 129:758–766. https://doi.org/10.1001/archophthalmol.2011.141
Sundelin SP, Nilsson SE (2001) Lipofuscin-formation in retinal pigment epithelial cells is reduced by antioxidants. Free Radic Biol Med 31:217–225
Lotery A, Griner R, Ferreira A et al (2017) Real-world visual acuity outcomes between ranibizumab and aflibercept in treatment of neovascular AMD in a large US data set. Eye 31:1697–1706. https://doi.org/10.1038/eye.2017.143
Eadie JA, Gottlieb JL, Ip MS et al (2014) Response to aflibercept in patients with persistent exudation despite prior treatment with bevacizumab or ranibizumab for age-related macular degeneration. Ophthalmic Surg Lasers Imaging Retina 45:394–397. https://doi.org/10.3928/23258160-20140909-03
Kimoto K, Kubota T (2012) Anti-VEGF agents for ocular angiogenesis and vascular permeability. J Ophthalmol 2012:852183. https://doi.org/10.1155/2012/852183
Tolentino M (2011) Systemic and ocular safety of intravitreal anti-VEGF therapies for ocular neovascular disease. Surv Ophthalmol 56:95–113. https://doi.org/10.1016/j.survophthal.2010.08.006
Pieramici DJ, Rabena MD (2008) Anti-VEGF therapy: comparison of current and future agents. Eye 22:1330–1336. https://doi.org/10.1038/eye.2008.88
Ba J, Peng R-S, Xu D et al (2015) Intravitreal anti-VEGF injections for treating wet age-related macular degeneration: a systematic review and meta-analysis. Drug Des Devel Ther 9:5397–5405. https://doi.org/10.2147/DDDT.S86269
Rush RB, Simunovic MP, Vandiver L et al (2014) Treat-and-extend bevacizumab for neovascular age-related macular degeneration: the importance of baseline characteristics. Retina 34:846–852. https://doi.org/10.1097/IAE.0000000000000033
Selım A, Koçak N, Aslankara H, Kaynak S (2014) Comparative study of photodynamic therapy monotherapy versus triple management in age-related macular degeneration. Turk J Med Sci 44:889–895
Kawczyk-Krupka A, Bugaj AM, Potempa M et al (2015) Vascular-targeted photodynamic therapy in the treatment of neovascular age-related macular degeneration: clinical perspectives. Photodiagnosis Photodyn Ther 12:161–175. https://doi.org/10.1016/j.pdpdt.2015.03.007
Amadio M, Govoni S, Pascale A (2016) Targeting VEGF in eye neovascularization: what's new? A comprehensive review on current therapies and oligonucleotide-based interventions under development. Pharmacol Res 103:253–269. https://doi.org/10.1016/j.phrs.2015.11.027
Biagi C, Conti V, Montanaro N et al (2014) Comparative safety profiles of intravitreal bevacizumab, ranibizumab and pegaptanib: the analysis of the WHO database of adverse drug reactions. Eur J Clin Pharmacol 70:1505–1512. https://doi.org/10.1007/s00228-014-1755-1
CATT Research Group, Martin DF, Maguire MG, et al (2011) Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med 364:1897–1908. doi: 10.1056/NEJMoa1102673
Krishnadev N, Meleth AD, Chew EY (2010) Nutritional supplements for age-related macular degeneration. Curr Opin Ophthalmol 21:184–189. https://doi.org/10.1097/ICU.0b013e32833866ee
Li Y, Ye D (2010) Cancer therapy by targeting hypoxia-inducible factor-1. Curr Cancer Drug Targets 10:782–796
Yang S-L, Wu C, Xiong Z-F, Fang X (2015) Progress on hypoxia-inducible factor-3: its structure, gene regulation and biological function (review). Mol Med Rep 12:2411–2416. https://doi.org/10.3892/mmr.2015.3689
Stroka DM, Burkhardt T, Desbaillets I et al (2001) HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. FASEB J 15:2445–2453. https://doi.org/10.1096/fj.01-0125com
Wiesener MS, Jürgensen JS, Rosenberger C et al (2003) Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J 17:271–273. https://doi.org/10.1096/fj.02-0445fje
Tian H, McKnight SL, Russell DW (1997) Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev 11:72–82. https://doi.org/10.1101/gad.11.1.72
Gu YZ, Moran SM, Hogenesch JB et al (1998) Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr 7:205–213
Duan C (2016) Hypoxia-inducible factor 3 biology: complexities and emerging themes. Am J Physiol 310:C260–C269. https://doi.org/10.1152/ajpcell.00315.2015
Jiang BH, Rue E, Wang GL et al (1996) Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J Biol Chem 271:17771–17778. https://doi.org/10.1074/jbc.271.30.17771
Ruas JL, Poellinger L, Pereira T (2002) Functional analysis of hypoxia-inducible factor-1 alpha-mediated transactivation. Identification of amino acid residues critical for transcriptional activation and/or interaction with CREB-binding protein. J Biol Chem 277:38723–38730. https://doi.org/10.1074/jbc.M205051200
Kallio PJ, Okamoto K, O'Brien S et al (1998) Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J 17:6573–6586. https://doi.org/10.1093/emboj/17.22.6573
Jiang BH, Zheng JZ, Leung SW et al (1997) Transactivation and inhibitory domains of hypoxia-inducible factor 1alpha. Modulation of transcriptional activity by oxygen tension. J Biol Chem 272:19253–19260. https://doi.org/10.1074/jbc.272.31.19253
Huang LE, Gu J, Schau M, Bunn HF (1998) Regulation of hypoxia-inducible factor 1 alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci USA 95:7987–7992
Depping R, Steinhoff A, Schindler SG et al (2008) Nuclear translocation of hypoxia-inducible factors (HIFs): involvement of the classical importin alpha/beta pathway. Biochim Biophys Acta 1783:394–404. https://doi.org/10.1016/j.bbamcr.2007.12.006
Kuschel A, Simon P, Tug S (2012) Functional regulation of HIF-1alpha under normoxia—is there more than post-translational regulation? J Cell Physiol 227:514–524. https://doi.org/10.1002/jcp.22798
Appelhoff RJ, Tian Y-M, Raval RR et al (2004) Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem 279:38458–38465. https://doi.org/10.1074/jbc.M406026200
Chowdhury R, McDonough MA, Mecinović J et al (2009) Structural basis for binding of hypoxia-inducible factor to the oxygen-sensing prolyl hydroxylases. Structure 17:981–989. https://doi.org/10.1016/j.str.2009.06.002
Mole DR, Pugh CW, Ratcliffe PJ, Maxwell PH (2002) Regulation of the HIF pathway: enzymatic hydroxylation of a conserved prolyl residue in hypoxia-inducible factor alpha subunits governs capture by the pVHL E3 ubiquitin ligase complex. Adv Enzyme Regul 42:333–347
Maxwell PH, Wiesener MS, Chang GW et al (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399:271–275. https://doi.org/10.1038/20459
Salceda S, Caro J (1997) Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 272:22642–22647. https://doi.org/10.1074/jbc.272.36.22642
Corn PG, McDonald ER, Herman JG, El-Deiry WS (2003) Tat-binding protein-1, a component of the 26S proteasome, contributes to the E3 ubiquitin ligase function of the von Hippel-Lindau protein. Nat Genet 35:229–237. https://doi.org/10.1038/ng1254
Hewitson KS, McNeill LA, Riordan MV et al (2002) Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem 277:26351–26355. https://doi.org/10.1074/jbc.C200273200
Ruas JL, Poellinger L (2005) Hypoxia-dependent activation of HIF into a transcriptional regulator. Semin Cell Dev Biol 16:514–522. https://doi.org/10.1016/j.semcdb.2005.04.001
Jeong JW, Bae MK, Ahn MY et al (2002) Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 111:709–720. https://doi.org/10.1016/s0092-8674(02)01085-1
Vichi A, Payne DM, Pacheco-Rodriguez G et al (2005) E3 ubiquitin ligase activity of the trifunctional ARD1 (ADP-ribosylation factor domain protein 1). Proc Natl Acad Sci USA 102:1945–1950. https://doi.org/10.1073/pnas.0409800102
Semenza GL, Jiang BH, Leung SW et al (1996) Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem 271:32529–32537. https://doi.org/10.1074/jbc.271.51.32529
Minchenko A, Salceda S, Bauer T, Caro J (1994) Hypoxia regulatory elements of the human vascular endothelial growth factor gene. Cell Mol Biol Res 40:35–39
O'Rourke JF, Dachs GU, Gleadle JM et al (1997) Hypoxia response elements. Oncol Res 9:327–332
Richard DE, Berra E, Gothié E et al (1999) p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J Biol Chem 274:32631–32637. https://doi.org/10.1074/jbc.274.46.32631
Khandrika L, Lieberman R, Koul S et al (2009) Hypoxia-associated p38 mitogen-activated protein kinase-mediated androgen receptor activation and increased HIF-1alpha levels contribute to emergence of an aggressive phenotype in prostate cancer. Oncogene 28:1248–1260. https://doi.org/10.1038/onc.2008.476
Liu W, Shen S-M, Zhao X-Y, Chen G-Q (2012) Targeted genes and interacting proteins of hypoxia inducible factor-1. Int J Biochem Mol Biol 3:165–178
Weidemann A, Johnson RS (2008) Biology of HIF-1α. Cell Death Differ 15:621–627. https://doi.org/10.1038/cdd.2008.12
Yang M-H, Wu M-Z, Chiou S-H et al (2008) Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol 10:295–305. https://doi.org/10.1038/ncb1691
Berghoff AS, Kovanda AK, Melchardt T et al (2014) αvβ3, αvβ5 and αvβ6 integrins in brain metastases of lung cancer. Clin Exp Metastasis 31:841–851. https://doi.org/10.1007/s10585-014-9675-0
Tang N-N, Zhu H, Zhang H-J et al (2014) HIF-1α induces VE-cadherin expression and modulates vasculogenic mimicry in esophageal carcinoma cells. World J Gastroenterol 20:17894–17904. https://doi.org/10.3748/wjg.v20.i47.17894
Wang B, Ding Y-M, Fan P et al (2014) Expression and significance of MMP2 and HIF-1α in hepatocellular carcinoma. Oncol Lett 8:539–546. https://doi.org/10.3892/ol.2014.2189
Osinsky SP, Ganusevich II, Bubnovskaya LN et al (2005) Hypoxia level and matrix metalloproteinases-2 and -9 activity in Lewis lung carcinoma: correlation with metastasis. Exp Oncol 27:202–205
Kimura D, Imaizumi T, Tamo W et al (2002) Hypoxia enhances the expression of plasminogen activator inhibitor-1 in human lung cancer cells, EBC-1. Tohoku J Exp Med 196:259–267
Görlach A, Berchner-Pfannschmidt U, Wotzlaw C et al (2003) Reactive oxygen species modulate HIF-1 mediated PAI-1 expression: involvement of the GTPase Rac1. Thromb Haemost 89:926–935
Hagiwara H, Sato H, Shirai S et al (2006) Connexin 32 down-regulates the fibrinolytic factors in metastatic renal cell carcinoma cells. Life Sci 78:2249–2254. https://doi.org/10.1016/j.lfs.2005.09.036
Guan G, Zhang Y, Lu Y et al (2015) The HIF-1α/CXCR4 pathway supports hypoxia-induced metastasis of human osteosarcoma cells. Cancer Lett 357:254–264. https://doi.org/10.1016/j.canlet.2014.11.034
Schioppa T, Uranchimeg B, Saccani A et al (2003) Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med 198:1391–1402. https://doi.org/10.1084/jem.20030267
Li Y, Qiu X, Zhang S et al (2009) Hypoxia induced CCR7 expression via HIF-1alpha and HIF-2alpha correlates with migration and invasion in lung cancer cells. Cancer Biol Ther 8:322–330. https://doi.org/10.4161/cbt.8.4.7332
Levy AP, Levy NS, Wegner S, Goldberg MA (1995) Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia. J Biol Chem 270:13333–13340. https://doi.org/10.1074/jbc.270.22.13333
Kurihara T, Westenskow PD, Friedlander M (2014) Hypoxia-inducible factor (HIF)/vascular endothelial growth factor (VEGF) signaling in the retina. Adv Exp Med Biol 801:275–281. https://doi.org/10.1007/978-1-4614-3209-8_35
Metzger CS, Koutsimpelas D, Brieger J (2015) Transcriptional regulation of the VEGF gene in dependence of individual genomic variations. Cytokine 76:519–526. https://doi.org/10.1016/j.cyto.2015.07.015
Nikitenko LL, Smith DM, Bicknell R, Rees MCP (2003) Transcriptional regulation of the CRLR gene in human microvascular endothelial cells by hypoxia. FASEB J 17:1499–1501. https://doi.org/10.1096/fj.02-0993fje
Han Z-B, Ren H, Zhao H et al (2008) Hypoxia-inducible factor (HIF)-1 alpha directly enhances the transcriptional activity of stem cell factor (SCF) in response to hypoxia and epidermal growth factor (EGF). Carcinogenesis 29:1853–1861. https://doi.org/10.1093/carcin/bgn066
Simon M-P, Tournaire R, Pouyssegur J (2008) The angiopoietin-2 gene of endothelial cells is up-regulated in hypoxia by a HIF binding site located in its first intron and by the central factors GATA-2 and Ets-1. J Cell Physiol 217:809–818. https://doi.org/10.1002/jcp.21558
Ren B-F, Deng L-F, Wang J et al (2008) Hypoxia regulation of facilitated glucose transporter-1 and glucose transporter-3 in mouse chondrocytes mediated by HIF-1alpha. Jt Bone Spine 75:176–181. https://doi.org/10.1016/j.jbspin.2007.05.012
Thamotharan S, Raychaudhuri N, Tomi M et al (2013) Hypoxic adaptation engages the CBP/CREST-induced coactivator complex of Creb-HIF-1α in transactivating murine neuroblastic glucose transporter. Am J Physiol Endocrinol Metab 304:E583–E598. https://doi.org/10.1152/ajpendo.00513.2012
Minchenko A, Leshchinsky I, Opentanova I et al (2002) Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) gene. Its possible role in the Warburg effect. J Biol Chem 277:6183–6187. https://doi.org/10.1074/jbc.M110978200
Li H, Ko HP, Whitlock JP (1996) Induction of phosphoglycerate kinase 1 gene expression by hypoxia. Roles of Arnt and HIF1alpha. J Biol Chem 271:21262–21267. https://doi.org/10.1074/jbc.271.35.21262
Li X, Jiang Y, Meisenhelder J et al (2016) Mitochondria-translocated PGK1 functions as a protein kinase to coordinate glycolysis and the TCA cycle in tumorigenesis. Mol Cell 61:705–719. https://doi.org/10.1016/j.molcel.2016.02.009
Luo W, Hu H, Chang R et al (2011) Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 145:732–744. https://doi.org/10.1016/j.cell.2011.03.054
Kim J-W, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3:177–185. https://doi.org/10.1016/j.cmet.2006.02.002
Kim J-W, Gao P, Liu Y-C et al (2007) Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol 27:7381–7393. https://doi.org/10.1128/MCB.00440-07
Narumi K, Kobayashi M, Otake S et al (2012) Regulation of human monocarboxylate transporter 4 in skeletal muscle cells: the role of protein kinase C (PKC). Int J Pharm 428:25–32. https://doi.org/10.1016/j.ijpharm.2012.02.021
Ezashi T, Das P, Roberts RM (2005) Low O2 tensions and the prevention of differentiation of hES cells. Proc Natl Acad Sci USA 102:4783–4788. https://doi.org/10.1073/pnas.0501283102
Kumar SM, Liu S, Lu H et al (2012) Acquired cancer stem cell phenotypes through Oct4-mediated dedifferentiation. Oncogene 31:4898–4911. https://doi.org/10.1038/onc.2011.656
Gustafsson MV, Zheng X, Pereira T et al (2005) Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell 9:617–628. https://doi.org/10.1016/j.devcel.2005.09.010
Rankin EB, Wu C, Khatri R et al (2012) The HIF signaling pathway in osteoblasts directly modulates erythropoiesis through the production of EPO. Cell 149:63–74. https://doi.org/10.1016/j.cell.2012.01.051
Baltaziak M, Wincewicz A, Kanczuga-Koda L et al (2013) The relationships between hypoxia-dependent markers: HIF-1alpha, EPO and EPOR in colorectal cancer. Folia Histochem Cytobiol 51:320–325. https://doi.org/10.5603/FHC.2013.0043
Kishi S, Abe H, Akiyama H et al (2011) SOX9 protein induces a chondrogenic phenotype of mesangial cells and contributes to advanced diabetic nephropathy. J Biol Chem 286:32162–32169. https://doi.org/10.1074/jbc.M111.244541
Zhou C-H, Zhang X-P, Liu F, Wang W (2015) Modeling the interplay between the HIF-1 and p53 pathways in hypoxia. Sci Rep 5:13834. https://doi.org/10.1038/srep13834
Wang X, Ma S, Qi G (2012) Effect of hypoxia-inducible factor 1-alpha on hypoxia/reoxygenation-induced apoptosis in primary neonatal rat cardiomyocytes. Biochem Biophys Res Commun 417:1227–1234. https://doi.org/10.1016/j.bbrc.2011.12.115
Nys K, Van Laethem A, Michiels C et al (2010) A p38(MAPK)/HIF-1 pathway initiated by UVB irradiation is required to induce Noxa and apoptosis of human keratinocytes. J Invest Dermatol 130:2269–2276. https://doi.org/10.1038/jid.2010.93
Li J, Zhang X, Sejas DP et al (2004) Hypoxia-induced nucleophosmin protects cell death through inhibition of p53. J Biol Chem 279:41275–41279. https://doi.org/10.1074/jbc.C400297200
Bühler K, Plaisance I, Dieterle T, Brink M (2009) The human urocortin 2 gene is regulated by hypoxia: identification of a hypoxia-responsive element in the 3'-flanking region. Biochem J 424:119–127. https://doi.org/10.1042/BJ20090311
Makino Y, Cao R, Svensson K et al (2001) Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature 414:550–554. https://doi.org/10.1038/35107085
Okabe K, Kobayashi S, Yamada T et al (2014) Neurons limit angiogenesis by titrating VEGF in retina. Cell 159:584–596. https://doi.org/10.1016/j.cell.2014.09.025
Lip PL, Blann AD, Hope-Ross M et al (2001) Age-related macular degeneration is associated with increased vascular endothelial growth factor, hemorheology and endothelial dysfunction. Ophthalmology 108:705–710
Hera R, Keramidas M, Peoc'h M et al (2005) Expression of VEGF and angiopoietins in subfoveal membranes from patients with age-related macular degeneration. Am J Ophthalmol 139:589–596. https://doi.org/10.1016/j.ajo.2004.11.064
Gerber HP, Condorelli F, Park J, Ferrara N (1997) Differential transcriptional regulation of the two vascular endothelial growth factor receptor genes. Flt-1, but not Flk-1/KDR, is up-regulated by hypoxia. J Biol Chem 272:23659–23667. https://doi.org/10.1074/jbc.272.38.23659
Rissanen TT, Vajanto I, Hiltunen MO et al (2002) Expression of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 (KDR/Flk-1) in ischemic skeletal muscle and its regeneration. Am J Pathol 160:1393–1403. https://doi.org/10.1016/S0002-9440(10)62566-7
Nomura M, Yamagishi S, Harada S et al (1998) Placenta growth factor (PlGF) mRNA expression in brain tumors. J Neurooncol 40:123–130
Yoshida D, Kim K, Noha M, Teramoto A (2006) Hypoxia inducible factor 1-alpha regulates of platelet derived growth factor-B in human glioblastoma cells. J Neurooncol 76:13–21. https://doi.org/10.1007/s11060-005-3279-0
Lannér MC, Raper M, Pratt WM, Rhoades RA (2005) Heterotrimeric G proteins and the platelet-derived growth factor receptor-beta contribute to hypoxic proliferation of smooth muscle cells. Am J Respir Cell Mol Biol 33:412–419. https://doi.org/10.1165/rcmb.2005-0004OC
Willam C, Koehne P, Jürgensen JS et al (2000) Tie2 receptor expression is stimulated by hypoxia and proinflammatory cytokines in human endothelial cells. Circ Res 87:370–377. https://doi.org/10.1161/01.res.87.5.370
Kim JH, Shin JP, Kim IT, Park DH (2018) Angiopoietin-like 4 correlates with response to intravitreal ranibizumab injections in neovascular age-related macular degeneration. Retina 38:523–530. https://doi.org/10.1097/IAE.0000000000001554
Martin SK, Diamond P, Williams SA et al (2010) Hypoxia-inducible factor-2 is a novel regulator of aberrant CXCL12 expression in multiple myeloma plasma cells. Haematologica 95:776–784. https://doi.org/10.3324/haematol.2009.015628
Lamalice L, Le Boeuf F, Huot J (2007) Endothelial cell migration during angiogenesis. Circ Res 100:782–794. https://doi.org/10.1161/01.RES.0000259593.07661.1e
Khaliq A, Patel B, Jarvis-Evans J et al (1995) Oxygen modulates production of bFGF and TGF-beta by retinal cells in vitro. Exp Eye Res 60:415–423
Arjamaa O, Nikinmaa M, Salminen A, Kaarniranta K (2009) Regulatory role of HIF-1alpha in the pathogenesis of age-related macular degeneration (AMD). Ageing Res Rev 8:349–358. https://doi.org/10.1016/j.arr.2009.06.002
Campochiaro PA (2013) Ocular neovascularization. J Mol Med 91:311–321. https://doi.org/10.1007/s00109-013-0993-5
André H, Tunik S, Aronsson M, Kvanta A (2015) Hypoxia-inducible factor-1α is associated with sprouting angiogenesis in the murine laser-induced choroidal neovascularization model. Invest Ophthalmol Vis Sci 56:6591–6604. https://doi.org/10.1167/iovs.15-16476
Lin M, Hu Y, Chen Y et al (2012) Impacts of hypoxia-inducible factor-1 knockout in the retinal pigment epithelium on choroidal neovascularization. Invest Ophthalmol Vis Sci 53:6197–6206. https://doi.org/10.1167/iovs.11-8936
Bhattacharya S, Chaum E, Johnson DA, Johnson LR (2012) Age-related susceptibility to apoptosis in human retinal pigment epithelial cells is triggered by disruption of p53-Mdm2 association. Invest Ophthalmol Vis Sci 53:8350–8366. https://doi.org/10.1167/iovs.12-10495
Li R, Du J, Chang Y (2016) Role of autophagy in hypoxia-induced angiogenesis of RF/6A cells in vitro. Curr Eye Res 41:1566–1570. https://doi.org/10.3109/02713683.2016.1145234
Wu J, Lei Z, Yu J (2015) Hypoxia induces autophagy in human vascular endothelial cells in a hypoxia-inducible factor 1-dependent manner. Mol Med Rep 11:2677–2682. https://doi.org/10.3892/mmr.2014.3093
Avery RL, Bakri SJ, Blumenkranz MS et al (2014) Intravitreal injection technique and monitoring: updated guidelines of an expert panel. Retina 34(Suppl 12):S1–S18. https://doi.org/10.1097/IAE.0000000000000399
Jager RD, Aiello LP, Patel SC, Cunningham ET (2004) Risks of intravitreous injection: a comprehensive review. Retina 24:676–698
Ventrice P, Leporini C, Aloe JF et al (2013) Anti-vascular endothelial growth factor drugs safety and efficacy in ophthalmic diseases. J Pharmacol Pharmacother 4:S38–42. https://doi.org/10.4103/0976-500X.120947
Coco RM, Sanabria MR, Castrejon M et al (2014) Funduscopic results after 4-year follow-up treatment with ranibizumab for age-related macular degeneration in a region of Spain. BMC Ophthalmol 14:138. https://doi.org/10.1186/1471-2415-14-138
Ding K, Eaton L, Bowley D et al (2017) Generation and characterization of ABBV642, a dual variable domain immunoglobulin molecule (DVD-Ig) that potently neutralizes VEGF and PDGF-BB and is designed for the treatment of exudative age-related macular degeneration. MAbs 9:269–284. https://doi.org/10.1080/19420862.2016.1268305
Jo N, Mailhos C, Ju MH et al (2006) Inhibition of platelet-derived growth factor B signaling enhances the efficacy of anti-vascular enclothelial growth factor therapy in multiple models of ocular neovascularization. Am J Pathol 168:2036–2053. https://doi.org/10.2353/ajpath.2006.050588
Lim JI, Niec M, Wong V (2015) One year results of a phase 1 study of the safety and tolerability of combination therapy using sustained release intravitreal triamcinolone acetonide and ranibizumab for subfoveal neovascular AMD. Br J Ophthalmol 99:618–623. https://doi.org/10.1136/bjophthalmol-2014-306002
Russo A, Costagliola C, Delcassi L et al (2013) A randomised controlled trial of ranibizumab with and without ketorolac eyedrops for exudative age-related macular degeneration. Br J Ophthalmol 97:1273–1276. https://doi.org/10.1136/bjophthalmol-2013-303417
Kim H, Csaky KG (2010) Nanoparticle-integrin antagonist C16Y peptide treatment of choroidal neovascularization in rats. J Control Release 142:286–293. https://doi.org/10.1016/j.jconrel.2009.10.031
Xia M, Bi K, Huang R et al (2009) Identification of small molecule compounds that inhibit the HIF-1 signaling pathway. Mol Cancer 8:117. https://doi.org/10.1186/1476-4598-8-117
Yeo E-J, Chun Y-S, Park J-W (2004) New anticancer strategies targeting HIF-1. Biochem Pharmacol 68:1061–1069. https://doi.org/10.1016/j.bcp.2004.02.040
Wang R, Zhou S, Li S (2011) Cancer therapeutic agents targeting hypoxia-inducible factor-1. Curr Med Chem 18:3168–3189
Jones DT, Harris AL (2012) Small-molecule inhibitors of the HIF pathway and synthetic lethal interactions. Expert Opin Ther Targets 16:463–480. https://doi.org/10.1517/14728222.2012.674516
Ma J, Sun Y, López FJ et al (2016) Blockage of PI3K/mTOR pathways inhibits laser-induced choroidal neovascularization and improves outcomes relative to VEGF-A suppression alone. Invest Ophthalmol Vis Sci 57:3138–3144. https://doi.org/10.1167/iovs.15-18795
Bertozzi D, Marinello J, Manzo SG et al (2014) The natural inhibitor of DNA topoisomerase I, camptothecin, modulates HIF-1α activity by changing miR expression patterns in human cancer cells. Mol Cancer Ther 13:239–248. https://doi.org/10.1158/1535-7163.MCT-13-0729
Toh Y-M, Li T-K (2011) Mitoxantrone inhibits HIF-1α expression in a topoisomerase II-independent pathway. Clin Cancer Res 17:5026–5037. https://doi.org/10.1158/1078-0432.CCR-11-0235
Yang H, Xia Q, Zou Y et al (2015) M410, a combretastatin A4 analogue, disrupts microtubules and inhibits HIF-1α in human breast cancer cells. Oncol Rep 34:334–340. https://doi.org/10.3892/or.2015.3975
Jo DH, An H, Chang D-J et al (2014) Hypoxia-mediated retinal neovascularization and vascular leakage in diabetic retina is suppressed by HIF-1α destabilization by SH-1242 and SH-1280, novel hsp90 inhibitors. J Mol Med 92:1083–1092. https://doi.org/10.1007/s00109-014-1168-8
Sharma V, Shaheen SS, Dixit D, Sen E (2012) Farnesyltransferase inhibitor manumycin targets IL1β-Ras-HIF-1α axis in tumor cells of diverse origin. Inflammation 35:516–519. https://doi.org/10.1007/s10753-011-9340-6
Tanaka T, Ikegami Y, Nakazawa H et al (2017) Low-dose farnesyltransferase inhibitor suppresses HIF-1α and snail expression in triple-negative breast cancer MDA-MB-231 cells in vitro. J Cell Physiol 232:192–201. https://doi.org/10.1002/jcp.25411
Hutt DM, Roth DM, Vignaud H et al (2014) The histone deacetylase inhibitor, Vorinostat, represses hypoxia inducible factor 1 alpha expression through translational inhibition. PLoS ONE 9:e106224. https://doi.org/10.1371/journal.pone.0106224
Welsh SJ, Williams RR, Birmingham A et al (2003) The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1alpha and vascular endothelial growth factor formation. Mol Cancer Ther 2:235–243
Yoshida T, Zhang H, Iwase T et al (2010) Digoxin inhibits retinal ischemia-induced HIF-1alpha expression and ocular neovascularization. FASEB J 24:1759–1767. https://doi.org/10.1096/fj.09-145664
Iwase T, Fu J, Yoshida T et al (2013) Sustained delivery of a HIF-1 antagonist for ocular neovascularization. J Control Release 172:625–633. https://doi.org/10.1016/j.jconrel.2013.10.008
Song S-J, Chung H, Yu HG (2008) Inhibitory effect of YC-1, 3-(5“-hydroxymethyl-2-”furyl)-1-benzylindazole, on experimental choroidal neovascularization in rat. Ophthalmic Res 40:35–40. https://doi.org/10.1159/000111157
Vavilala DT, O'Bryhim BE, Ponnaluri VKC et al (2013) Honokiol inhibits pathological retinal neovascularization in oxygen-induced retinopathy mouse model. Biochem Biophys Res Commun 438:697–702. https://doi.org/10.1016/j.bbrc.2013.07.118
Carriero MV, Bifulco K, Minopoli M et al (2014) UPARANT: a urokinase receptor-derived peptide inhibitor of VEGF-driven angiogenesis with enhanced stability and in vitro and in vivo potency. Mol Cancer Ther 13:1092–1104. https://doi.org/10.1158/1535-7163.MCT-13-0949
Cammalleri M, Dal Monte M, Locri F et al (2016) The urokinase receptor-derived peptide UPARANT mitigates angiogenesis in a mouse model of laser-induced choroidal neovascularization. Invest Ophthalmol Vis Sci 57:2586–2597. https://doi.org/10.1167/iovs.15-18758
Cammalleri M, Dal Monte M, Locri F et al (2019) The urokinase-type plasminogen activator system as drug target in retinitis pigmentosa: new pre-clinical evidence in the rd10 mouse model. J Cell Mol Med 1074:185. https://doi.org/10.1111/jcmm.14391
Dal Monte M, Rezzola S, Cammalleri M et al (2015) Antiangiogenic effectiveness of the urokinase receptor-derived peptide UPARANT in a model of oxygen-induced retinopathy. Invest Ophthalmol Vis Sci 56:2392–2407. https://doi.org/10.1167/iovs.14-16323
Kong D, Park EJ, Stephen AG et al (2005) Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res 65:9047–9055. https://doi.org/10.1158/0008-5472.CAN-05-1235
Takei A, Ekström M, Mammadzada P et al (2017) Gene transfer of prolyl hydroxylase domain 2 inhibits hypoxia-inducible angiogenesis in a model of choroidal neovascularization. Sci Rep 7:42546. https://doi.org/10.1038/srep42546
Barben M, Ail D, Storti F et al (2018) Hif1a inactivation rescues photoreceptor degeneration induced by a chronic hypoxia-like stress. Cell Death Differ 25:2071–2085. https://doi.org/10.1038/s41418-018-0094-7
Darrow JJ (2019) Luxturna: FDA documents reveal the value of a costly gene therapy. Drug Discov Today 24:949–954. https://doi.org/10.1016/j.drudis.2019.01.019
Xue K, Groppe M, Salvetti AP, MacLaren RE (2017) Technique of retinal gene therapy: delivery of viral vector into the subretinal space. Eye 31:1308–1316. https://doi.org/10.1038/eye.2017.158
Trapani I, Puppo A, Auricchio A (2014) Vector platforms for gene therapy of inherited retinopathies. Prog Retin Eye Res 43:108–128. https://doi.org/10.1016/j.preteyeres.2014.08.001
Kymäläinen H, Appelt JU, Giordano FA et al (2014) Long-term episomal transgene expression from mitotically stable integration-deficient lentiviral vectors. Hum Gene Ther 25:428–442. https://doi.org/10.1089/hum.2013.172
Martin KRG, Klein RL, Quigley HA (2002) Gene delivery to the eye using adeno-associated viral vectors. Methods 28:267–275
Le Meur G, Weber M, Péréon Y et al (2005) Postsurgical assessment and long-term safety of recombinant adeno-associated virus-mediated gene transfer into the retinas of dogs and primates. Arch Ophthalmol 123:500–506. https://doi.org/10.1001/archopht.123.4.500
Bainbridge JWB, Mistry A, Schlichtenbrede FC et al (2003) Stable rAAV-mediated transduction of rod and cone photoreceptors in the canine retina. Gene Ther 10:1336–1344. https://doi.org/10.1038/sj.gt.3301990
Li Q, Miller R, Han P-Y et al (2008) Intraocular route of AAV2 vector administration defines humoral immune response and therapeutic potential. Mol Vis 14:1760–1769
Wan X, Pei H, Zhao M-J et al (2016) Efficacy and safety of rAAV2-ND4 treatment for Leber's hereditary optic neuropathy. Sci Rep 6:21587. https://doi.org/10.1038/srep21587
Constable IJ, Pierce CM, Lai C-M et al (2016) Phase 2a Randomized clinical trial: safety and post hoc analysis of subretinal rAAV.sFLT-1 for wet age-related macular degeneration. EBioMedicine 14:168–175. https://doi.org/10.1016/j.ebiom.2016.11.016
Mori K, Gehlbach P, Yamamoto S et al (2002) AAV-mediated gene transfer of pigment epithelium-derived factor inhibits choroidal neovascularization. Invest Ophthalmol Vis Sci 43:1994–2000
Lai CC, Wu WC, Chen SL et al (2001) Suppression of choroidal neovascularization by adeno-associated virus vector expressing angiostatin. Invest Ophthalmol Vis Sci 42:2401–2407
Campochiaro PA, Nguyen QD, Shah SM et al (2006) Adenoviral vector-delivered pigment epithelium-derived factor for neovascular age-related macular degeneration: results of a phase I clinical trial. Hum Gene Ther 17:167–176. https://doi.org/10.1089/hum.2006.17.167
Campochiaro PA, Lauer AK, Sohn EH et al (2017) Lentiviral vector gene transfer of endostatin/angiostatin for macular degeneration (GEM) study. Hum Gene Ther 28:99–111. https://doi.org/10.1089/hum.2016.117
Campochiaro PA (2012) Gene transfer for ocular neovascularization and macular edema. Gene Ther 19:121–126. https://doi.org/10.1038/gt.2011.164
Boye SE, Boye SL, Lewin AS, Hauswirth WW (2013) A comprehensive review of retinal gene therapy. Mol Ther 21:509–519. https://doi.org/10.1038/mt.2012.280
Corydon TJ (2015) Antiangiogenic eye gene therapy. Hum Gene Ther 26:525–537. https://doi.org/10.1089/hum.2015.064
Cascio S, D'Andrea A, Ferla R et al (2010) miR-20b modulates VEGF expression by targeting HIF-1α and STAT3 in MCF-7 breast cancer cells. J Cell Physiol. https://doi.org/10.1002/jcp.22126
Lei Z, Li B, Yang Z et al (2009) Regulation of HIF-1alpha and VEGF by miR-20b tunes tumor cells to adapt to the alteration of oxygen concentration. PLoS ONE 4:e7629. https://doi.org/10.1371/journal.pone.0007629
McDonough MA, Li V, Flashman E et al (2006) Cellular oxygen sensing: Crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2). Proc Natl Acad Sci USA 103:9814–9819. https://doi.org/10.1073/pnas.0601283103
Jazwa A, Florczyk U, Jozkowicz A, Dulak J (2013) Gene therapy on demand: site specific regulation of gene therapy. Gene 525:229–238. https://doi.org/10.1016/j.gene.2013.03.093
Dougherty CJ, Smith GW, Dorey CK et al (2008) Robust hypoxia-selective regulation of a retinal pigment epithelium-specific adeno-associated virus vector. Mol Vis 14:471–480
Biswal MR, Prentice HM, Smith GW et al (2018) Cell-specific gene therapy driven by an optimized hypoxia-regulated vector reduces choroidal neovascularization. J Mol Med 96:1107–1118. https://doi.org/10.1007/s00109-018-1683-0
Stout JT, Francis PJ (2011) Surgical approaches to gene and stem cell therapy for retinal disease. Hum Gene Ther 22:531–535. https://doi.org/10.1089/hum.2011.060
Dalkara D, Byrne LC, Klimczak RR, et al (2013) In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med 5:189ra76–189ra76. doi: 10.1126/scitranslmed.3005708
Lukason M, DuFresne E, Rubin H et al (2011) Inhibition of choroidal neovascularization in a nonhuman primate model by intravitreal administration of an AAV2 vector expressing a novel anti-VEGF molecule. Mol Ther 19:260–265. https://doi.org/10.1038/mt.2010.230
Collins M, Thrasher A (2015) Gene therapy: progress and predictions. Proc Biol Sci 282:20143003. https://doi.org/10.1098/rspb.2014.3003
Auricchio A (2003) Pseudotyped AAV vectors for constitutive and regulated gene expression in the eye. Vis Res 43:913–918
Boye SL, Bennett A, Scalabrino ML et al (2016) Impact of Heparan sulfate binding on transduction of retina by recombinant adeno-associated virus vectors. J Virol 90:4215–4231. https://doi.org/10.1128/JVI.00200-16
Acknowledgements
Open access funding provided by Karolinska Institute. The authors thank Anders Kvanta and Flavia Plastino for support and valuable input on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mammadzada, P., Corredoira, P.M. & André, H. The role of hypoxia-inducible factors in neovascular age-related macular degeneration: a gene therapy perspective. Cell. Mol. Life Sci. 77, 819–833 (2020). https://doi.org/10.1007/s00018-019-03422-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-019-03422-9