
Abstract
Metastasis is the most frequent cause of death in cancer patients. Epithelial-to-mesenchymal transition (EMT) is the process in which cells lose epithelial integrity and become motile, a critical step for cancer cell invasion, drug resistance and immune evasion. The transforming growth factor-β (TGFβ) signaling pathway is a major driver of EMT. Increasing evidence demonstrates that metabolic reprogramming is a hallmark of cancer and extensive metabolic changes are observed during EMT. The aim of this review is to summarize and interconnect recent findings that illustrate how changes in glycolysis, mitochondrial, lipid and choline metabolism coincide and functionally contribute to TGFβ-induced EMT. We describe TGFβ signaling is involved in stimulating both glycolysis and mitochondrial respiration. Interestingly, the subsequent metabolic consequences for the redox state and lipid metabolism in cancer cells are found to be in favor of EMT as well. Combined we illustrate that a better understanding of the mechanistic links between TGFβ signaling, cancer metabolism and EMT holds promising strategies for cancer therapy, some of which are already actively being explored in the clinic.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Background
Increasing evidence shows that the metabolic dysregulation of cancer cells is related to the epithelial-to-mesenchymal transition (EMT) program [1, 2]. In this review, we address the current understanding of metabolic alterations during transforming growth factor-β (TGFβ)-induced EMT and cancer metastasis. In particular, we focus on documented changes in glycolysis, the metabolism of lipids, mitochondrial and choline-associated pathways, and their functional roles.
The TGFβ family is a family of cytokines that act on many different cell types in a pleiotropic manner [3, 4]. TGFβ is secreted by many cell types, and high levels are present in platelets and bone. Three TGFβ isoforms, i.e., TGFβ1, TGFβ2 and TGFβ3, are present in mammals. Differences in their biological functions are predominantly attributed to different spatiotemporal expression patterns [5], and exogenous ligand addition to the three TGFβ isoforms has redundant cellular effects. Therefore, in this review, we will refer to TGFβ and not mention a specific isoform unless required. TGFβ signaling is involved in embryonic development and adult tissue homeostasis through the regulation of cell proliferation, survival and differentiation [6, 7]. TGFβ elicits its cellular effects via single transmembrane-spanning TGFβ type I and type II receptors (TGFβR1 and TGFβR2) [8, 9]. Both receptors are endowed with intrinsic serine/threonine kinase activity. TGFβ induces the formation of heteromeric complexes in which the TGFβR2 kinase phosphorylates TGFβR1 [10]. The activated TGFβR1 transduces the signal into the cell by phosphorylating receptor-regulated (R-)SMADs, SMAD2 and SMAD3. These R-SMADs are canonical intracellular effectors of TGFβ. Activated SMAD2 and SMAD3 partner with the SMAD4 transcription factor, and upon their nuclear translocation, heteromeric SMAD complexes orchestrate the regulation of a plethora of targets (Fig. 1). SMAD3 and SMAD4, but not SMAD2, bind directly to DNA [11]. The multifunctionality of TGFβ/SMAD signaling events is mediated in part by cell type-dependent interactions with other DNA-binding transcription factors, co-activators and repressors [3, 12]. Canonical target genes include the extracellular matrix protein plasminogen activator inhibitor-1 (PAI-1) and inhibitory SMAD7. SMAD7 regulates the intensity and duration of TGFβ signaling through negative feedback. SMAD7 recruits the E3 ubiquitin ligase SMURF2 to activated TGFβR1/2 receptors and subsequently targets the receptors for degradation [13, 14]. In addition to the canonical SMAD pathway, TGFβ can also activate non-SMAD signaling pathways, such as the Rho GTPase family, the phosphatidyl inositol 3 kinase (PI3K/AKT), and mitogen-activated protein kinase (MAPK) signaling pathways [15,16,17] (Fig. 1).

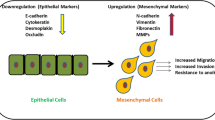
TGFβ-induced epithelial-to-mesenchymal transition. TGFβ signaling can be activated by TGFβ ligands and activate both canonical (blue) and non-canonical pathways (grey). In the canonical SMAD pathway, TGFβR2 activated TGFβR1 phosphorylates SMAD2 and SMAD3, which subsequently form a complex with SMAD4 and translocate to the nucleus. Upon DNA binding, the SMAD complex activates drivers of EMT (SNAI1/2, ZEB1/2 and TWIST1/2) as well as negative feedback component SMAD7. SMAD7 negative feedback regulates TGFβ signaling via recruiting the E3 ubiquitin ligase SMURF2 to activated TβR1/2 receptors and then targets the receptors for degradation. SMAD-independent activation of PI3K/AKT, RHO and RAS/MAPK pathways by TGFβ are non-canonical effectors also involved in EMT and survival. Cancer cells undergoing EMT lose epithelial properties, including cell–cell adhesion, apical–basal polarity and the decrease expression of epithelial genes, i.e. E-cadherin, ZO-1, laminin-1, occludin, claudin. Subsequently, cancer cells acquire migratory mesenchymal characteristics, involving tight-junction dissociation and increased expression of mesenchymal markers, i.e. N-cadherin, vimentin and fibronectin
TGFβ signaling plays a dual role in cancer progression [18, 19]. In the early stages of cancer, TGFβ acts as a tumor suppressor by inhibiting proliferation and the induction of apoptosis in healthy and premalignant cells [20,21,22]. Stimulation of TGFβ signaling represses the cell cycle in G1 phase by the inhibition of cyclin-dependent kinases (CDKs). TGFβ also downregulates c-MYC expression, a pro-oncogenic transcription factor that activates the expression of many pro-proliferative genes [23, 24]. In later stages of tumorigenesis, TGFβ-induced cytostatic responses are mitigated by activation of oncogenes or loss of tumor suppressor genes. TGFβ, frequently secreted in high amounts by cancer and surrounding cells of the tumor microenvironment, then switches its action into a tumor promoter not only by stimulating angiogenesis and immune evasion but also by stimulating a process called epithelial–mesenchymal transition (EMT) in cancer cells [25,26,27]. Also, increased expression of factors such as 14-3-3ζ and PSPC1 mediate the switch of TGFβ signaling from tumor suppressing to promoting cancer progression [28, 29].
TGFβ-induced epithelial–mesenchymal transition
EMT is characterized by morphological changes in which apical–basal polarized epithelial cells are converted into cells with a mesenchymal spindle-shaped phenotype [30]. EMT is a process of key importance for embryogenesis, and organ development. EMT has been causally linked to cancer invasion and metastasis [31,32,33] as well as chemotherapy resistance [34] and TGFβ is a pivotal driver of these multistep processes.
Numerous studies have shown that the activation of EMT is fundamental to malignant progression and metastatic dissemination [32, 35]. EMT is defined by the loss of cell–cell adhesion accompanied by the decreased expression of E-cadherin, cytokeratin, ZO-1, laminin-1, occludin and claudin, and the acquisition of a motile and migratory phenotype with the increased expression of N-cadherin, vimentin, and fibronectin, ultimately facilitating the migration of cancer cells [31, 36]. EMT is a dynamic and multistep process closely linked to the interaction between multiple cytokine and inflammatory factors, transcription factors and cellular pathways. Expression of genes that inhibit an epithelial cell phenotype and activate a mesenchymal phenotype involves the transcription factors SNAI1/2, ZEB1/2 and TWIST1/2. In the early stages of EMT, these transcription factors are activated, bind to the E-box in the E-cadherin promoter region and downregulate the expression level of E-cadherin, subsequently reducing epithelial identity [37] (Fig. 1). Increasing evidence demonstrated that EMT is not a binary process, but that partial states exist in which epithelial and mesenchymal markers are co-expressed by cancer cells. These hybrid states are associated with an aggressive phenotype as they confer high plasticity and stemness properties to cancer cells [38,39,40]. While in most cancer cells, TGFβ-induced EMT is associated with pro-oncogenic response, in pancreatic cancer cells with an intact TGFβ/SMAD4 pathway, it was found associated with apoptosis in a KLF5 repression-dependent manner. In the absence of SMAD4, KLF5 cooperates with SOX4 to promote tumor progression in pancreatic cancer cells [41].
The canonical TGFβ/SMAD pathway plays a pivotal role in mediating EMT. TGFβ-induced SMADs bind to the promotors of SNAI1, TWIST1/2 and ZEB1 and increase their expression [42, 43]. In addition, SMADs can interact and cooperate with SNAI1/2 in a common transcriptional repressive complex that promotes EMT [44]. Epigenetic changes induced by TGFβ/SMAD signaling also contribute to EMT [45, 46]. The non-SMAD signaling pathways of TGFβ can also facilitate epithelial plasticity, sometimes in collaboration with the SMAD pathway [47] (Fig. 1). For example, activation of the PI3K/AKT pathway was required for TGFβ-induced EMT, inhibition of mTOR, a downstream protein kinase of PI3K/AKT signaling, reduced cell migration, adhesion, and invasion that accompany TGFβ-induced EMT of namru murine mammary gland (NMuMG) cells [48, 49]. Moreover, AKT-induced TWIST phosphorylation promoted TGFβ2 transcription and TGFβ receptor activation, and stimulates EMT [50].
It is worth noting that TGFβ-induced EMT can also be a reversible process in cell culture. Upon TGFβ removal, mesenchymal cells can revert back to an epithelial phenotype. Recent findings indicated that a chronic TGFβ treatment induced a stable mesenchymal state in mammary epithelial and breast cancer cells that is different to the reversible EMT upon short-term TGFβ exposure. This stable EMT phenotype was associated with an increased tumor stemness and cancer drug resistance that is susceptible to mTOR inhibition [51].
Metabolic reprogramming in tumorigenesis and EMT
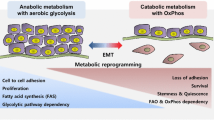
Metabolic reprogramming is a hallmark of cancer that contributes to tumorigenesis and disease progression [52]. Cancer cells rewire metabolic pathways to satisfy their requirement for ATP production, biomass generation and redox balance. The Warburg effect is the most recognized metabolic phenotype observed in cancers. Cancer cells upregulate the uptake of glucose and shift their metabolism from oxidative phosphorylation towards glycolysis, even under aerobic conditions [53, 54]. Although ATP production from glycolysis is very inefficient (2 mol ATP per mol glucose compared to 36 mol ATP per mol glucose in glycolysis and oxidative phosphorylation, respectively), tumors experience advantages in their growth and development from high levels of glycolysis for several reasons. First, high glycolytic rates can increase the tolerance of cancer cells to oxygen fluctuations. Second, as lactate, the final product in glycolysis, can contribute to tumor acidity, the accumulation of lactate promotes immune escape and tumor invasion [55, 56]. Third and most importantly, aerobic glycolysis satisfies the demand of rapidly proliferating cancer cells for macromolecular anabolism as large amounts of intermediate metabolites from glycolysis are shunted into different biosynthetic pathways [53, 57, 58].
A recent study found that the Warburg effect contributed to cancer anoikis resistance, which is a prerequisite for tumor metastasis. The shift of ATP generation from oxidative phosphorylation to that from glycolysis protects cancer cells against reactive oxygen species (ROS)-mediated anoikis [59, 60]. As mentioned above, the aberrant activity of oncogenes and tumor suppressors, such as hypoxia-inducible factor 1 (HIF-1), AKT, MYC, p53 and phosphatase and tensin homolog (PTEN), directly affect metabolic pathways, particularly glycolysis [58, 61, 62]. In addition, enhanced glycolysis accompanied by increased lactate fermentation and alleviated mitochondrial respiration protects cancer cells against oxidative stress, favoring tumor metastasis.
The molecular mechanisms of metabolic reprogramming in cancer cells are complex. Metabolic alterations in cancer have been found to be related to the mutation or abnormal expression of oncogenes or tumor suppressors. For instance, KRAS mutations can alter the metabolic flux of pancreatic cancer cells, selectively decompose glucose through the non-redox pentose phosphate pathway, and promote pentose production and nucleic acid synthesis [63]. Aberrant expression of metabolic enzymes is also a key factor for metabolic reprogramming in cancer that is often regulated by certain oncogenes or tumor suppressor genes [64]. For example, PI3K, KRAS and hypoxia-inducible factor (HIF) are responsible for the upregulation of glucose transporter 1 (GLUT1) [65,66,67]. While it remains to be experimentally tested, it is interesting to take into account that PI3K/AKT and KRAS/MEK/ERK pathways can also be triggered as part of non-canonical TGFβ-signaling and, therefore, might contribute to TGFβ-associated metabolic effects (Fig. 1).
Moreover, metabolic enzyme mutation and dysregulated metabolic enzyme activity can affect cellular metabolism [68]. As cancer cells rely on altered metabolism to support cell proliferation and survival, metabolic pathways are potential therapeutic targets. Recent findings indicate that metabolic changes and EMT are intertwined. While metabolic alterations possibly induce EMT, EMT may also lead to metabolic changes [1, 2]. Notably, a group of 44 metabolic genes named the “mesenchymal metabolic signature” (MMS) genes were found to exhibit increased expression levels in high-grade carcinoma cell lines. Expression of these MMS genes is correlated tightly with mesenchymal markers and was shown to be upregulated upon the activation of EMT by TWIST1 expression in human mammary epithelial cells. Among these genes, dihydropyridine dehydrogenase (DPYD), an enzyme involved in pyrimidine degradation, was found to be essential for EMT. The role of TGFβ in regulating DPYD remains to be investigated. Importantly, this finding implicates that EMT and metabolic reprogramming are intertwined [69]. Increasing evidence indicates that metabolic alterations influence the EMT process through affecting epigenetic modifications [62]. In this context, the intermediary metabolites of glycolysis and oxidative phosphorylation, such as acetyl-CoA, nicotinamide adenine dinucleotide; NAD+/NADH and flavin adenine dinucleotide (FAD), are used as cofactors to promote epigenetic modifications [62, 70]. While it is clear that EMT and cellular metabolism are interlinked, the knowledge on the role of TGFβ signaling, as well as alternative pathways that can induce EMT, in metabolic reprogramming during EMT is still limited and scattered over various cancer models. In the following sections, we describe the emerging relationships between TGFβ-induced EMT and reprogramming of glycolysis, mitochondrial respiration, lipid and choline metabolism.
TGFβ signaling stimulates glycolysis
Aerobic glycolysis enhances the proliferation and survival of tumor cells and provides the basis for tumor cell metastasis and invasion by supplying a large number of metabolic precursors. Simultaneously, the shift from oxidative phosphorylation to glycolysis reduces mitochondrial metabolism, accompanied by decreased ROS production, and was shown to result in anoikis resistance and tumor metastasis [59, 60]. Moreover, the preferential use of aerobic glycolysis leads to the accumulation of lactate in the tumor microenvironment, facilitating the stabilization of HIF-1 and subsequently upregulating the expression of vascular endothelial growth factor (VEGF), resulting in angiogenesis and the stimulation of cell migration [71]. A recent study suggested that extracellular lactate induces SNAI1 and EMT through directly remodeling the extracellular matrix and releasing activated TGFβ1. Furthermore, high extracellular lactate levels can contribute to immune evasion, thereby promoting tumor growth and metastasis [72].
Glycolysis has been found to be upregulated in malignant cancer cells. For instance, highly metastatic MDA-MB-231 breast cancer cells have a much higher glycolytic efficiency than nonmetastatic MCF-7 breast cancer cells [55]. Enhanced glycolysis is mainly due to the increased expression or activity of key enzymes. In recent years, tumor targeting by inhibiting the activity of key enzymes in the glycolytic pathway has been explored to decrease proliferation, invasion and metastasis. The levels of glucose transporter 1 (GLUT1) have been demonstrated to increase upon TGFβ stimulation in mesangial cells, breast cancer cells, glioma cells and pancreatic ductal adenocarcinoma [73,74,75,76] (Fig. 2). GLUT1 expression was correlated with EMT markers, including E-cadherin and vimentin, and accompanied by increased glucose uptake during TGFβ-induced EMT in breast cancer cells [74]. Furthermore, glucose transporter 3 (GLUT3) also showed higher expression in mesenchymal cells compared to that in epithelial cells among non-small-cell lung carcinoma (NSCLC) cell lines and was also upregulated during TGFβ-induced EMT in H2122 lung cancer cells (Fig. 2).

Glycolysis is enhanced during TGFβ-induced EMT. Increased activity of glycolytic enzymes, GLUT1, hexokinase 2 (HK2), 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), 6-phosphofructo-1-kinase (PFK1), pyruvate kinase M2 (PKM2), lactate dehydrogenase type A (LDHA) result in a high glycolytic rate during TGFβ-triggered EMT in cancer cells. While glycolysis is stimulated and lactate is produced, pyruvate dehydrogenase kinase 4 (PDK4), a negative regulator of pyruvate dehydrogenase (PDH) is inhibited by TGFβ signaling to also allow pyruvate into the TCA cycle
GLUT3 was identified as a transcriptional target of ZEB1 that facilitates increased cancer cell motility and invasion [77]. TGFβ1 was also shown to upregulate the expression of the gene-encoding hexokinase 2 (HK2), which phosphorylates glucose to produce glucose-6-phosphate (G6P) in the first step in the glycolytic pathway [73] (Fig. 2). HK2 was recently reported to promote pancreatic ductal adenocarcinoma (PDAC) metastasis by mediating lactate production. Moreover, HK2 was also shown to play a key role in the metastatic progression of neuroblastoma [78, 79]. Another enzyme, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), is involved in glycolytic alteration upon TGFβ-induced EMT. One of the rate-limiting steps in glycolysis is the 6-phosphofructo-1-kinase (PFK1) catalyzed conversion of fructose-6-phosphate (F6P) to fructose-1,6-bisphosphate (F1,6P2). PFKFB3 is responsible for the synthesis of fructose-2,6-bisphosphate (F2,6P2), a potent allosteric activator of PFK1, which stimulates glycolysis [80, 81]. TGFβ1 enhanced PFKFB3 expression and stimulated glycolysis in Panc1 pancreatic cells. PFKFB3 silencing inhibited TGFβ-induced invasion in this cell line by repressing the expression of SNAI1 [82]. In addition, PFKFB3 was also upregulated by TGFβ1 in glioma cells, resulting in an increase in fructose 2,6-bisphosphate, glucose uptake, glycolytic flux and lactate production [73] (Fig. 2). Pyruvate kinase M2 (PKM2) is overexpressed in more than 70% of human cancers [83]. This enzyme catalyzes the last step of glycolysis, in which phosphoenolpyruvate (PEP) is dephosphorylated to pyruvate with ATP production. Upon TGFβ-induced EMT of A549 lung adenocarcinoma cells, the expression of PKM2 increases. When levels of PKM2 were decreased by aspirin-triggered resolvin D1 (AT-RvD1), which is well known to be associated with inflammation, TGFβ1-induced EMT was mitigated [84]. Moreover, in colon cancer cells, PKM2 interacts with TGFβ-induced factor homeobox 2 (TGIF2) during TGFβ-induced EMT (Fig. 2). TGFIF is a transcriptional repressor of TGFβ signaling, and the complex between PKM2 and TGIF2 promotes histone H3K9 deacetylation, resulting in a decrease in E-cadherin transcription [85].
The Warburg effect is characterized by increased glucose uptake and glycolytic activity and the accumulation of lactate in cancer [53]. Interestingly, lactate levels are directly related to cancer metastasis [72, 86]. Cancer cells can metabolize lactate as an energy source and also obtain lactate from cells in the tumor microenvironment, thereby maintaining their acidic environment, leading to apoptosis resistance and increased survival and proliferation [87, 88]. The increased production of lactate occurs during TGFβ-induced EMT [73, 76, 82]. Lactate dehydrogenase type A (LDHA) plays an important role in catalyzing the conversion of pyruvate into lactate. Lactate was shown to promote glioma migration by the TGFβ2-dependent regulation of matrix metalloproteinase-2 (MMP2) [89].
At the end of the glycolytic pathway, pyruvate dehydrogenase kinase (PDK) is an enzyme that inactivates pyruvate dehydrogenase (PDH), which is responsible for converting pyruvate to acetyl-CoA in the mitochondria. Acetyl-CoA then enters the citric acid cycle (TCA) to generate ATP and electron donors, including NADH. In lung cancer cells, downregulation of PDK4 resulted in a shift from glycolysis to oxidative phosphorylation (OXPHOS). TGFβ represses PDK4 expression, and PDK4 inhibition was sufficient to drive EMT and promote erlotinib resistance in epidermal growth factor receptor (EGFR) mutant lung cancer cells (Fig. 2). Consistent with these results, the overexpression of PDK4 partially blocked TGFβ-induced EMT [90].
Collectively, these results show that TGFβ stimulates glycolysis while inducing EMT transcriptional programs in a synergistic manner. The transition from oxidative metabolism to glycolysis may benefit tumor metastasis due to the accumulation of cellular building blocks, the rapid generation of ATP and resistance to oxygen fluctuations. The observation that PDK4 is antagonized by TGFβ signaling, however, implicates that flexibility in metabolic routing is a prerequisite for EMT as pyruvate is still allowed, and essential, to shunt into the TCA cycle for EMT to complete.
The TCA cycle is active during EMT
Mitochondria not only are the main energizing organelles in eukaryotes but also play a crucial role in anabolic metabolism by providing intermediates to produce glucose, amino acids, lipids and nucleotides. Therefore, mitochondrial dysfunction is closely related to many diseases, including cancer. Accumulating evidence indicates that disordered mitochondrial metabolism is associated with tumor invasion and metastasis [91,92,93]. The TCA cycle, one of the main pathways within mitochondria, plays a key role in cell proliferation and provides important intermediates for amino acid, nucleic acid and lipid biosynthesis. Some enzymes involved in the TCA cycle are mutated or dysfunctional in different cancers, i.e. fumarate hydratase (FH), succinate dehydrogenase (SDH), and isocitrate dehydrogenase (IDH). Although these mutations have been directly linked to EMT, the role of TGFβ in regulating TCA intermediates during EMT induction remains to be fully explored [2, 94,95,96,97,98,99].
FH is a metabolic enzyme responsible for converting fumarate to malate. FH mutations have been discovered in hereditary leiomyomatosis, renal cell cancer, pheochromocytoma and paragangliomas [97, 100]. A recent study demonstrated the relationship between FH and EMT in nasopharyngeal carcinoma (NPC). The chromatin-remodeling factor lymphoid-specific helicase (LSH), the expression of which is controlled by the Epstein–Barr virus-encoded protein LMP1, was increased in NPC. LSH can promote EMT via recruiting the epigenetic silencer factor G9a to repress FH transcription. FH dysfunction resulted in decreased malate and increased α-ketoglutarate (α-KG) levels. Increased α-KG levels altered nuclear factor κ-B kinase α (IKKα)-dependent EMT gene expression by inducing the binding of IKKα to the promoter region of EMT-related genes. This leads to migration and invasion in vitro and tumor growth and metastasis in vivo [101]. Furthermore, fumarate accumulation in FH-deficient renal cancer cells elicited EMT by inhibiting the ten–eleven translocation (TET)-dependent demethylation of the anti-metastatic mRNA miR-200, which suppressed the expression of the EMT transcription factors SNAI2, ZEB1 and ZEB2 [96, 102, 103] (Fig. 3).

The TCA cycle is active in TGFβ-induced EMT. The TCA cycle occurs in the matrix of mitochondria and is a key metabolic pathway for providing intermediates to generate glucose, amino acids, lipids, nucleotides and most importantly, ATP. Altered activity of fumarate hydratase (FH), succinate dehydrogenase (SDH), and isocitrate dehydrogenase (IDH) are connected to EMT. The chromatin-remodeling factor lymphoid-specific helicase (LSH) can promote EMT through recruiting the epigenetic silencer factor G9a to suppress FH expression. Furthermore, the accumulation of fumarate also enhances EMT by inhibiting miR-200. Reduced levels of SDHB stimulate EMT by activating the TGFβ signaling pathway. IDH converts isocitrate to α-ketoglutarate but the oncogenic IDHR132H leads to the production of the oncometabolite 2HG. 2HG interferes with chromatin remodeling enzymes and downregulates miR-200 expression via inhibition of TET, subsequently stimulating EMT
Another TCA cycle enzyme linked with EMT is SDH, which is involved in the conversion of succinate to fumarate in the respiratory chain. The SDH complex is composed of four subunits: SDHA (a flavoprotein), SDHB (an iron–sulfur protein), and the two membrane anchor subunits SDHC and SDHD. Mutations in gene-encoding subunits of this enzyme complex have been reported in a subset of human cancers [104, 105]. Decreased SDHB expression has been reported in both human colorectal cancer (CRC) samples and CRC cell lines, and reduced SDHB activity has been correlated with a more advanced clinical lymphatic and distant metastatic phenotype. Further research found that the knockdown of SDHB contributed to cell invasion and migration via EMT by activating the TGFβ signaling pathway through upregulation of the tight junction transcriptional repression complex SNAI1-SMAD3/SMAD4 [106]. The knockdown of SDHB resulted in a hypermethylated epigenome, which was sufficient to trigger EMT in mouse ovarian cancer cells. In addition, SDHB downregulation resulted in succinate accumulation, altered center carbon metabolism and induced mitochondrial dysfunction [94] (Fig. 3).
IDH has also been associated with cancer development and EMT. IDH catalyzes the oxidative decarboxylation of isocitrate into α-ketoglutarate (α-KG). Furthermore, IDH exists as three isoforms: cytosolic IDH1 and mitochondrial IDH2, which are both NADP+ -dependent enzymes, and mitochondrial IDH3, which is an NAD+ -dependent enzyme. Mutations in IDH are associated with the development of multiple cancers [107,108,109]. Mutations in IDH1 and IDH2 lead to the production of R-2-hydroxyglutarate (2HG) from isocitrate instead of α-KG. 2HG and α-KG are highly similar in structure [110], and 2HG is a competitive inhibitor of α-KG-dependent dioxygenases which mainly induces the histone demethylase and DNA demethylase 10–11 TET proteins. The cellular accumulation of 2HG is closely related to the occurrence and development of cancer [111]. The production of 2HG inhibits the activity of α-KG-dependent dioxygenase, leading to impaired histone modification and DNA demethylation [112]. 2HG promotes EMT by increasing histone H3 methylation of the ZEB1 promoter region to upregulate the expression of ZEB1 and downregulate miR-200 expression [95, 113]. Together, ZEB1, miR-200 and TGFβ are part of an autocrine signaling network that stimulates EMT [114]. It might, therefore, be expected that TGFβ signaling synergizes with IDH mutations in the stimulation of invasive cancers (Fig. 3).
Taken together, there is sufficient evidence that TCA cycle metabolites are involved in EMT, but determining the causal relation between metabolite accumulation in one step of the TCA cycle to EMT is challenging. For example, accumulation of fumarate in FH mutant cells is also expected to increase the levels of its precursors such as succinate and α-ketoglutarate. Changes in TCA metabolites can have broad epigenetic effects; therefore, it becomes hard to exactly pin point the exact event that triggers EMT and elucidate the mechanism behind this. It is clear, however, that elucidation of TCA metabolite effects in TGFβ-induced EMT holds promising insights into precision-targeting metastatic cancers in the future. Interestingly, TGFβ-induced EMT seems to trigger both glycolysis and mitochondrial activity. This is expected to be a requirement to provide the cell with sufficient ATP to become motile. Indeed, by performing single-cell ATP assays, it was shown that TGFβ-induced EMT results in a spike in ATP levels [115]. Putting ATP production in overdrive comes with a cost, however, as it coincides with increased production of ROS and reduced lipogenesis for which TCA intermediates are required.
Redox regulation of TGFβ-mediated EMT
ROS are highly reactive molecules and the most common forms of cellular ROS are hydrogen peroxide (H2O2), superoxide (O •−2 ) and hydroxyl radicals (HO•). Especially, H2O2 can function as a second messenger by oxidizing target proteins, hereby altering their function or binding partners. The main source of ROS is the ATP-producing electron transport chain in mitochondria. ROS play an important role in cell signaling and homeostasis [116]. ROS levels in tumor tissues are higher compared to normal tissue and ROS affect many aspects of tumorigenesis [117]. It has been suggested that ROS production is induced by TGFβ to mediate cell proliferation, apoptosis and EMT in cancer [118].
The primary source from which ROS production is stimulated during TGFβ-induced EMT is the mitochondria and found essential for this process [119, 120]. Conversely, it has been demonstrated that mitochondrial thioredoxin (TXN2) repressed TGFβ-induced EMT via different mechanisms. First, TXN interferes with ROS directly as these are important players in the antioxidant pathways that reduce free radicals. Second, TXN antioxidant responses counteract expression of high-mobility group AT-hook 2 (HMGA2), which is a positive regulator of EMT [119, 120]. Furthermore, the mitochondrial enzyme superoxide dismutase 2 (SOD2), which catalyzes O •−2 radicals to H2O2 and oxygen, has been found upregulated and required in TGFβ-induced EMT in human oral and esophageal epithelial cell lines [121]. This implicates that the generation of O •−2 and subsequent H2O2 by mitochondria is indeed actively stimulated by TGFβ during EMT, but kept within a concentration range that is not harmful for the cell (Fig. 4).

Redox regulation of TGFβ-mediated EMT. TGFβ increases O •−2 production by mitochondria, which stimulates EMT. H2O2 dismutation from O •−2 released from mitochondrial by superoxide dismutase 2 (SOD2) is counter balanced by thioredoxin (TXN) pathways. TXN interferes with TGFβ-induced EMT through reduction of H2O2 and suppression of high-mobility group AT-hook 2 (HMGA2) transcription factor expression. In addition, TGFβ also induces NADPH oxidase 4 (NOX4) to produce O •−2 followed by dismutation into H2O2. H2O2 derived from NOX4 inhibits protein–tyrosine phosphatase 1B (PTP1B), a negative regulator of EMT and mitogenic receptor tyrosine kinase (RTK) signaling. Additionally, NOX4 derived H2O2 stimulates TGFβ-induced p38MAPK activation, which enhances EMT by promoting SNAI1 expression
An additional source of TGFβ-induced ROS is NADPH oxidase 4 (NOX4), which belongs to the family of NADPH oxidase enzymes. NOX4 is localized at the cell membrane and NOX4-produced ROS is considered an important redox signal transducer in TGFβ-induced EMT in pancreatic cancer and glioblastoma cells [122,123,124]. ROS derived from NOX4 oxidize and thereby inhibit protein-tyrosine phosphatase 1B (PTP1B), a known negative regulator of mitogenic signaling and cell migration. Prevention of NOX4-generated ROS by chemical inhibition repressed TGFβ-induced EMT [123]. Additionally, NOX4 stimulates TGFβ-induced p38MAPK activation, which enhances EMT by promoting SNAI1 expression [123]. Others have shown that NOX4 is increased during TGFβ-induced EMT in a SMAD3-dependent manner in breast cancer cells [125] (Fig. 4).
Combined, the generation of ROS from mitochondria and NOX4 in response to TGFβ-induced EMT increases the cellular redox state towards a more oxidative milieu. The consequential redox signaling effects on molecular signal transduction subsequently give rise to an EMT permissive state. Increased redox signaling in renal tubular epithelial cells stimulates phosphorylation of SMAD2 and MAPK, and the expression of mesenchymal markers, which could be counteracted by addition of antioxidants [120]. Similarly, extracellular signal-regulated kinase (ERK) inhibition could repress TGFβ and H2O2-induced EMT, further supporting the important role of ROS in TGFβ-induced EMT via the receptor tyrosine kinase pathways [120]. In accordance, another recent finding indicated H2O2-enhanced TGFβ-mediated EMT via SMAD and MAPK kinase (MEK)/ERK signaling in MCF-10A human mammary epithelial cells [126] (Fig. 4).
Nuclear factor E2-related factor 2 (NRF2) is a critical transcriptional factor that plays a key role in cellular responses to high levels of ROS via stimulating many antioxidant proteins including glutathione S-transferase (GST), NAD(P)H, and quinone oxidoreductase 1 (NQO1) [127]. NRF2 activation contributed to EMT by mediating a decrease in E-cadherin expression and stimulates TGF-β1-induced SMAD2/3 activity in a pancreatic ductal adenocarcinoma cell line [128]. In addition, NRF2 knockdown promoted TGF-β1-induced EMT via upregulating SNAIL expression in pulmonary fibrosis [129].
Due to the complex nature of redox signaling on the level of signal transduction and limited experimental tools to study it, the mechanism on how these signals become specific and mechanistically influence signaling proteins is still in the process to be fully elucidated. It is clear, however, that redox signaling and changing the cellular redox state is not only simply a negative consequence of increased ATP production, but also an important aspect of TGFβ-induced EMT. Altering the redox balance is a cancer-targeting strategy that is currently intensively studied and understanding redox regulation of TGFβ-induced EMT might unveil promising targets to interfere metastasis formation.
Lipid metabolism in TGFβ-induced EMT
Lipids have traditionally been defined as molecules that are insoluble in water and soluble in organic solvents. As this property can be found in a number of biological molecules, a classification system based on chemical and biochemical principles has been established [130]. According to their function, lipids can roughly be divided into storage lipids, such as triglycerides and cholesterol esters, and signaling lipids, such as eicosanoids or steroid hormones. Among the metabolic pathways that are reprogrammed in cancer, lipid metabolism has received less attention than glycolysis and mitochondrial metabolism, but interest in lipid metabolism in cancer has steadily been increasing over the past few years. Abnormal lipid metabolism is closely associated with tumorigenesis; specifically, abnormal lipid metabolism contributes to invasion and metastasis [131, 132]. The morphological change from an epithelial apical–basal polarized phenotype to a spindle-shaped phenotype, which is closely related to cell membrane fluidity, is the most recognized feature of EMT [30]. Increased plasma membrane fluidity and the destabilization of lipid rafts have been observed during EMT [133] and were ascribed to abnormal lipogenesis and changes in the structural components of lipid rafts modulated by EMT inducers [132, 134].
Dysfunctional lipid metabolism in cancer is mainly characterized by enhanced de novo fatty acid synthesis and reduced fatty acid decomposition due to rapid proliferation. Furthermore, enhanced fatty acid metabolism is essential for providing lipids for membrane formation and energy production. For example, expression of fatty acid translocase CD36 elevates intracellular fatty acid levels and promotes EMT of hepatocellular carcinoma (HCC) cells [135]. Fatty acid uptake-related genes, such as caveolin-1 (CAV1) and CD36, are abundantly present in metastatic tumors and have been associated with EMT in multiple cancers [136]. Moreover, CD36-positive cells were essential for metastasis formation in human oral cancer cells [137] and the TGFβ signaling pathway was activated upon free fatty acid addition in hepatocellular carcinoma cells, resulting in EMT induction [135]. Fatty acid synthase (FASN), adenosine triphosphate citrate lyase (ACLY) and acetyl-CoA carboxylase (ACC) are the three main enzymes involved in de novo fatty acid synthesis [138,139,140]. Increased FASN has been demonstrated in cisplatin-resistant NSCLC cells compared to its expression in parental cells; this difference in expression was deemed responsible for the increased EMT and metastatic potential of cisplatin-resistant cells. FASN suppression impaired EMT with the downregulation of mesenchymal markers via the FASN-TGFβ1-FASN positive loop specifically in cisplatin-resistant cells [141]. However, the opposite observation has also been described. Decreased levels of FASN and ACC were accompanied by the downregulation of carbohydrate-responsive element-binding protein (ChREBP) and sterol regulatory element-binding proteins (SREBP) in NSCLC cells upon TGFβ-induced EMT, two key transcriptional regulators of lipogenesis. Furthermore, SNAI1 suppressed lipogenesis during EMT via inhibiting ChREBP and SREBP and FASN silencing was sufficient to drive EMT and enhance cancer cell metastasis in vivo. In addition, increased oxygen consumption and ATP generation were observed during TGFβ-induced EMT. These effects might be ascribed to the entry of increased acetyl-CoA into the TCA cycle or OXPHOS instead of lipogenesis, generating sufficient ATP for cancer cell migration [142] (Fig. 5).

Lipid and choline metabolism in TGFβ-induced EMT. During lipogenesis, citrate from the TCA cycle is transported into the cytoplasm and cleaved by ATP citrate lyase (ACLY) to acetyl-CoA, which is carboxylated to malonyl-CoA by acetyl-CoA carboxylase (ACC). Subsequently, fatty acid synthase (FASN) catalyzes malonyl-CoA into palmitate, a substrate for triglyceride, phospholipid and cholesterol ester synthesis. Sterol regulatory element-binding proteins (SREBP) and carbohydrate-responsive element-binding protein (ChREBP) are important regulators of ACLY, ACC and FASN transcription. Expression of these enzymes and nuclear factors is repressed during TGFβ-induced EMT. Lipid rafts are formed by cholesterol and sphingolipids, including sphingomyelin (SM), sphingosine-1-phosphate (S1P) and glycosphingolipid (GSL). TGFβ-induced EMT correlates with reduced expression of sphingomyelin synthase (SMS), which converts ceramide to SM. This interferes with EMT induction by TGFβ via inhibition of TGFβR1 and SMAD2 phosphorylation. S1P upregulates matrix metalloproteinase 7 (MMP-7) by stimulating the PI3K/AKT signaling pathway. MMP7 is linked to EMT through the regulation of MMP-7/syndecan-1/TGFβ autocrine loop. Furthermore, TGFβ-signaling decreases Gg4, an important component of GSL required for EMT. ZEB1 promotes the expression of a-series glycosphingolipids including GM3 synthase connecting glycosphingolipid metabolism and epithelial cell adhesion during TGFβ-induced EMT. Choline kinase alpha (CHKα) is a key enzyme in the cytidine 5′-diphosphocholine (CDP)-choline pathway (known as the Kennedy pathway) to generate phosphatidylcholine, an essential component of cell membranes. Abnormal expression and activity of CHKα is associated with tumor metastasis. The EMT activator ZEB1-altered choline metabolism by stimulating CHKα
Except for their de novo fatty acid synthesis, sphingolipids have also been correlated with EMT. Sphingolipids are structural components of the cell membrane with vital roles in cell recognition and signaling [143, 144]. Glycosphingolipids (GSLs) were shown to be involved in TGFβ-induced EMT in NMuMG and HCV29 human normal bladder cells [145]. TGFβ treatment changed the GSL composition and, in particular, downregulated the content of Gg4, which is an important glycosphingolipid that participates in the EMT process. In addition, exogenous supplementation with Gg4 inhibited cell motility and mesenchymal marker expression and elevated the expression of epithelial markers. Furthermore, Gg4 was reported to mediate EMT through its interactions with epithelial molecules such as E-cadherin and β-catenin to maintain epithelial cell membrane organization [146]. The mechanism by which gangliosides regulate EMT may be related to the expression of transcriptional repressors. For instance, ZEB1 upregulates the expression of a-series glycosphingolipids by stimulating the expression of GM3 synthase (St3gal5). Repression of ZEB1 decreased the expression of a-series GSLs and St3gal5, leading to the increased expression of junction-associated proteins such as E-cadherin and plakoglobin [147] (Fig. 5).
Sphingosine-1-phosphate (S1P) is a signaling sphingolipid that is also associated with the EMT process. S1P mediated EMT by regulating the autocrine production of TGFβ1, which contributed to a fibrotic response in A549 lung cancer cells [148]. High levels of serum S1P in hepatocellular carcinoma were found to be associated with poor clinical prognosis [149]. In a recent study, S1P was found to activate PI3K/AKT signaling pathway, resulting in increased MMP-7 expression. MMP-7 regulates the shedding of syndecan-1, a transmembrane heparan sulfate proteoglycan that mediates cell adhesion and migration. The loss of syndecan-1 led to an increase in autocrine TGFβ1, which enabled EMT. Meanwhile, the endogenous expression of TGFβ1 was tightly mediated by MMP-7, thus constituting an MMP-7/syndecan-1/TGFβ1 autocrine loop to mediate hepatocellular carcinoma (HCC) metastasis [150].
Sphingomyelin synthase (SMS), of which there are two isoforms (SMS1/2), are vital enzymes for the production of sphingomyelin (SM) [151, 152]. SM can mediate signaling responses, apoptosis and lipid rafts [153, 154]. The decreased expression of SMS1 was found during TGFβ1-induced EMT in the MDA-MB-231 cell line. In contrast, the overexpression of SMS1-inhibited EMT induced by TGFβ1 and coincides with the upregulation of E-cadherin and downregulation of vimentin [155]. Another study indicated that SMS1 blocks EMT via repressing the expression of TGFβR1 and SMAD2 phosphorylation [155]. Overall, these findings imply that sphingolipids play important roles in EMT induced by TGFβ, and a deeper understanding of the mechanisms by which sphingolipids regulate cancer cell signaling and metastasis will be beneficial to improve cancer treatment.
Choline metabolism supports EMT
Lipid synthesis is essential for cell growth, proliferation and differentiation, and although many mutations in oncogenes, such as KRAS and MYC, control lipid metabolism [156, 157], few lipid enzymes have been reported to drive tumorigenesis [158]. Choline kinase α (CHKα), a key enzyme involved in choline metabolism, is a notable exception [159]. Aberrant choline metabolism has been observed in multiple cancers. An increasing amount of research has focused on the molecular mechanism underlying abnormal choline metabolism, providing potential targets in cancer therapy [158, 160].
CHKα belongs to the choline kinase protein family, a group of enzymes with a choline kinase/ethanolamine kinase active domain encoded by the CHKA and CHKB genes [159]. CHKα, the first enzyme in the cytidine 5′-diphosphocholine (CDP)-choline pathway (also known as the Kennedy pathway), catalyzes the phosphorylation of free choline using ATP as a phosphate donor to generate phosphocholine. Phosphocholine is a substrate in the synthesis of phosphatidylcholine, the major phospholipid in cell membranes. Numerous studies have confirmed that the abnormal expression and activity of CHKα are apparent in many cancers and correlated with metastasis [161,162,163].
Although direct regulation of CHKα by TGFβ-signaling has not been reported to date, the expression of EMT transcription factors, including ZEB1, TWIST1 and SNAI1, and the EMT target genes vimentin and N-cadherin were found to be associated with dysfunctional choline metabolism in glioblastoma cells. Also, inhibition of CHKα significantly reduced cell viability, invasiveness, clonogenicity, and the expression of EMT-associated genes [164]. In ovarian cancer cells, downregulation of CHKα impaired aggressiveness and enhanced cellular sensitivity to drug treatment [165]. Furthermore, a combination of CHKA loss and 5-fluorouracil (5-FU) treatment resulted in a larger reduction of cell viability and proliferation in breast cancer cell lines compared to that following 5-FU treatment alone [166]. Overexpression of CHKα induced an aggressive phenotype and increased resistance to 5-FU treatment in MCF-7 breast cancer cells [167]. In addition, increased levels of CHKα significantly reduced the survival of mice with bladder cancer, while the inhibition of CHKα suppressed tumor growth and resulted in a relevant increase in in vivo survival [168]. Further research found that the EGFR/PI3K/AKT pathway was essential for mediating CHKα function in colorectal carcinoma [169]. Inhibition of CHKα by EB-3D, a selective CHKα inhibitor, induced senescence in breast cancer cell lines through stimulation of the 5′ AMP-activated protein kinase (AMPK)-mTOR pathway and impaired breast cancer cell proliferation, migration, and invasion accompanied by the loss of mesenchymal markers [170].
Due to the increased expression and activity of CHKα in many different cancers described above, CHKα has become a promising target in cancer therapy. Among many CHKα inhibitors under development, RSM-932A is the first inhibitor to be tested in humans in a phase I clinical trial conducted by TCD Pharma (Valladolid, Spain) at two U.S. clinical centers (NCT01215864) in patients with advanced solid tumors.
Conclusion and perspectives
EMT is an important stage of cancer development. Cancer cells undergoing EMT acquire invasive characteristics and infiltrate into the surrounding matrix to form a microenvironment that promotes tumor growth and metastasis. As summarized above, increasing evidence supports that EMT is accompanied by complex metabolic rewiring. Changes in metabolites and pathways might be used to improve cancer diagnosis, and the targeting of cancer metabolism to prevent EMT may hold promising new therapeutic strategies for cancer patients.
Some molecules have successfully been applied in clinical therapy to target metabolic pathways. For instance, a group of antisense nucleoside analogs, 5-fluorouracil, gemcitabine, methotrexate and fludarabine, which target different enzymes involved in nuclei acid synthesis, acted as an effective therapy for diverse cancers in patients [171]. Another example is l-asparaginase, an agent approved to treat acute lymphoblastic leukemia (ALL) and non-Hodgkin’s lymphoma via catalyzing the conversion of l-asparagine to l-aspartic acid and ammonia, limiting the asparagine available for the tumor [64]. In addition to the successful use of molecules targeting metabolism in the clinic, several metabolic enzymes are evolving as potential candidate targets. Examples include but are not limited to proteins closely linked to TGFβ-induced EMT, such as GLUT1, HK2, PFKFB3, PKM2, LDHA and PDK4, proteins that control glycolysis [172,173,174,175,176,177,178,179,180], and FASN and ACC, which are involved in lipid synthesis [181,182,183,184] (Table 1). Overall, a large number of preclinical research reports provide alternative therapeutic strategies that target EMT and tumor cell migration.
Targeting TGFβ directly during EMT and accompanying metabolic rewiring might provide a potential window for cancer treatment as recent studies have shown that TGFβ inhibition reverses therapy resistance, enhances combinatorial synergy and sensitizes for radiotherapy in cell and mouse models [18]. Systemic treatment with TGFβ inhibitors, however, comes with side effects due to the pleiotropic nature of the pathway and, therefore, is unsuitable or challenging for use in the clinic [18]. In turn, there is a need to find the exact tumor supporting downstream effects of TGFβ signaling to provide promising opportunities for targeting and with this review, we put metabolic reprogramming in the spotlight. However, targeting metabolism comes with a new set of challenges.
Rather than a single metabolite, multiple metabolic pathways are changed simultaneously in tumorigenesis, and combined these metabolic pathways provide potent support for tumor progression. Tumor cells possess high metabolic plasticity in their use of metabolic pathways which also depend on a dynamically changing tumor microenvironment. Normal tissues also depend on the same metabolic pathways for survival and proliferation. These characteristics represent a big challenge for the therapeutic targeting of tumor metabolism. Increased glycolysis as part of the Warburg effect is common in cancer cells but no inhibitors of glycolysis have been approved for therapy, as these also target glycolysis in all healthy cells [171]. In addition, glutamine metabolism can compensate for the decreased glycolysis in tumor cells. Thus, partial and/or combinational targeting might be a more effective strategy as it can be expected that only the highly dependent tumor cells will be affected. Alternatively, instead of focusing on blocking main metabolic routes, targeting synthesis of tumor-specific metabolites that are rare in healthy cells could be more promising. Metabolic changes during EMT are interesting in this respect as EMT is an almost unique property of cancer cells in the adult body, with small cell populations during wound healing and angiogenesis as exceptions.
Finally, next to metabolic changes in cancer cells during TGFβ-induced EMT, the metabolic microenvironment will be affected. This not only contributes to promote tumor progression and therapy resistance, but also connects to immune suppression. Nutrient deficiency caused by hyperactive tumor cell metabolism and metabolites released from tumor cells into the microenvironment affect immune cell function and facilitate immune suppression [185, 186]. Increased glycolysis in tumor cells results in low glucose levels in the microenvironment and is linked to inhibition of glycolysis in tumor-infiltrating lymphocytes [187]. Additionally, accumulation of lactate in tumor microenvironment impairs proliferation and mobility of cytotoxic T lymphocytes and natural killer (NK) cells [56]. Conclusion, this review illustrates that the understanding and targeting of TGFβ-induced metabolic changes during EMT is emerging and provides basis for novel cancer treatments including targeted and immune therapy.
Abbreviations
- 2HG:
-
R-2-Hydroxyglutarate
- 5-FU:
-
5-Fluorouracil
- ACC:
-
Acetyl-CoA carboxylase
- ACLY:
-
Adenosine triphosphate citrate lyase
- ALL:
-
Acute lymphoblastic leukemia
- AMPK:
-
5′ AMP-activated protein kinase
- AT-RvD1:
-
Aspirin-triggered resolvin D1
- CAV1:
-
Caveolin-1
- CDKs:
-
Cyclin-dependent kinases
- CDP:
-
Cytidine 5′-difosfocholine
- CHKα:
-
Choline kinase
- ChREBP:
-
Carbohydrate-responsive element-binding protein
- CRC:
-
Colorectal cancer
- E-cadherin:
-
Epithelial cadherin
- DPYD:
-
Dihydropyridine dehydrogenase
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Epithelial-to-mesenchymal transition
- ERK:
-
Extracellular signal-regulated kinase
- F1,6P2:
-
Fructose-1,6-bisphosphate
- F2,6P2:
-
Fructose-2,6-bisphosphate
- F6P:
-
Fructose-6-phosphate
- FASN:
-
Fatty acid synthase
- FAD:
-
Flavin adenine dinucleotide
- FH:
-
Fumarate hydratase
- G6P:
-
Glucose-6-phosphate
- GLUT1:
-
Glucose transporter 1
- GLUT3:
-
Glucose transporter 3
- GSLs:
-
Glycosphingolipids
- GST:
-
Glutathione S-transferase
- HCC:
-
Hepatocellular carcinoma
- HMGA2:
-
High-mobility group AT-hook 2
- HIF:
-
Hypoxia-inducible factor
- HK2:
-
Hexokinase 2
- IDH:
-
Isocitrate dehydrogenase
- KG:
-
Ketoglutarate
- LDHA:
-
Lactate dehydrogenase type A
- LSH:
-
Lymphoid-specific helicase
- MAPK:
-
Mitogen-activating protein kinase
- MEK:
-
MAPK kinase
- MMP:
-
Matrix metalloproteinase
- mTOR:
-
Mammalian target of rapamycin
- NADH:
-
Nicotinamide adenine dinucleotide
- NF-κB:
-
Nuclear factor-κB
- NK:
-
Natural killer cells
- NMuMG:
-
Namru murine mammary gland
- NOX4:
-
NADPH oxidase 4
- NPC:
-
Nasopharyngeal carcinoma
- NQO1:
-
Quinone oxidoreductase 1
- NRF2:
-
Nuclear factor E2-related factor 2
- NSCLC:
-
Non-small cell lung carcinoma
- OXPHOS:
-
Oxidative phosphorylation
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PDH:
-
Pyruvate dehydrogenase
- PDK:
-
Pyruvate dehydrogenase kinase
- PEP:
-
Phosphoenolpyruvate
- PFK1:
-
6-Phosphofructo-1-kinase
- PFKFB3:
-
6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 3
- PI3K:
-
Phosphatidyl inosititol 3-kinase
- PKM2:
-
Pyruvate kinase M2
- PTEN:
-
Phosphatase and tensin homolog
- PTP1B:
-
Protein–tyrosine phosphatase 1B
- ROS:
-
Reactive oxygen species
- SDH:
-
Succinate dehydrogenase
- SMAD:
-
Sma and Mad related
- SMS:
-
Sphingomyelin synthase
- SOD:
-
Superoxide dismutase
- S1P:
-
Sphingosine-1-phosphate
- SREBP:
-
Sterol regulatory element-binding protein
- St3gal5:
-
GM3 synthase
- TCA:
-
Citric acid cycle
- TET:
-
Ten–eleven translocation
- TGFβ:
-
Transforming growth factor β
- TGFβR1:
-
TGF-β type I receptor
- TGFβR2:
-
TGF-β type II receptor
- TGIF2:
-
TGFβ-induced factor homeobox 2
- TXN2:
-
Mitochondrial thioredoxin
- VEGF:
-
Vascular endothelial growth factor
References
Morandi A, Taddei ML, Chiarugi P, Giannoni E (2017) Targeting the metabolic reprogramming that controls epithelial-to-mesenchymal transition in aggressive tumors. Front Oncol. https://doi.org/10.3389/fonc.2017.00040
Sciacovelli M, Frezza C (2017) Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J 284(19):3132–3144. https://doi.org/10.1111/febs.14090
David CJ, Massague J (2018) Contextual determinants of TGFβ action in development, immunity and cancer. Nat Rev Mol Cell Biol 19(7):419–435. https://doi.org/10.1038/s41580-018-0007-0
Zhang YE (2018) Mechanistic insight into contextual TGF-β signaling. Curr Opin Cell Biol 51:1–7. https://doi.org/10.1016/j.ceb.2017.10.001
Gatherer D, Ten Dijke P, Baird DT, Akhurst RJ (1990) Expression of TGF-β isoforms during first trimester human embryogenesis. Development (Cambridge, England) 110(2):445–460
Massagué J (2012) TGFβ signalling in context. Nat Rev Mol Cell Biol 13(10):616–630. https://doi.org/10.1038/nrm3434
Akhurst RJ, Hata A (2012) Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov 11(10):790–811. https://doi.org/10.1038/nrd3810
Derynck R, Budi EH (2019) Specificity, versatility, and control of TGF-β family signaling. Sci Signal. https://doi.org/10.1126/scisignal.aav5183
Heldin CH, Moustakas A (2016) Signaling receptors for TGF-β family members. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a022053
Wrana JL, Attisano L, Wieser R, Ventura F, Massague J (1994) Mechanism of activation of the TGF-β receptor. Nature 370(6488):341–347. https://doi.org/10.1038/370341a0
Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM (1998) Direct binding of Smad3 and Smad4 to critical TGF β-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. Embo J 17(11):3091–3100. https://doi.org/10.1093/emboj/17.11.3091
Hill CS (2016) Transcriptional control by the SMADs. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a022079
Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL (2000) Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF β receptor for degradation. Mol Cell 6(6):1365–1375. https://doi.org/10.1016/s1097-2765(00)00134-9
Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten Dijke P (1997) Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature 389(6651):631–635. https://doi.org/10.1038/39369
Katsuno Y, Lamouille S, Derynck R (2013) TGF-β signaling and epithelial–mesenchymal transition in cancer progression. Curr Opin Oncol 25(1):76–84. https://doi.org/10.1097/CCO.0b013e32835b6371
Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME, Arteaga CL, Moses HL (2001) Transforming growth factor-β1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell 12(1):27–36. https://doi.org/10.1091/mbc.12.1.27
Derynck R, Zhang YE (2003) Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 425(6958):577–584. https://doi.org/10.1038/nature02006
Colak S, Ten Dijke P (2017) Targeting TGF-β signaling in cancer. Trends Cancer 3(1):56–71. https://doi.org/10.1016/j.trecan.2016.11.008
Zhang Y, Alexander PB, Wang XF (2017) TGF-β family signaling in the control of cell proliferation and survival. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a022145
Massague J, Blain SW, Lo RS (2000) TGFβ signaling in growth control, cancer, and heritable disorders. Cell 103(2):295–309. https://doi.org/10.1016/s0092-8674(00)00121-5
Schuster N, Krieglstein K (2002) Mechanisms of TGF-β-mediated apoptosis. Cell Tissue Res 307(1):1–14. https://doi.org/10.1007/s00441-001-0479-6
Ten Dijke P, Goumans MJ, Itoh F, Itoh S (2002) Regulation of cell proliferation by Smad proteins. J Cell Physiol 191(1):1–16. https://doi.org/10.1002/jcp.10066
Liu F, Matsuura I (2005) Inhibition of Smad antiproliferative function by CDK phosphorylation. Cell Cycle (Georgetown, Tex) 4(1):63–66. https://doi.org/10.4161/cc.4.1.1366
Pardali K, Moustakas A (2007) Actions of TGF-β as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta 1775(1):21–62. https://doi.org/10.1016/j.bbcan.2006.06.004
Drabsch Y, ten Dijke P (2012) TGF-β signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev 31(3–4):553–568. https://doi.org/10.1007/s10555-012-9375-7
Inman GJ (2011) Switching TGFβ from a tumor suppressor to a tumor promoter. Curr Opin Genet Dev 21(1):93–99. https://doi.org/10.1016/j.gde.2010.12.004
Massague J (2008) TGFβ in cancer. Cell 134(2):215–230. https://doi.org/10.1016/j.cell.2008.07.001
Lu J, Guo H, Treekitkarnmongkol W, Li P, Zhang J, Shi B, Ling C, Zhou X, Chen T, Chiao PJ, Feng X, Seewaldt VL, Muller WJ, Sahin A, Hung MC, Yu D (2009) 14-3-3zeta Cooperates with ErbB2 to promote ductal carcinoma in situ progression to invasive breast cancer by inducing epithelial–mesenchymal transition. Cancer Cell 16(3):195–207. https://doi.org/10.1016/j.ccr.2009.08.010
Yeh HW, Hsu EC, Lee SS, Lang YD, Lin YC, Chang CY, Lee SY, Gu DL, Shih JH, Ho CM, Chen CF, Chen CT, Tu PH, Cheng CF, Chen RH, Yang RB, Jou YS (2018) PSPC1 mediates TGF-β1 autocrine signalling and Smad2/3 target switching to promote EMT, stemness and metastasis. Nat Cell Biol 20(4):479–491. https://doi.org/10.1038/s41556-018-0062-y
Derynck R, Weinberg RA (2019) EMT and cancer: more than meets the eye. Dev Cell 49(3):313–316. https://doi.org/10.1016/j.devcel.2019.04.026
Kalluri R, Weinberg RA (2009) The basics of epithelial–mesenchymal transition. J Clin Investig 119(6):1420–1428. https://doi.org/10.1172/JCI39104
Thiery JP (2002) Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer 2(6):442–454. https://doi.org/10.1038/nrc822
Yang J, Weinberg RA (2008) Epithelial–mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 14(6):818–829. https://doi.org/10.1016/j.devcel.2008.05.009
van Staalduinen J, Baker D, Ten Dijke P, van Dam H (2018) Epithelial-mesenchymal-transition-inducing transcription factors: new targets for tackling chemoresistance in cancer? Oncogene. https://doi.org/10.1038/s41388-018-0378-x
Savagner P (2010) The epithelial–mesenchymal transition (EMT) phenomenon. Ann Oncol. https://doi.org/10.1093/annonc/mdq292
Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol 15(3):178–196. https://doi.org/10.1038/nrm3758
Puisieux A, Brabletz T, Caramel J (2014) Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol 16(6):488–494. https://doi.org/10.1038/ncb2976
Correction for Kroger et al (2019) Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc Natl Acad Sci USA 116(23):11553–11554. https://doi.org/10.1073/pnas.1907473116
Pastushenko I, Blanpain C (2019) EMT transition states during tumor progression and metastasis. Trends Cell Biol 29(3):212–226. https://doi.org/10.1016/j.tcb.2018.12.001
Santamaria PG, Moreno-Bueno G, Cano A (2019) Contribution of epithelial plasticity to therapy resistance. J Clin Med 8(5):89. https://doi.org/10.3390/jcm8050676
David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, Iacobuzio-Donahue CA, Massague J (2016) TGF-β tumor suppression through a lethal EMT. Cell 164(5):1015–1030. https://doi.org/10.1016/j.cell.2016.01.009
Tsubakihara Y, Moustakas A (2018) Epithelial–mesenchymal transition and metastasis under the control of transforming growth factor β. Int J Mol Sci 19(11):3672. https://doi.org/10.3390/ijms19113672
Hao Y, Baker D, Ten Dijke P (2019) TGF-β-Mediated epithelial–mesenchymal transition and cancer metastasis. Int J Mol Sci 20(11):2767. https://doi.org/10.3390/ijms20112767
Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL, Crystal RG, de Herreros AG, Moustakas A, Pettersson RF, Fuxe J (2009) A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-β mediated epithelial–mesenchymal transition. Nat Cell Biol 11(8):943–950. https://doi.org/10.1038/ncb1905
Du D, Katsuno Y, Meyer D, Budi EH, Chen SH, Koeppen H, Wang H, Akhurst RJ, Derynck R (2018) Smad3-mediated recruitment of the methyltransferase SETDB1/ESET controls Snail1 expression and epithelial–mesenchymal transition. EMBO Rep 19(1):135–155. https://doi.org/10.15252/embr.201744250
Suriyamurthy S, Baker D, Ten Dijke P, Iyengar PV (2019) Epigenetic reprogramming of TGF-β signaling in breast cancer. Cancers 11(5):726. https://doi.org/10.3390/cancers11050726
Derynck R, Muthusamy BP, Saeteurn KY (2014) Signaling pathway cooperation in TGF-β-induced epithelial–mesenchymal transition. Curr Opin Cell Biol 31:56–66. https://doi.org/10.1016/j.ceb.2014.09.001
Lamouille S, Derynck R (2007) Cell size and invasion in TGF-β-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol 178(3):437–451. https://doi.org/10.1083/jcb.200611146
Xu W, Yang Z, Lu N (2015) A new role for the PI3K/Akt signaling pathway in the epithelial–mesenchymal transition. Cell Adhes Migr 9(4):317–324. https://doi.org/10.1080/19336918.2015.1016686
Xue G, Restuccia DF, Lan Q, Hynx D, Dirnhofer S, Hess D, Ruegg C, Hemmings BA (2012) Akt/PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-β signaling axes. Cancer Discov 2(3):248–259. https://doi.org/10.1158/2159-8290.CD-11-0270
Katsuno Y, Meyer DS, Zhang Z, Shokat KM, Akhurst RJ, Miyazono K, Derynck R (2019) Chronic TGF-β exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci Signal. https://doi.org/10.1126/scisignal.aau8544
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. https://doi.org/10.1016/j.cell.2011.02.013
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (New York, NY) 324(5930):1029–1033. https://doi.org/10.1126/science.1160809
Liberti MV, Locasale JW (2016) The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci 41(3):211–218. https://doi.org/10.1016/j.tibs.2015.12.001
Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4(11):891–899. https://doi.org/10.1038/nrc1478
Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, Kastenberger M, Bogdan C, Schleicher U, Mackensen A, Ullrich E, Fichtner-Feigl S, Kesselring R, Mack M, Ritter U, Schmid M, Blank C, Dettmer K, Oefner PJ, Hoffmann P, Walenta S, Geissler EK, Pouyssegur J, Villunger A, Steven A, Seliger B, Schreml S, Haferkamp S, Kohl E, Karrer S, Berneburg M, Herr W, Mueller-Klieser W, Renner K, Kreutz M (2016) LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab 24(5):657–671. https://doi.org/10.1016/j.cmet.2016.08.011
Kroemer G, Pouyssegur J (2008) Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell 13(6):472–482. https://doi.org/10.1016/j.ccr.2008.05.005
Cairns RA, Harris IS, Mak TW (2011) Regulation of cancer cell metabolism. Nat Rev Cancer 11(2):85–95. https://doi.org/10.1038/nrc2981
Kamarajugadda S, Stemboroski L, Cai Q, Simpson NE, Nayak S, Tan M, Lu J (2012) Glucose oxidation modulates anoikis and tumor metastasis. Mol Cell Biol 32(10):1893–1907. https://doi.org/10.1128/MCB.06248-11
Lu J, Tan M, Cai Q (2015) The Warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett 356(2):156–164. https://doi.org/10.1016/j.canlet.2014.04.001
Agathocleous M, Harris WA (2013) Metabolism in physiological cell proliferation and differentiation. Trends Cell Biol 23(10):484–492. https://doi.org/10.1016/j.tcb.2013.05.004
Li L, Li W (2015) Epithelial–mesenchymal transition in human cancer: comprehensive reprogramming of metabolism, epigenetics, and differentiation. Pharmacol Ther 150:33–46. https://doi.org/10.1016/j.pharmthera.2015.01.004
Ying H, Kimmelman Alec C, Lyssiotis Costas A, Hua S, Chu Gerald C, Fletcher-Sananikone E, Locasale Jason W, Son J, Zhang H, Coloff Jonathan L, Yan H, Wang W, Chen S, Viale A, Zheng H, Paik J-h, Lim C, Guimaraes Alexander R, Martin Eric S, Chang J, Hezel Aram F, Perry Samuel R, Hu J, Gan B, Xiao Y, Asara John M, Weissleder R, Wang YA, Chin L, Cantley Lewis C, DePinho Ronald A (2012) Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149(3):656–670. https://doi.org/10.1016/j.cell.2012.01.058
Counihan JL, Grossman EA, Nomura DK (2018) Cancer metabolism: current understanding and therapies. Chem Rev 118(14):6893–6923. https://doi.org/10.1021/acs.chemrev.7b00775
Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, Diaz LA Jr, Velculescu VE, Lengauer C, Kinzler KW, Vogelstein B, Papadopoulos N (2009) Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science (New York, NY) 325(5947):1555–1559. https://doi.org/10.1126/science.1174229
Semenza GL (2010) HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev 20(1):51–56. https://doi.org/10.1016/j.gde.2009.10.009
Wieman HL, Wofford JA, Rathmell JC, Margolis B (2007) Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell 18(4):1437–1446. https://doi.org/10.1091/mbc.e06-07-0593
Vander Heiden MG, DeBerardinis RJ (2017) Understanding the intersections between metabolism and cancer biology. Cell 168(4):657–669. https://doi.org/10.1016/j.cell.2016.12.039
Shaul YD, Freinkman E, Comb WC, Cantor JR, Tam WL, Thiru P, Kim D, Kanarek N, Pacold ME, Chen WW, Bierie B, Possemato R, Reinhardt F, Weinberg RA, Yaffe MB, Sabatini DM (2014) Dihydropyrimidine accumulation is required for the epithelial–mesenchymal transition. Cell 158(5):1094–1109. https://doi.org/10.1016/j.cell.2014.07.032
Sabari BR, Zhang D, Allis CD, Zhao Y (2017) Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol 18(2):90–101. https://doi.org/10.1038/nrm.2016.140
Goodwin ML, Gladden LB, Nijsten MW, Jones KB (2014) Lactate and cancer: revisiting the warburg effect in an era of lactate shuttling. Front Nutr 1:27. https://doi.org/10.3389/fnut.2014.00027
Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, Renner K, Timischl B, Mackensen A, Kunz-Schughart L, Andreesen R, Krause SW, Kreutz M (2007) Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 109(9):3812–3819. https://doi.org/10.1182/blood-2006-07-035972
Rodriguez-Garcia A, Samso P, Fontova P, Simon-Molas H, Manzano A, Castano E, Rosa JL, Martinez-Outshoorn U, Ventura F, Navarro-Sabate A, Bartrons R (2017) TGF-β1 targets Smad, p38 MAPK, and PI3K/Akt signaling pathways to induce PFKFB3 gene expression and glycolysis in glioblastoma cells. FEBS J 284(20):3437–3454. https://doi.org/10.1111/febs.14201
Li W, Wei Z, Liu Y, Li H, Ren R, Tang Y (2010) Increased 18F-FDG uptake and expression of Glut1 in the EMT transformed breast cancer cells induced by TGF-β. Neoplasma 57(3):234–240. https://doi.org/10.4149/neo_2010_03_234
Inoki K, Haneda M, Maeda S, Koya D, Kikkawa R (1999) TGF-β 1 stimulates glucose uptake by enhancing GLUT1 expression in mesangial cells. Kidney Int 55(5):1704–1712. https://doi.org/10.1046/j.1523-1755.1999.00438.x
Liu M, Quek LE, Sultani G, Turner N (2016) Epithelial–mesenchymal transition induction is associated with augmented glucose uptake and lactate production in pancreatic ductal adenocarcinoma. Cancer Metab 4:19. https://doi.org/10.1186/s40170-016-0160-x
Masin M, Vazquez J, Rossi S, Groeneveld S, Samson N, Schwalie PC, Deplancke B, Frawley LE, Gouttenoire J, Moradpour D, Oliver TG, Meylan E (2014) GLUT3 is induced during epithelial–mesenchymal transition and promotes tumor cell proliferation in non-small cell lung cancer. Cancer Metab 2:11. https://doi.org/10.1186/2049-3002-2-11
Botzer LE, Maman S, Sagi-Assif O, Meshel T, Nevo I, Yron I, Witz IP (2016) Hexokinase 2 is a determinant of neuroblastoma metastasis. Br J Cancer 114(7):759–766. https://doi.org/10.1038/bjc.2016.26
Anderson M, Marayati R, Moffitt R, Yeh JJ (2017) Hexokinase 2 promotes tumor growth and metastasis by regulating lactate production in pancreatic cancer. Oncotarget 8(34):56081–56094. https://doi.org/10.18632/oncotarget.9760
De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquiere B, Cauwenberghs S, Eelen G, Phng LK, Betz I, Tembuyser B, Brepoels K, Welti J, Geudens I, Segura I, Cruys B, Bifari F, Decimo I, Blanco R, Wyns S, Vangindertael J, Rocha S, Collins RT, Munck S, Daelemans D, Imamura H, Devlieger R, Rider M, Van Veldhoven PP, Schuit F, Bartrons R, Hofkens J, Fraisl P, Telang S, Deberardinis RJ, Schoonjans L, Vinckier S, Chesney J, Gerhardt H, Dewerchin M, Carmeliet P (2013) Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154(3):651–663. https://doi.org/10.1016/j.cell.2013.06.037
Van Schaftingen E, Lederer B, Bartrons R, Hers HG (1982) A kinetic study of pyrophosphate: fructose-6-phosphate phosphotransferase from potato tubers. Application to a microassay of fructose 2,6-bisphosphate. Eur J Biochem 129(1):191–195. https://doi.org/10.1111/j.1432-1033.1982.tb07039.x
Yalcin A, Solakoglu TH, Ozcan SC, Guzel S, Peker S, Celikler S, Balaban BD, Sevinc E, Gurpinar Y, Chesney JA (2017) 6-Phosphofructo-2-kinase/fructose 2,6-bisphosphatase-3 is required for transforming growth factor β1-enhanced invasion of Panc1 cells in vitro. Biochem Biophys Res Commun 484(3):687–693. https://doi.org/10.1016/j.bbrc.2017.01.178
Altenberg B, Greulich KO (2004) Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics 84(6):1014–1020. https://doi.org/10.1016/j.ygeno.2004.08.010
Liu Y, Yuan X, Li W, Cao Q, Shu Y (2016) Aspirin-triggered resolvin D1 inhibits TGF-β1-induced EMT through the inhibition of the mTOR pathway by reducing the expression of PKM2 and is closely linked to oxidative stress. Int J Mol Med 38(4):1235–1242. https://doi.org/10.3892/ijmm.2016.2721
Hamabe A, Konno M, Tanuma N, Shima H, Tsunekuni K, Kawamoto K, Nishida N, Koseki J, Mimori K, Gotoh N, Yamamoto H, Doki Y, Mori M, Ishii H (2014) Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial–mesenchymal transition. Proc Natl Acad Sci 111(43):15526–15531. https://doi.org/10.1073/pnas.1407717111
Gatenby RA, Gawlinski ET, Gmitro AF, Kaylor B, Gillies RJ (2006) Acid-mediated tumor invasion: a multidisciplinary study. Cancer Res 66(10):5216–5223. https://doi.org/10.1158/0008-5472.CAN-05-4193
Hirschhaeuser F, Sattler UG, Mueller-Klieser W (2011) Lactate: a metabolic key player in cancer. Cancer Res 71(22):6921–6925. https://doi.org/10.1158/0008-5472.CAN-11-1457
Doherty JR, Cleveland JL (2013) Targeting lactate metabolism for cancer therapeutics. J Clin Investig 123(9):3685–3692. https://doi.org/10.1172/JCI69741
Baumann F, Leukel P, Doerfelt A, Beier CP, Dettmer K, Oefner PJ, Kastenberger M, Kreutz M, Nickl-Jockschat T, Bogdahn U, Bosserhoff AK, Hau P (2009) Lactate promotes glioma migration by TGF-β2-dependent regulation of matrix metalloproteinase-2. Neuro Oncol 11(4):368–380. https://doi.org/10.1215/15228517-2008-106
Sun Y, Daemen A, Hatzivassiliou G, Arnott D, Wilson C, Zhuang G, Gao M, Liu P, Boudreau A, Johnson L, Settleman J (2014) Metabolic and transcriptional profiling reveals pyruvate dehydrogenase kinase 4 as a mediator of epithelial-mesenchymal transition and drug resistance in tumor cells. Cancer Metab 2(1):20. https://doi.org/10.1186/2049-3002-2-20
Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, Dhup S, Tardy M, Vazeille T, Bouzin C, Feron O, Michiels C, Gallez B, Sonveaux P (2014) A mitochondrial switch promotes tumor metastasis. Cell Rep 8(3):754–766. https://doi.org/10.1016/j.celrep.2014.06.043
Cha YH, Yook JI, Kim HS, Kim NH (2015) Catabolic metabolism during cancer EMT. Arch Pharm Res 38(3):313–320. https://doi.org/10.1007/s12272-015-0567-x
Guerra F, Guaragnella N, Arbini AA, Bucci C, Giannattasio S, Moro L (2017) Mitochondrial dysfunction: a novel potential driver of epithelial-to-mesenchymal transition in cancer. Front Oncol 7:295. https://doi.org/10.3389/fonc.2017.00295
Aspuria PP, Lunt SY, Varemo L, Vergnes L, Gozo M, Beach JA, Salumbides B, Reue K, Wiedemeyer WR, Nielsen J, Karlan BY, Orsulic S (2014) Succinate dehydrogenase inhibition leads to epithelial–mesenchymal transition and reprogrammed carbon metabolism. Cancer Metab 2:21. https://doi.org/10.1186/2049-3002-2-21
Grassian AR, Lin F, Barrett R, Liu Y, Jiang W, Korpal M, Astley H, Gitterman D, Henley T, Howes R, Levell J, Korn JM, Pagliarini R (2012) Isocitrate dehydrogenase (IDH) mutations promote a reversible ZEB1/microRNA (miR)-200-dependent epithelial–mesenchymal transition (EMT). J Biol Chem 287(50):42180–42194. https://doi.org/10.1074/jbc.M112.417832
Sciacovelli M, Goncalves E, Johnson TI, Zecchini VR, da Costa AS, Gaude E, Drubbel AV, Theobald SJ, Abbo SR, Tran MG, Rajeeve V, Cardaci S, Foster S, Yun H, Cutillas P, Warren A, Gnanapragasam V, Gottlieb E, Franze K, Huntly B, Maher ER, Maxwell PH, Saez-Rodriguez J, Frezza C (2016) Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 537(7621):544–547. https://doi.org/10.1038/nature19353
Castro-Vega LJ, Buffet A, De Cubas AA, Cascon A, Menara M, Khalifa E, Amar L, Azriel S, Bourdeau I, Chabre O, Curras-Freixes M, Franco-Vidal V, Guillaud-Bataille M, Simian C, Morin A, Leton R, Gomez-Grana A, Pollard PJ, Rustin P, Robledo M, Favier J, Gimenez-Roqueplo AP (2014) Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet 23(9):2440–2446. https://doi.org/10.1093/hmg/ddt639
Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW 3rd, Cornelisse CJ, Devilee P, Devlin B (2000) Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science (New York, NY) 287(5454):848–851. https://doi.org/10.1126/science.287.5454.848
Gimenez-Roqueplo AP, Favier J, Rustin P, Rieubland C, Crespin M, Nau V, Van Kien PK, Corvol P, Plouin PF, Jeunemaitre X (2003) Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res 63(17):5615–5621
Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, Roylance RR, Olpin S, Bevan S, Barker K, Hearle N, Houlston RS, Kiuru M, Lehtonen R, Karhu A, Vilkki S, Laiho P, Eklund C, Vierimaa O, Aittomaki K, Hietala M, Sistonen P, Paetau A, Salovaara R, Herva R, Launonen V, Aaltonen LA, Multiple Leiomyoma C (2002) Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 30(4):406–410. https://doi.org/10.1038/ng849
He X, Yan B, Liu S, Jia J, Lai W, Xin X, Tang CE, Luo D, Tan T, Jiang Y, Shi Y, Liu Y, Xiao D, Chen L, Liu S, Mao C, Yin G, Cheng Y, Fan J, Cao Y, Muegge K, Tao Y (2016) Chromatin remodeling factor LSH drives cancer progression by suppressing the activity of fumarate hydratase. Cancer Res 76(19):5743–5755. https://doi.org/10.1158/0008-5472.CAN-16-0268
De Craene B, Berx G (2013) Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer 13(2):97–110. https://doi.org/10.1038/nrc3447
Sciacovelli M, Frezza C (2017) Fumarate drives EMT in renal cancer. Cell Death Differ 24(1):1–2. https://doi.org/10.1038/cdd.2016.137
Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P, Buffet A, Marcaillou C, Bertherat J, Amar L, Rustin P, De Reynies A, Gimenez-Roqueplo AP, Favier J (2013) SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 23(6):739–752. https://doi.org/10.1016/j.ccr.2013.04.018
Bardella C, Pollard PJ, Tomlinson I (2011) SDH mutations in cancer. Biochim Biophys Acta 1807(11):1432–1443. https://doi.org/10.1016/j.bbabio.2011.07.003
Wang H, Chen Y, Wu G (2016) SDHB deficiency promotes TGFβ-mediated invasion and metastasis of colorectal cancer through transcriptional repression complex SNAIL1-SMAD3/4. Transl Oncol 9(6):512–520. https://doi.org/10.1016/j.tranon.2016.09.009
Waitkus MS, Diplas BH, Yan H (2016) Isocitrate dehydrogenase mutations in gliomas. Neuro Oncol 18(1):16–26. https://doi.org/10.1093/neuonc/nov136
Saha SK, Gordan JD, Kleinstiver BP, Vu P, Najem MS, Yeo JC, Shi L, Kato Y, Levin RS, Webber JT, Damon LJ, Egan RK, Greninger P, McDermott U, Garnett MJ, Jenkins RL, Rieger-Christ KM, Sullivan TB, Hezel AF, Liss AS, Mizukami Y, Goyal L, Ferrone CR, Zhu AX, Joung JK, Shokat KM, Benes CH, Bardeesy N (2016) Isocitrate dehydrogenase mutations confer dasatinib hypersensitivity and SRC dependence in intrahepatic cholangiocarcinoma. Cancer Discov 6(7):727–739. https://doi.org/10.1158/2159-8290.CD-15-1442
Chan SM, Thomas D, Corces-Zimmerman MR, Xavy S, Rastogi S, Hong WJ, Zhao F, Medeiros BC, Tyvoll DA, Majeti R (2015) Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med 21(2):178–184. https://doi.org/10.1038/nm.3788
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of αa-ketoglutarate-dependent dioxygenases. Cancer Cell 19(1):17–30. https://doi.org/10.1016/j.ccr.2010.12.014
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O’Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, Thompson CB (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483(7390):474–478. https://doi.org/10.1038/nature10860
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462(7274):739–744. https://doi.org/10.1038/nature08617
Colvin H, Nishida N, Konno M, Haraguchi N, Takahashi H, Nishimura J, Hata T, Kawamoto K, Asai A, Tsunekuni K, Koseki J, Mizushima T, Satoh T, Doki Y, Mori M, Ishii H (2016) Oncometabolite D-2-hydroxyglurate directly induces epithelial–mesenchymal transition and is associated with distant metastasis in colorectal cancer. Sci Rep 6:36289. https://doi.org/10.1038/srep36289
Gregory PA, Bracken CP, Smith E, Bert AG, Wright JA, Roslan S, Morris M, Wyatt L, Farshid G, Lim YY, Lindeman GJ, Shannon MF, Drew PA, Khew-Goodall Y, Goodall GJ (2011) An autocrine TGF-β/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell 22(10):1686–1698. https://doi.org/10.1091/mbc.E11-02-0103
Zadran S, Arumugam R, Herschman H, Phelps ME, Levine RD (2014) Surprisal analysis characterizes the free energy time course of cancer cells undergoing epithelial-to-mesenchymal transition. Proc Natl Acad Sci USA 111(36):13235–13240. https://doi.org/10.1073/pnas.1414714111
Chio IIC, Tuveson DA (2017) ROS in cancer: the burning question. Trends Mol Med 23(5):411–429. https://doi.org/10.1016/j.molmed.2017.03.004
Hornsveld M, Dansen TB (2016) The hallmarks of cancer from a redox perspective. Antioxid Redox Signal 25(6):300–325. https://doi.org/10.1089/ars.2015.6580
Liu RM, Desai LP (2015) Reciprocal regulation of TGF-β and reactive oxygen species: a perverse cycle for fibrosis. Redox Biol 6:565–577. https://doi.org/10.1016/j.redox.2015.09.009
Ishikawa F, Kaneko E, Sugimoto T, Ishijima T, Wakamatsu M, Yuasa A, Sampei R, Mori K, Nose K, Shibanuma M (2014) A mitochondrial thioredoxin-sensitive mechanism regulates TGF-β-mediated gene expression associated with epithelial–mesenchymal transition. Biochem Biophys Res Commun 443(3):821–827. https://doi.org/10.1016/j.bbrc.2013.12.050
Rhyu DY, Yang Y, Ha H, Lee GT, Song JS, Uh ST, Lee HB (2005) Role of reactive oxygen species in TGF-β1-induced mitogen-activated protein kinase activation and epithelial–mesenchymal transition in renal tubular epithelial cells. J Am Soc Nephrol 16(3):667–675. https://doi.org/10.1681/ASN.2004050425
Kinugasa H, Whelan KA, Tanaka K, Natsuizaka M, Long A, Guo A, Chang S, Kagawa S, Srinivasan S, Guha M, Yamamoto K, St Clair DK, Avadhani NG, Diehl JA, Nakagawa H (2015) Mitochondrial SOD2 regulates epithelial–mesenchymal transition and cell populations defined by differential CD44 expression. Oncogene 34(41):5229–5239. https://doi.org/10.1038/onc.2014.449
Lambeth JD (2004) NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 4(3):181–189. https://doi.org/10.1038/nri1312
Hiraga R, Kato M, Miyagawa S, Kamata T (2013) Nox4-derived ROS signaling contributes to TGF-β-induced epithelial–mesenchymal transition in pancreatic cancer cells. Anticancer Res 33(10):4431–4438
Mondol AS, Tonks NK, Kamata T (2014) Nox4 redox regulation of PTP1B contributes to the proliferation and migration of glioblastoma cells by modulating tyrosine phosphorylation of coronin-1C. Free Radic Biol Med 67:285–291. https://doi.org/10.1016/j.freeradbiomed.2013.11.005
Boudreau HE, Casterline BW, Rada B, Korzeniowska A, Leto TL (2012) Nox4 involvement in TGF-β and SMAD3-driven induction of the epithelial-to-mesenchymal transition and migration of breast epithelial cells. Free Radic Biol Med 53(7):1489–1499. https://doi.org/10.1016/j.freeradbiomed.2012.06.016
Iizuka D, Sasatani M, Barcellos-Hoff MH, Kamiya K (2017) Hydrogen peroxide enhances TGFβ-mediated epithelial-to-mesenchymal transition in human mammary epithelial MCF-10A cells. Anticancer Res 37(3):987–995. https://doi.org/10.21873/anticanres.11408
Ma Q (2013) Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53:401–426. https://doi.org/10.1146/annurev-pharmtox-011112-140320
Arfmann-Knubel S, Struck B, Genrich G, Helm O, Sipos B, Sebens S, Schafer H (2015) The crosstalk between Nrf2 and TGF-β1 in the epithelial–mesenchymal transition of pancreatic duct epithelial cells. PLoS One 10(7):e0132978. https://doi.org/10.1371/journal.pone.0132978
Zhou W, Mo X, Cui W, Zhang Z, Li D, Li L, Xu L, Yao H, Gao J (2016) Nrf2 inhibits epithelial–mesenchymal transition by suppressing snail expression during pulmonary fibrosis. Sci Rep 6:38646. https://doi.org/10.1038/srep38646
Fahy E, Cotter D, Sud M, Subramaniam S (2011) Lipid classification, structures and tools. Biochim Biophys Acta (BBA) Mol Cell Biol Lipids 1811(11):637–647. https://doi.org/10.1016/j.bbalip.2011.06.009
Santos CR, Schulze A (2012) Lipid metabolism in cancer. FEBS J 279(15):2610–2623. https://doi.org/10.1111/j.1742-4658.2012.08644.x
Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L, Cao Y (2017) Emerging roles of lipid metabolism in cancer metastasis. Mol Cancer 16(1):76. https://doi.org/10.1186/s12943-017-0646-3
Tisza MJ, Zhao W, Fuentes JS, Prijic S, Chen X, Levental I, Chang JT (2016) Motility and stem cell properties induced by the epithelial-mesenchymal transition require destabilization of lipid rafts. Oncotarget 7(32):51553. https://doi.org/10.18632/oncotarget.9928
Li J, Dong L, Wei D, Wang X, Zhang S, Li H (2014) Fatty acid synthase mediates the epithelial–mesenchymal transition of breast cancer cells. Int J Biol Sci 10(2):171
Nath A, Li I, Roberts LR, Chan C (2015) Elevated free fatty acid uptake via CD36 promotes epithelial–mesenchymal transition in hepatocellular carcinoma. Sci Rep 5:14752. https://doi.org/10.1038/srep14752
Nath A, Chan C (2016) Genetic alterations in fatty acid transport and metabolism genes are associated with metastatic progression and poor prognosis of human cancers. Sci Rep 6:18669. https://doi.org/10.1038/srep18669
Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, Berenguer A, Prats N, Toll A, Hueto JA, Bescos C, Di Croce L, Benitah SA (2017) Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541(7635):41–45. https://doi.org/10.1038/nature20791
Singh R, Yadav V, Kumar S, Saini N (2015) MicroRNA-195 inhibits proliferation, invasion and metastasis in breast cancer cells by targeting FASN, HMGCR, ACACA and CYP27B1. Sci Rep 5:17454. https://doi.org/10.1038/srep17454
Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA, Thompson CB (2005) ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 8(4):311–321. https://doi.org/10.1016/j.ccr.2005.09.008
Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, Verhoeven G, Swinnen JV (2007) Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res 67(17):8180–8187. https://doi.org/10.1158/0008-5472.Can-07-0389
Yang L, Zhang F, Wang X, Tsai Y, Chuang KH, Keng PC, Lee SO, Chen Y (2016) A FASN-TGF-β1-FASN regulatory loop contributes to high EMT/metastatic potential of cisplatin-resistant non-small cell lung cancer. Oncotarget 7(34):55543–55554. https://doi.org/10.18632/oncotarget.10837
Jiang L, Xiao L, Sugiura H, Huang X, Ali A, Kuro-o M, Deberardinis RJ, Boothman DA (2015) Metabolic reprogramming during TGFβ1-induced epithelial-to-mesenchymal transition. Oncogene 34(30):3908–3916. https://doi.org/10.1038/onc.2014.321
Hannun YA, Obeid LM (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9(2):139–150. https://doi.org/10.1038/nrm2329
Ogretmen B (2018) Sphingolipid metabolism in cancer signalling and therapy. Nat Rev Cancer 18(1):33–50. https://doi.org/10.1038/nrc.2017.96
Guan F, Handa K, Hakomori SI (2009) Specific glycosphingolipids mediate epithelial-to-mesenchymal transition of human and mouse epithelial cell lines. Proc Natl Acad Sci USA 106(18):7461–7466. https://doi.org/10.1073/pnas.0902368106
Guan F, Schaffer L, Handa K, Hakomori SI (2010) Functional role of gangliotetraosylceramide in epithelial-to-mesenchymal transition process induced by hypoxia and by TGF-β. FASEB J 24(12):4889–4903. https://doi.org/10.1096/fj.10-162107
Mathow D, Chessa F, Rabionet M, Kaden S, Jennemann R, Sandhoff R, Grone HJ, Feuerborn A (2015) Zeb1 affects epithelial cell adhesion by diverting glycosphingolipid metabolism. EMBO Rep 16(3):321–331. https://doi.org/10.15252/embr.201439333
Milara J, Navarro R, Juan G, Peiro T, Serrano A, Ramon M, Morcillo E, Cortijo J (2012) Sphingosine-1-phosphate is increased in patients with idiopathic pulmonary fibrosis and mediates epithelial to mesenchymal transition. Thorax 67(2):147–156. https://doi.org/10.1136/thoraxjnl-2011-200026
Wang C, Mao J, Redfield S, Mo Y, Lage JM, Zhou X (2014) Systemic distribution, subcellular localization and differential expression of sphingosine-1-phosphate receptors in benign and malignant human tissues. Exp Mol Pathol 97(2):259–265. https://doi.org/10.1016/j.yexmp.2014.07.013
Zeng Y, Yao X, Chen L, Yan Z, Liu J, Zhang Y, Feng T, Wu J, Liu X (2016) Sphingosine-1-phosphate induced epithelial–mesenchymal transition of hepatocellular carcinoma via an MMP-7/syndecan-1/TGF-β autocrine loop. Oncotarget 7(39):63324–63337. https://doi.org/10.18632/oncotarget.11450
Huitema K, van den Dikkenberg J, Brouwers JF, Holthuis JC (2004) Identification of a family of animal sphingomyelin synthases. EMBO J 23(1):33–44. https://doi.org/10.1038/sj.emboj.7600034
Yamaoka S, Miyaji M, Kitano T, Umehara H, Okazaki T (2004) Expression cloning of a human cDNA restoring sphingomyelin synthesis and cell growth in sphingomyelin synthase-defective lymphoid cells. J Biol Chem 279(18):18688–18693. https://doi.org/10.1074/jbc.M401205200
Lingwood D, Simons K (2010) Lipid rafts as a membrane-organizing principle. Science (New York, NY) 327(5961):46–50. https://doi.org/10.1126/science.1174621
Taniguchi M, Okazaki T (2014) The role of sphingomyelin and sphingomyelin synthases in cell death, proliferation and migration—from cell and animal models to human disorders. Biochim Biophys Acta (BBA) Mol Cell Biol Lipids 1841(5):692–703. https://doi.org/10.1016/j.bbalip.2013.12.003
Liu S, Hou H, Zhang P, Wu Y, He X, Li H, Yan N (2018) Sphingomyelin synthase 1 regulates the epithelial-to-mesenchymal transition mediated by the TGF-β/Smad pathway in MDA-MB-231 cells. Mol Med Rep. https://doi.org/10.3892/mmr.2018.9722
Gouw AM, Eberlin LS, Margulis K, Sullivan DK, Toal GG, Tong L, Zare RN, Felsher DW (2017) Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma. Proc Natl Acad Sci USA 114(17):4300–4305. https://doi.org/10.1073/pnas.1617709114
Edmunds LR, Sharma L, Kang A, Lu J, Vockley J, Basu S, Uppala R, Goetzman ES, Beck ME, Scott D, Prochownik EV (2014) c-Myc programs fatty acid metabolism and dictates acetyl-CoA abundance and fate. J Biol Chem 289(36):25382–25392. https://doi.org/10.1074/jbc.M114.580662
Arlauckas SP, Popov AV, Delikatny EJ (2016) Choline kinase α—putting the ChoK-hold on tumor metabolism. Prog Lipid Res 63:28–40. https://doi.org/10.1016/j.plipres.2016.03.005
Aoyama C, Liao H, Ishidate K (2004) Structure and function of choline kinase isoforms in mammalian cells. Prog Lipid Res 43(3):266–281. https://doi.org/10.1016/j.plipres.2003.12.001
Huang C, Freter C (2015) Lipid metabolism, apoptosis and cancer therapy. Int J Mol Sci 16(1):924–949. https://doi.org/10.3390/ijms16010924
Bertilsson H, Tessem MB, Flatberg A, Viset T, Gribbestad I, Angelsen A, Halgunset J (2012) Changes in gene transcription underlying the aberrant citrate and choline metabolism in human prostate cancer samples. Clin Cancer Res 18(12):3261–3269. https://doi.org/10.1158/1078-0432.Ccr-11-2929
Ramirez de Molina A, Rodriguez-Gonzalez A, Gutierrez R, Martinez-Pineiro L, Sanchez J, Bonilla F, Rosell R, Lacal J (2002) Overexpression of choline kinase is a frequent feature in human tumor-derived cell lines and in lung, prostate, and colorectal human cancers. Biochem Biophys Res Commun 296(3):580–583. https://doi.org/10.1016/s0006-291x(02)00920-8
Ramirez de Molina A, Gutierrez R, Ramos MA, Silva JM, Silva J, Bonilla F, Sanchez JJ, Lacal JC (2002) Increased choline kinase activity in human breast carcinomas: clinical evidence for a potential novel antitumor strategy. Oncogene 21(27):4317–4322. https://doi.org/10.1038/sj.onc.1205556
Koch K, Hartmann R, Schroter F, Suwala AK, Maciaczyk D, Kruger AC, Willbold D, Kahlert UD, Maciaczyk J (2016) Reciprocal regulation of the cholinic phenotype and epithelial–mesenchymal transition in glioblastoma cells. Oncotarget 7(45):73414–73431. https://doi.org/10.18632/oncotarget.12337
Granata A, Nicoletti R, Tinaglia V, De Cecco L, Pisanu ME, Ricci A, Podo F, Canevari S, Iorio E, Bagnoli M, Mezzanzanica D (2014) Choline kinase-α by regulating cell aggressiveness and drug sensitivity is a potential druggable target for ovarian cancer. Br J Cancer 110(2):330–340. https://doi.org/10.1038/bjc.2013.729
Mori N, Glunde K, Takagi T, Raman V, Bhujwalla ZM (2007) Choline kinase down-regulation increases the effect of 5-fluorouracil in breast cancer cells. Cancer Res 67(23):11284–11290. https://doi.org/10.1158/0008-5472.Can-07-2728
Shah T, Wildes F, Penet MF, Winnard PT Jr, Glunde K, Artemov D, Ackerstaff E, Gimi B, Kakkad S, Raman V, Bhujwalla ZM (2010) Choline kinase overexpression increases invasiveness and drug resistance of human breast cancer cells. NMR Biomed 23(6):633–642. https://doi.org/10.1002/nbm.1510
Hernando E, Sarmentero-Estrada J, Koppie T, Belda-Iniesta C, Ramirez de Molina V, Cejas P, Ozu C, Le C, Sanchez JJ, Gonzalez-Baron M, Koutcher J, Cordon-Cardo C, Bochner BH, Lacal JC, Ramirez de Molina A (2009) A critical role for choline kinase-α in the aggressiveness of bladder carcinomas. Oncogene 28(26):2425–2435. https://doi.org/10.1038/onc.2009.91
Hu L, Wang RY, Cai J, Feng D, Yang GZ, Xu QG, Zhai YX, Zhang Y, Zhou WP, Cai QP (2016) Overexpression of CHKA contributes to tumor progression and metastasis and predicts poor prognosis in colorectal carcinoma. Oncotarget 7(41):66660–66678. https://doi.org/10.18632/oncotarget.11433
Mariotto E, Viola G, Ronca R, Persano L, Aveic S, Bhujwalla ZM, Mori N, Accordi B, Serafin V, Lopez-Cara LC, Bortolozzi R (2018) Choline kinase Α inhibition by EB-3D triggers cellular senescence, reduces tumor growth and metastatic dissemination in breast cancer. Cancers (Basel) 10(10):391. https://doi.org/10.3390/cancers10100391
Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP (2017) Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol 14(1):11–31. https://doi.org/10.1038/nrclinonc.2016.60
Ooi AT, Gomperts BN (2015) Molecular pathways: targeting cellular energy metabolism in cancer via inhibition of SLC2A1 and LDHA. Clin Cancer Res 21(11):2440–2444. https://doi.org/10.1158/1078-0432.CCR-14-1209
Ma Y, Wang W, Idowu MO, Oh U, Wang XY, Temkin SM, Fang X (2018) Ovarian cancer relies on glucose transporter 1 to fuel glycolysis and growth: anti-tumor activity of BAY-876. Cancers (Basel) 11(1):33. https://doi.org/10.3390/cancers11010033
Maschek G, Savaraj N, Priebe W, Braunschweiger P, Hamilton K, Tidmarsh GF, Young LRD, Lampidis TJ (2004) 2-Deoxy-d-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res 64(1):31–34. https://doi.org/10.1158/0008-5472.can-03-3294
Papaldo P, Lopez M, Cortesi E, Cammilluzzi E, Antimi M, Terzoli E, Lepidini G, Vici P, Barone C, Ferretti G, Di Cosimo S, Nistico C, Carlini P, Conti F, Di Lauro L, Botti C, Vitucci C, Fabi A, Giannarelli D, Marolla P (2003) Addition of either lonidamine or granulocyte colony-stimulating factor does not improve survival in early breast cancer patients treated with high-dose epirubicin and cyclophosphamide. J Clin Oncol 21(18):3462–3468. https://doi.org/10.1200/JCO.2003.03.034
Mondal S, Roy D, Sarkar Bhattacharya S, Jin L, Jung D, Zhang S, Kalogera E, Staub J, Wang Y, Xuyang W, Khurana A, Chien J, Telang S, Chesney J, Tapolsky G, Petras D, Shridhar V (2019) Therapeutic targeting of PFKFB3 with a novel glycolytic inhibitor PFK158 promotes lipophagy and chemosensitivity in gynecologic cancers. Int J Cancer 144(1):178–189. https://doi.org/10.1002/ijc.31868
Craig D, Logsdon WL, Abbruzzese JL (2013) Cancer metabolism and its therapeutic implications. J Cell Sci Ther 4(2):143. https://doi.org/10.4172/2157-7013.1000143
Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G (2013) Metabolic targets for cancer therapy. Nat Rev Drug Discov 12(11):829–846. https://doi.org/10.1038/nrd4145
Boudreau A, Purkey HE, Hitz A, Robarge K, Peterson D, Labadie S, Kwong M, Hong R, Gao M, Del Nagro C, Pusapati R, Ma S, Salphati L, Pang J, Zhou A, Lai T, Li Y, Chen Z, Wei B, Yen I, Sideris S, McCleland M, Firestein R, Corson L, Vanderbilt A, Williams S, Daemen A, Belvin M, Eigenbrot C, Jackson PK, Malek S, Hatzivassiliou G, Sampath D, Evangelista M, O’Brien T (2016) Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat Chem Biol 12(10):779–786. https://doi.org/10.1038/nchembio.2143
Michelakis ED, Webster L, Mackey JR (2008) Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 99(7):989–994. https://doi.org/10.1038/sj.bjc.6604554
Beloribi-Djefaflia S, Vasseur S, Guillaumond F (2016) Lipid metabolic reprogramming in cancer cells. Oncogenesis 5:e189. https://doi.org/10.1038/oncsis.2015.49
Flavin R, Peluso S, Nguyen PL, Loda M (2010) Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol 6(4):551–562
Menendez JA, Lupu R (2017) Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin Ther Targets 21(11):1001–1016. https://doi.org/10.1080/14728222.2017.1381087
Loomba R, Kayali Z, Noureddin M, Ruane P, Lawitz EJ, Bennett M, Wang L, Harting E, Tarrant JM, McColgan BJ, Chung C, Ray AS, Subramanian GM, Myers RP, Middleton MS, Lai M, Charlton M, Harrison SA (2018) GS-0976 reduces hepatic steatosis and fibrosis markers in patients with nonalcoholic fatty liver disease. Gastroenterology 155(5):1463–1473. https://doi.org/10.1053/j.gastro.2018.07.027
Prado-Garcia H, Sanchez-Garcia FJ (2017) Editorial: Immuno-metabolism in tumor microenvironment. Front Immunol 8:374. https://doi.org/10.3389/fimmu.2017.00374
Yu YR, Ho PC (2019) Sculpting tumor microenvironment with immune system: from immunometabolism to immunoediting. Clin Exp Immunol 197(2):153–160. https://doi.org/10.1111/cei.13293
Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, Tonc E, Schreiber RD, Pearce EJ, Pearce EL (2015) Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162(6):1229–1241. https://doi.org/10.1016/j.cell.2015.08.016
Acknowledgements
We thank our colleagues for valuable discussion.
Funding
Our research on metabolic reprogramming in cancer is supported by CGC.nl and by Chinese Scholarship Council.
Author information
Authors and Affiliations
Contributions
All authors contributed to writing the review. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Consent for publication
All authors read and approved the final manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hua, W., ten Dijke, P., Kostidis, S. et al. TGFβ-induced metabolic reprogramming during epithelial-to-mesenchymal transition in cancer. Cell. Mol. Life Sci. 77, 2103–2123 (2020). https://doi.org/10.1007/s00018-019-03398-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-019-03398-6