
Abstract
Rotary ATPases are unique rotary molecular motors that function as energy conversion machines. Among all known rotary ATPases, F1-ATPase is the best characterized rotary molecular motor. There are many high-resolution crystal structures and the rotation dynamics have been investigated in detail by extensive single-molecule studies. In contrast, knowledge on the structure and rotation dynamics of V1-ATPase, another rotary ATPase, has been limited. However, recent high-resolution structural studies and single-molecule studies on V1-ATPase have provided new insights on how the catalytic sites in this molecular motor change its conformation during rotation driven by ATP hydrolysis. In this review, we summarize recent information on the structural features and rotary dynamics of V1-ATPase revealed from structural and single-molecule approaches and discuss the possible chemomechanical coupling scheme of V1-ATPase with a focus on differences between rotary molecular motors.
Similar content being viewed by others
Introduction
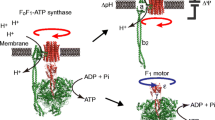
All living cells use a chemical fuel called adenosine triphosphate (ATP) to maintain the functions required for life, such as protein biosynthesis, muscle contraction, and brain activity. Therefore, ATP is often referred to as the energy currency for the cell. Under aerobic conditions, the majority of ATP is produced by the F-type ATP synthase [1]. F-type ATP synthases (also known as F-ATPases) are ubiquitous rotary motor enzymes found in the inner membrane of mitochondria, the thylakoid membrane of chloroplasts, and the plasma membranes of bacteria. They catalyze ATP synthesis from ADP and inorganic phosphate using the energy of ion (proton or sodium) translocation caused by transmembrane electrochemical potential (proton or sodium ions) and, when operating in reverse, they also generate an electrochemical potential difference of ions using the energy released by ATP hydrolysis [2,3,4].
Eukaryotic vacuolar-type ATPases (V-ATPases) are also rotary motor enzymes; they are evolutionarily and functionally related to F-type ATP synthases [5, 6]. They function in reverse of F-type ATP synthases, that is, they transport ions (protons or sodium ions) across the membrane using the energy derived from ATP hydrolysis. Acidification of vesicles by intracellular V-ATPases is important for various cellular processes, including receptor-mediated endocytosis, membrane trafficking, and protein processing and degradation. They also function on the plasma membrane of certain cells, such as tumor cells, renal intercalated cells, and osteoclasts. Aberrant function of V-ATPase is associated with a number of human diseases including tumor metastasis, distal renal tubular acidosis, and osteoporosis. Therefore, they are considered as potential drug targets [5, 7]. V-ATPases are found in some bacteria, such as Thermus thermophilus (T. thermophilus) and Enterococcus hirae (E. hirae). V-ATPase from T. thermophilus functions as an ATP synthase under physiological conditions [8, 9]. Therefore, this enzyme is sometimes called A-type ATPase found in Archeae (A-ATPase). A-ATPases function as ATP synthases similar to the F-type ATP synthase, although the structure and subunit composition of A-ATPases are more similar to those of V-ATPases (Fig. 1a) [10,11,12]. V-ATPase from E. hirae functions as a sodium ion pump similar in nature to eukaryotic V-ATPase, and plays an important role in maintaining sodium homeostasis in cells under an alkaline environment [13,14,15].

Schematic illustrations of bacterial rotary ATPases. a A/V-type ATPases/synthases are found in Archeae and some bacterial taxa. They are composed of a soluble catalytic core A1/V1 motor (A3B3DF) and a membrane integral Ao/Vo motor (ac n d; where n is the copy number of the c subunits). The A1/V1 and Ao/Vo motors are connected by the two peripheral stalks (EG). The N- and C-terminal domains of a subunit are shown as aNT and aCT, respectively. b Bacterial F-type ATPases/synthases are structurally different from bacterial A/V-type ATPases/synthases. The F1 (α3β3γδε) and Fo motors (ab2c n ) are connected by only one peripheral stalk (b2)
All rotary ATPases are unique rotary molecular motors that function as energy conversion machines with a similar architecture and rotary catalytic mechanism [5, 11, 12, 16]. They are large, multi-subunit complexes composed of a hydrophilic F1/V1/A1 motor for ATP synthesis/hydrolysis and a membrane-embedded Fo/Vo/Ao motor for ion transport. The bacterial F1/V1/A1 and Fo/Vo/Ao motors are connected by one central stalk and one or two peripheral stalks (Fig. 1). Interestingly, V-ATPases in eukaryotes have three peripheral stalks, although there is only one peripheral stalk in eukaryotic F-ATPases [16,17,18]. The peripheral stalks of eukaryotic V-ATPases play an important role in reversible dissociation of V1 motor from Vo motor with silencing of the ATP hydrolysis activity of the free V1 motor [18], in contrast to F1 motor which will rapidly hydrolyze ATP when isolated. This implies the fact that it was necessary for V-ATPases to evolve a regulatory mechanism for keeping the dissociated V1 motors catalytically inactive to prevent wasteful energy consumption.
Among all known rotary ATPases, the hydrophilic portion of F-ATPase (F1-motor or F1-ATPase) is the best characterized; there are high-resolution crystal structures of several rotational states [19,20,21,22,23,24,25,26,27,28] and the chemomechanical coupling scheme has been revealed in detail by extensive single-molecule studies [29,30,31,32,33,34,35,36,37,38,39,40]. Relatively little is known about the structure and rotation scheme of V1-motors (V1-ATPase) [41,42,43]. Recently, high-resolution crystal structures of several rotational states of V1-ATPase from E. hirae have been determined [44, 45]. Furthermore, basic rotary dynamics of this V1-ATPase have been revealed by single-molecule studies [46,47,48,49]. These studies have provided new insights into the rotation mechanism of V1-ATPase. In the present review, we discuss recent findings on the structural features and rotary dynamics of bacterial V1-ATPase revealed from structural and single-molecule studies with a focus on differences from the properties of F1-ATPase.
V1-ATPase: structural studies
In the bacterial V-ATPase, the catalytic V1 moiety is composed of A, B, D, and F subunits, in which three alternately arranged A and B subunits form a hexameric stator A3B3 ring. The central rotary shaft of D and F subunits penetrates the central cavity of the A3B3 ring and rotates using the energy of ATP hydrolysis [41,42,43,44]. Unlike the isolated eukaryotic V1 moiety in which subunit H inhibits its activity [18], the isolated bacterial V1 moiety can generally catalyze ATP hydrolysis and hence is called V1-ATPase. Structural studies have been conducted using a V1-ATPase from the thermophilic eubacterium T. thermophilus, which has high stability. The crystal structure of the A3B3 subcomplex from T. thermophilus was determined at 2.8 Å resolution [41]. The diameter of the A3B3 subcomplex is larger than that of the α3β3 subcomplex in F1-ATPase, because it includes an outward protrusion domain in the A subunit (Fig. 2a, green circles), termed the “non-homologous region” that is absent from β subunit; the structure also provides molecular information about the B–A interface. The catalytic sites are located at the interfaces of the A and B subunits (Fig. 2a, red arrows), with the majority of the catalytic residues residing in the A subunits, similar to the catalytic β subunits and the non-catalytic α subunits in F1-ATPase [19, 27]. The overall structure of V1-ATPase (A3B3DF complex) from T. thermophilus was first determined at 4.5–4.8 Å resolution (Fig. 2b) [42]. This structure provided the initial information about the position and orientation of the rotor DF subunits in the A3B3 ring and revealed structural similarities and differences between V1- and F1-ATPase. However, the lack of high-resolution structural information for the overall V1-ATPase from T. thermophilus limits our understanding of its molecular architecture and operation. Meanwhile, high-resolution crystal structures of the A3B3 ring and entire V1-ATPase from E. hirae have recently been solved with and without bound nucleotides [44, 45].

Crystal structures of A3B3 subcomplex and V1-ATPase from T. thermophilus. a Crystal structure of the nucleotide-free A3B3 subcomplex from T. thermophilus determined at 2.8 Å resolution (PDB ID: 3GQB) [41]. Side view (left) and Top view (right) from the membrane side. Only the C-terminal domains of the A3B3 ring are shown in top view to clarify the distinct conformations of individual A or B subunits. The A and B subunits are shown in blue and magenta, respectively. Each A and B subunit consists of the N-terminal β-barrel (N), the central α/β domain (α/β), and the C-terminal helical domain (C). Green circles indicate the “non-homologous region” in the A subunit. Red arrows indicate the catalytic sites. b Overall structure of V1-ATPase (A3B3DF) from T. thermophilus determined at 4.5 Å resolution (PDB ID: 3A5C) [42]. Side view (left) and top view (right) from the membrane side. Only the C-terminal domains of the A3B3 ring and the D subunit are shown in top view. The A, B, D, and F subunits are shown in blue, magenta, green, and red, respectively. AW, BW pair shows a wide-open conformation, as observed in a αEβE pair in F1-ATPase [19]. ANBN and AN′BN′ pairs show a narrowly closed conformation, as do the αTPβTP and αDPβDP pairs in F1-ATPase [19]. The crystals were obtained by co-crystallization with Mg2+ ADP and aluminium fluoride. Strong electron densities which presumably correspond to the phosphate groups of bound-ADP were found in the ANBN and AN’BN’ pairs (orange arrowheads)
Structure of A3B3 ring from E. hirae: asymmetric structure
The high-resolution crystal structures of the A3B3 ring from E. hirae were solved with and without a non-hydrolyzable ATP analog [adenosine 5′-(β,γ-imido)triphosphate or AMPPNP] at 3.4 and 2.8 Å resolution, respectively (Fig. 3) [44]. The overall architecture of A3B3 from E. hirae is similar to that of α3β3 in F1-ATPase, but the structures show some differences.

Crystal structures of A3B3 subcomplex from E. hirae. a Crystal structure of the nucleotide-free A3B3 subcomplex from E. hirae determined at 2.8 Å resolution (PDB ID: 3VR2) [44]. Each A and B subunit consists of the N-terminal β-barrel (N), the central α/β domain (α/β), and the C-terminal helical domain (C) as seen in A3B3 from T. thermophilus (Fig. 2a). b Crystal structure of the nucleotide-bound A3B3 subcomplex from E. hirae determined at 3.4 Å resolution (PDB ID: 3VR3) [44]. Two AMPPNP molecules are bound to the ‘bound’ sites (indicated by red arrowheads). Top views from the membrane side are shown on the left and center. Only the C-terminal domains of the A3B3 rings are shown in the center to clarify the distinct conformations of individual A or B subunits and the different structures of the three catalytic sites. On the right, conformations of the individual A and B subunits superimposed at the N-terminal β-barrel domain (white) are shown. O and O′ open conformation, C closed conformation
Each A and B subunit consists of an N-terminal β-barrel, central α/β domain, and C-terminal helical domain (Fig. 3a, right). Superimposition of the N-terminal β-barrel part of three A or B subunits shows that the conformations of each A and B subunit are not identical. If one of the A subunits is in the closed conformation (C), the other two A subunits show open conformations (O or O′). Similarly, one of the B subunits takes the closed form (C) and the other two take open conformations (O or O′) (Fig. 3a), resulting in the asymmetry of the A3B3 ring. Interestingly, even in the absence of nucleotides and central rotor DF subunits, each catalytic site shows the three distinct states. In contrast, F1-ATPase shows a threefold symmetric structure with three identical catalytic sites in the absence of bound nucleotides [50, 51]; accordingly, the asymmetric structure of the stator ring is not observed in the case of F1-ATPase. In the presence of high concentration of AMPPNP (5 mM), the A3B3 ring binds two AMPPNP molecules (Fig. 3b), resulting in changes in the conformation of A (O′ to C) and B (O to O′) subunits, but the ACBO′ pair shows little conformational change upon AMPPNP binding. Therefore, the three catalytic sites (AOBC, AO′BO, and ACBO′) are termed ‘empty’, ‘bindable’, and ‘bound’ sites, respectively (Fig. 3a, b).
Two AMPPNP-bound structure of V1-ATPase: catalytic dwell state
In addition to the A3B3 ring structures, the nucleotide-free entire V1-ATPase from E. hirae (eV1) was also determined at 2.2 Å resolution [44]. Insertion of the rotor DF subunits into the stator A3B3 ring induces conformational changes in the A and B subunits, even in the absence of bound nucleotides (Fig. 4a). This results in changes in the conformations of A and B subunits from the closed (C) to the more closed ‘closer’ (CR) conformation and from the open (O') to closer (CR) conformation, respectively. Consequently, eV1 shows three different catalytic sites termed ‘empty’, ‘bound’, and ‘tight’ sites (Fig. 4a). Furthermore, by soaking eV1 in AMPPNP, the structure of nucleotide-bound V1-ATPase (bV1 or 2ATPV1) was also determined at 2.7 Å resolution (Fig. 4b) [44, 45]. Two AMPPNP molecules were bound to the binding sites of the ‘bound’ and ‘tight’ sites of eV1, but the overall structure was very similar to that of eV1 (Fig. 4a, b). Even in the presence of high AMPPNP (2 mM), no electron density peak for AMPPNP was found in the ‘empty’ site, indicating that it has a very low affinity for AMPPNP. Comparison of the ‘tight’ and ‘bound’ sites in bV1 revealed the movement of the Arg-finger (Arg-350) in the ‘tight’ site closer to the γ-phosphate relative to the ‘bound’ site (Fig. 4c). This γ-phosphate moved closer to Glu-261 in the A subunit which is essential for ATPase activity in yeast V1-ATPase [52]. The corresponding Glu-188 of the β subunit in bovine mitochondrial F1-ATPase is an essential residue for ATP hydrolysis and interacts with the γ-phosphate of the nucleotide and lytic water molecules [19, 28, 53]. The closer proximity of the Arg-finger to the γ-phosphate may enhance the ATP hydrolysis reaction. Therefore, it is possible that the ‘tight’ site corresponds to the catalytic site waiting for ATP hydrolysis and this structure corresponds to the catalytic dwell state (Fig. 5a). Similar nucleotide-free and nucleotide-bound structures of yeast mitochondrial F1-ATPase have been reported [25]. These results suggest that interactions between the rotor and stator are as crucial as nucleotide binding for determining the structure of the catalytic sites of rotary ATPases.

Crystal structures of entire V1-ATPase from E. hirae. a Crystal structure of the nucleotide-free V1-ATPase from E. hirae determined at 2.2 Å resolution (PDB ID: 3VR4) [44]. b Crystal structure of the nucleotide-bound V1-ATPase from E. hirae determined at 2.7 Å resolution (bV1, PDB ID: 3VR6) [44]. Two AMPPNP molecules are bound to the ‘bound’ and the ‘tight’ sites (indicated by red arrowheads). Top views from the membrane side are shown on the left. Only the C-terminal domains of the A3B3 rings and the α-helical coiled-coil portion of the D subunit are shown. On the right, conformations of the individual A subunit superimposed at the N-terminal β-barrel domain (white) are shown. O open conformation, C closed conformation, CR more closed ‘closer’ conformation. Two AMPPNP-bound V1-ATPase shows an almost identical structure to the nucleotide-free V1-ATPase. c Nucleotide-binding site of the bV1 (PDB ID: 3VR6). Superposition of the ‘bound’ site (transparent grey) and the ‘tight’ site (colored) of the bV1

Crystal structures of two AMPPNP-bound V1-ATPase (bV1), two ADP-bound V1-ATPase (2ADPV1), and three ADP-bound V1-ATPase (3ADPV1) from E. hirae. a Crystal structure of two AMPPNP-bound V1-ATPase (bV1, 2.7 Å resolution, PDB ID: 3VR6) [44]. b Crystal structure of two ADP-bound V1-ATPase (2ADPV1, 3.3 Å resolution, PDB ID: 5KNB) [45]. c Crystal structure of three ADP-bound V1-ATPase (3ADPV1, 3.0 Å resolution, PDB ID: 5KNC) [45]. Top views of the C-terminal domain of A3B3 rings and central rotor D subunit (green) viewed from the membrane side are shown on the left. Red, orange, and magenta arrowheads indicate the catalytic sites that bind to AMPPNP, ADP, and sulfate, respectively. Conformations of the individual A and B subunits superimposed at the N-terminal β-barrel domain (white) are shown on the right. O and O′ open conformation, HC half-closed conformation, C closed conformation, CR more closed ‘closer’ conformation
Two ADP-bound structure of V1-ATPase: ATP-binding dwell state
More recently, the crystal structures of two other nucleotide-bound states were reported [45], i.e., the two ADP-bound structure (2ADPV1) and the three ADP-bound structure (3ADPV1). When soaking the eV1 crystals in 20 µM ADP, it binds to the ‘bound’ and ‘tight’ sites of eV1, as in the case of bV1, and the two ADP-bound structure was solved at 3.3 Å resolution (Fig. 5b). ADP binding to the ‘tight’ site in eV1 induces changes in the A and B subunits to more open conformations (CR to C in A subunit; CR to C′ in B subunit), but the ‘bound’ site in eV1 shows no conformational change upon ADP binding. The observed conformational changes result in the tilting of rotor DF subunits towards the ADP-bound site (ACBC′) (Fig. 5b). Interestingly, the ‘empty’ site shows a cooperative conformational change without ADP binding. The newly found catalytic sites (ACBC′ and AO′BO′′) are termed ‘ADP-bound’ and ‘bindable-like’ sites, respectively. The ‘bindable-like’ site is similar to the ‘bindable’ site in the A3B3 structure (Fig. 3a) and takes a more open conformation than that of the ‘empty’ site. Therefore, the ‘bindable-like’ site is considered to be the catalytic site waiting for ATP binding, and the 2ADPV1 structure is regarded as the ATP-binding dwell state.
Three ADP-bound structure of V1-ATPase: ADP-release dwell state
The 3ADPV1 structure was solved at 3.0 Å resolution by soaking the eV1 crystals in 2 mM ADP [45]. In this structure, all three catalytic sites are occupied by ADP and, in addition, a sulfate is bound to one catalytic site (Fig. 5c, magenta arrowhead). A comparison between 2ADPV1 and 3ADPV1 structures shows that ADP (and sulfate) binding to the ‘bindable-like’ site in 2ADPV1 induces a conformational change in the A subunit (O′) to the ‘half-closed’ conformation (HC), whereas the B subunit (O′′) shows no conformational change. This catalytic site (AHCBO′′) in 3ADPV1 is termed a ‘half-closed’ site. The conformational change in the catalytic site from the ‘bindable-like’ site to the ‘half-closed’ site induces a conformational change of the ‘ADP-bound’ site to a more tight-like conformation (ACR′BCR′). This shifted ‘ADP-bound’ site in 3ADPV1 is termed a ‘tight-like’ site, because the nucleotide-binding site is more similar to that of the ‘tight’ site than to that of the ‘ADP-bound’ site (Fig. 6). The β-phosphate of ADP in the ‘tight-like’ site is more distant from the surrounding interacting residues compared to that in the ‘ADP-bound’ site, suggesting that an ADP will be easily released from the ‘tight-like’ site (Fig. 6) [45]. Therefore, the “tight-like” site is considered to be a dwelling state before ADP release and the 3ADPV1 structure is regarded as the ADP-release dwell state.

Nucleotide-binding site of the three ADP-bound V1-ATPase (3ADPV1). Nucleotide-binding site of the “tight-like” site in 3ADPV1 (colored) is superimposed at the adenosine onto those (transparent grey) of the “ADP-bound” site in 2ADPV1 (left) and the “tight” site in bV1 (right)
Structural difference between F1-and V1-ATPase: conformational features
A comparison of the two AMPPNP-bound structures of V1-ATPase from E. hirae [44, 45] and F1-ATPase from bovine mitochondria [28] is shown in Fig. 7. Both nucleotide-binding sites show very similar arrangements of catalytically important residues and nucleotides (Fig. 7a). However, the overall structures of A and B subunits in V1-ATPase show some differences from those of the β and α subunits in F1-ATPase. The non-catalytic B subunit of this V1-ATPase does not bind to a nucleotide, whereas the non-catalytic α subunit of F1-ATPase binds to a nucleotide (Fig. 7b). Superimposition of the N-terminal β-barrel part of three A and B subunits in V1-ATPase and β and α subunits in F1-ATPase reveals conformational differences between AC and ACR, and BC and BCR of V1-ATPase, but the conformations of βTP and βDP as well as αTP and αDP of F1-ATPase are very similar (Fig. 7c). These differences are also evidenced by the positional displacement of residues between two of the three A and B subunits in V1-ATPase and β and α subunits in F1-ATPase (Fig. 8), which also shows that the structures of AC and ACR are largely different from that of AO (Fig. 8, top left). In contrast, the central portions (residues 180–320) of the βTP and βDP structures show very similar conformations to that of the βE structure (Fig. 8, top right). These results suggest that the A subunit in V1-ATPase from E. hirae undergoes the whole conformational change upon nucleotide binding, whereas the β subunit in F1-ATPase undergoes the conformational change mainly in the P-loop and C-terminal domains.

Structural differences between F1-and V1-ATPase. a Comparison of the nucleotide-binding site of the “tight” site in E. hirae V1-ATPase (bV1, PDB ID: 3VR6) with that of the “ADP-bound” site in bovine F1-ATPase (PDB ID: 2JDI) [28]. b Top views from the membrane side of bV1 (left) and bovine F1-ATPase (right). The catalytic and non-catalytic sites that bind to AMPPNP molecules are indicated by red arrowheads. c Superimposed structures at the N-terminal β-barrel (white) of three structures of A and B subunits in bV1 compared with the β and α subunits in F1-ATPase

Comparison of the conformational differences between F1- and V1-ATPase. Positional displacement of residues (Cα atoms) between two of the three A subunits (top left) and B subunits (bottom left) in bV1, and β subunits (top right) and α subunits (bottom right) in bovine F1-ATPase (PDB ID: 2JDI), which are superimposed at the N-terminal β-barrel domains (see Fig. 7c)
Thus, the conformational features of the A and B subunits in V1-ATPase from E. hirae are apparently different from those of the β and α subunits in bovine F1-ATPase, despite the highly similar nucleotide-binding sites of these ATPases. These structural differences and similarities may be related to the ‘unique’ and ‘common’ mechanisms of rotary catalysis between these rotary ATPases, such as the chemomechanical coupling scheme of the rotation [40, 46].
Dynamics of rotary V1-ATPase: single-molecule studies
After the establishment of the single-molecule rotation assay of F1-ATPase in 1997 [29], the dynamics of rotary ATPases from various species have been studied at the single-molecule level [38,39,40, 46, 54,55,56,57]. The first rotation of V1-ATPase has been directly visualized under an optical microscope by the attachment of large beads to the rotor DF subunits in V1-ATPase from T. thermophilus [58]. V1-ATPase from T. thermophilus rotates stepwise in a counterclockwise direction, consuming one ATP molecule at each step when viewed from the membrane side. The basic step size is 120°, which is similar to that of F1-ATPase, and no substeps have been resolved in the rotation, even when using a slowly hydrolyzable ATP analog (adenosine 5′-O-(3-thio)triphosphate or ATPγS) and high-speed imaging of gold nanoparticles [48, 59]. These results indicate that ATP binding and ATP cleavage (and/or phosphate release) occur at the same angle in this V1-ATPase. In the case of F1-ATPases, some or all of these elementary reaction steps occur at different angles and the basic 120° step is further divided into two or three substeps [31, 32, 39, 40], i.e., 80° and 40° substeps in thermophilic Bacillus PS3 F1-ATPase [31, 32], 85° and 35° substeps in Escherichia coli F1-ATPase [39], and 65°, 25°, and 30° substeps in human F1-ATPase [40]. The 80°, 85°, and 65° substeps are triggered by ATP binding and ADP release, while the 40° and 35° substeps are triggered by ATP cleavage and phosphate release. In human F1-ATPase, it is proposed that ATP cleavage and phosphate release trigger different substeps of 30° and 25°, respectively.
Recently, rotary dynamics of V1-ATPase from E. hirae have been characterized using single-molecule analyses at a submillisecond temporal resolution employing gold nanoparticles and an objective-type total internal reflection dark field microscope (Fig. 9a) [46,47,48]. V1-ATPase from E. hirae rotates in basically the same manner as that from T. thermophilus which functions as ATP synthase [55, 59]. This V1-ATPase also shows only three pauses separated by 120° at all concentrations ranging from below to above the Michaelis constant (Km), where distinct elementary reaction steps of ATP hydrolysis (ATP binding, ATP cleavage, or product release) become the rate-limiting step (Fig. 9b), suggesting that 120° stepping rotation without substeps is a common property of V1-ATPase [46], despite the difference in physiological function between V-ATPase from E. hirae (ion pump) and that from T. thermophilus (ATP synthesis). These results imply that the basic properties of rotary dynamics of F-ATPases and V-ATPases are determined by their overall structures and that the difference in physiological function derives from regulatory mechanisms such as Mg2+ ADP inhibition and inhibitor proteins [9, 18, 23, 26].

Single-molecule rotation of E. hirae V1-ATPase. a Schematic illustration of the single-molecule rotation assay of E. hirae V1-ATPase. The A3B3 ring is immobilized on a glass surface via a His-tag on the A subunit, and an optical probe (gold nanoparticle, 40 nm in diameter) is attached to the D subunit to visualize the rotary motion of rotor DF subunits using an optical microscope [46, 68]. b Rotations of E. hirae V1-ATPase for various concentrations of ATP. Left: 40 mM ATP, considerably higher than the Michaelis constant (Km, 154 µM). Center: 100 µM ATP, near the Km. Right: 10 mM ATP, considerably lower than the Km [46]
Chemomechanical coupling scheme of V1-ATPase
Figure 10a shows a proposed chemomechanical coupling scheme of V1-ATPase from E. hirae based on recently determined structural features and rotary dynamics [44,45,46,47]. As mentioned above, three distinct structures of E. hirae V1-ATPase have been solved (bV1 = 2ATPV1, 2ADPV1, and 3ADPV1) [44, 45]. These three structures are regarded as the three different dwelling states in the rotation waiting for the elementary reaction steps of ATP hydrolysis corresponding, respectively, to the catalytic (ATP cleavage) dwell (bV1 = 2ATPV1), ATP-binding dwell (2ADPV1), and ADP-release dwell (3ADPV1) states.

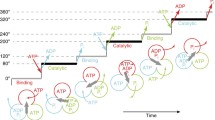
Chemomechanical coupling scheme of V1-ATPase. a Proposed rotation model of E. hirae V1-ATPase for 120° rotation [45]. The structure models are based on the crystal structures of 2ADPV1 (ATP-binding dwell), 3ADPV1 (ADP-release dwell), and bV1 (catalytic dwell). The catalytic sites that bind to nucleotides are indicated by arrowheads (top). Each blue circle represents the chemical state of each catalytic site, and the central red arrow represents the orientation of the rotor DF subunits. ATP* represents the pre- or post-hydrolysis state of ATP (middle). Correspondence table for all catalytic sites observed in the crystal structures of the A3B3 and V1-ATPase (bottom). b Possible chemomechanical coupling scheme of E. hirae V1-ATPase [44,45,46, 48, 49] (left) and human F1-ATPase [40] (right) for 360° rotation. 0° is set as the ATP-binding angle for the catalytic site at the 12 O’clock position (green). In the model of E. hirae V1-ATPase, ATP bound at 0° is cleaved into ADP and Pi at 240°. Among these, phosphate first dissociates at 240°, and then, ADP release occurs at 240°. Other catalytic sites also obey the same reaction scheme offset by 120° and 240°. In the model of human F1-ATPase, ATP bound at 0° is cleaved into ADP and Pi at 210°, ADP dissociates at 240°, and then, phosphate release occurs at 305°
In this model, ATP binds to the ‘bindable-like’ site in 2ADPV1, which induces conformational changes of ‘bindable-like’ and ‘ADP-bound’ sites to ‘half-closed’ and ‘tight-like’ sites, respectively. This results in a slight shift of the rotor DF subunits toward the ‘tight-like’ site, but does not induce the obvious rotation of the rotor DF subunits. Then, ADP release occurs at the ‘tight-like’ site in 3ADPV1, and all three catalytic sites change the conformations from ‘half-closed’ to ‘bound’, from ‘bound’ to ‘tight’, and from ‘tight-like’ to ‘empty’. Coupled with these conformational changes, the DF subunits rotate 120°. After this rotation, ATP bound at the ‘tight’ site in bV1 (= 2ATPV1) is cleaved into ADP and phosphate. The release of phosphate, which has a lower affinity than ADP, is coupled with a conformational change from a ‘tight’ site to ‘ADP-bound’ site and from an ‘empty’ site to ‘bindable-like’ site. Consequently, the rotor DF subunits tilt toward the ‘ADP-bound’ site, without showing obvious rotation. Finally, it returns to the initial rotational state with 120° rotation (Fig. 10a).
Currently, there are no single-molecule studies on V1-ATPase from E. hirae that directly demonstrate the dwell angles for ATP cleavage, ADP release, and phosphate release at a single catalytic site over one revolution. However, considering the results of single-molecule studies and structural studies, the model for 360° rotation cycle is conceivable [48].
If the ATP-binding angle is defined as 0° in the 360° rotation cycle (Fig. 10b, left), ATP is cleaved at 240°. Then, phosphate is released first at 240°, and finally, ADP is released at 240°. In comparison, the proposed chemomechanical coupling scheme of F1-ATPase is more complicated. In the case of thermophilic Bacillus PS3 F1-ATPase, ATP cleavage, ADP release, and phosphate release occur at 200°, 240°, and 320° [37], respectively, although the timing of phosphate release is controversial. Furthermore, in the case of human F1-ATPase, ATP cleavage, ADP release, and phosphate release occur at 210°, 240°, and 305°, respectively (Fig. 10b, right) [40]. The coupling scheme of human F1-ATPase can be considered a variation of that of thermophilic Bacillus PS3 F1-ATPase, in which the ATP cleavage and the phosphate-release dwells are split into different angles. Of course, it is possible that V1-ATPase also shows the substeps, because the angles waiting for ADP release and phosphate release have not been directly demonstrated by advanced single-molecule experiments, as has been performed in the case of F1-ATPase [34, 35, 37]. Furthermore, multiscale molecular dynamics simulations predict the formation of rotational intermediate states of this V1-ATPase that have not yet been resolved [60]. Interestingly, the recent information-based soft clustering method revealed that thermophilic Bacillus PS3 F1-ATPase makes a small rotational movement during the catalytic dwell triggered by the ATP hydrolysis reaction; this had not been previously resolved using the conventional analysis methods [61]. Such advanced single-molecule techniques and analysis methods may reveal the unresolved reaction scheme and movements of V1-ATPase.
Future prospects
The recent high-resolution structural studies and single-molecule studies reviewed have begun to clarify the rotation mechanism of V1-ATPase. By comparing the differences and similarities in the rotation mechanism between V1-ATPase and F1-ATPase, we can determine the ‘unique’ and ‘common’ mechanisms by which these rotary ATPases function and thereby establish the working principle of rotary ATPases. However, to fully understand the rotation mechanism of rotary ATPases, it is necessary to further improve our ‘knowledge and understanding’ by designing rotary ATPases with improved, modified, or novel functions based on our current ‘knowledge and understanding,’ and by experimentally verifying designed proteins. Such a synthetic approach has become a trend in biology and nanobiotechnology [62,63,64,65,66,67], and this approach will be extremely helpful to understand the mechanisms by which rotary ATPases operate.
Abbreviations
- AMPPNP:
-
Adenosine 5′-(β,γ-imido)triphosphate
- ATPγS:
-
Adenosine 5′-O-(3-thio)triphosphate
References
Mitchell P (1966) Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biol Rev Camb Philos Soc 41:445–502
Boyer PD (1997) The ATP synthase—a splendid molecular machine. Annu Rev Biochem 66:717–749
Yoshida M, Muneyuki E, Hisabori T (2001) ATP synthase—a marvellous rotary engine of the cell. Nat Rev Mol Cell Biol 2:669–677
von Ballmoos C, Wiedenmann A, Dimroth P (2009) Essentials for ATP synthesis by F1F0 ATP synthases. Annu Rev Biochem 78:649–672
Forgac M (2007) Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol 8:917–929
Mulkidjanian AY, Makarova KS, Galperin MY, Koonin EV (2007) Inventing the dynamo machine: the evolution of the F-type and V-type ATPases. Nat Rev Microbiol 5:892–899
Bowman EJ, Bowman BJ (2005) V-ATPases as drug targets. J Bioenerg Biomembr 37:431–435
Nakano M, Imamura H, Toei M, Tamakoshi M, Yoshida M, Yokoyama K (2008) ATP hydrolysis and synthesis of a rotary motor V-ATPase from Thermus thermophilus. J Biol Chem 283:20789–20796
Yokoyama K, Muneyuki E, Amano T, Mizutani S, Yoshida M, Ishida M, Ohkuma S (1998) V-ATPase of Thermus thermophilus is inactivated during ATP hydrolysis but can synthesize ATP. J Biol Chem 273:20504–20510
Gruber G, Manimekalai MS, Mayer F, Muller V (2014) ATP synthases from archaea: the beauty of a molecular motor. Biochim Biophys Acta 1837:940–952
Cross RL, Muller V (2004) The evolution of A-, F-, and V-type ATP synthases and ATPases: reversals in function and changes in the H+/ATP coupling ratio. FEBS Lett 576:1–4
Muench SP, Trinick J, Harrison MA (2011) Structural divergence of the rotary ATPases. Q Rev Biophys 44:311–356
Murata T, Igarashi K, Kakinuma Y, Yamato I (2000) Na+ binding of V-type Na+ -ATPase in Enterococcus hirae. J Biol Chem 275:13415–13419
Murata T, Yamato I, Kakinuma Y, Leslie AG, Walker JE (2005) Structure of the rotor of the V-Type Na+ -ATPase from Enterococcus hirae. Science 308:654–659
Murata T, Yamato I, Kakinuma Y, Shirouzu M, Walker JE, Yokoyama S, Iwata S (2008) Ion binding and selectivity of the rotor ring of the Na+ -transporting V-ATPase. Proc Natl Acad Sci USA 105:8607–8612
Stewart AG, Sobti M, Harvey RP, Stock D (2013) Rotary ATPases: models, machine elements and technical specifications. Bioarchitecture 3:2–12
Kühlbrandt W, Davies KM (2016) Rotary ATPases: a new twist to an ancient machine. Trends Biochem Sci 41:106–116
Oot RA, Kane PM, Berry EA, Wilkens S (2016) Crystal structure of yeast V1-ATPase in the autoinhibited state. EMBO J 35:1694–1706
Abrahams JP, Leslie AG, Lutter R, Walker JE (1994) Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature 370:621–628
Menz RI, Walker JE, Leslie AG (2001) Structure of bovine mitochondrial F(1)-ATPase with nucleotide bound to all three catalytic sites: implications for the mechanism of rotary catalysis. Cell 106:331–341
Kagawa R, Montgomery MG, Braig K, Leslie AG, Walker JE (2004) The structure of bovine F1-ATPase inhibited by ADP and beryllium fluoride. EMBO J 23:2734–2744
Rees DM, Montgomery MG, Leslie AG, Walker JE (2012) Structural evidence of a new catalytic intermediate in the pathway of ATP hydrolysis by F1-ATPase from bovine heart mitochondria. Proc Natl Acad Sci USA 109:11139–11143
Gledhill JR, Montgomery MG, Leslie AG, Walker JE (2007) How the regulatory protein, IF(1), inhibits F(1)-ATPase from bovine mitochondria. Proc Natl Acad Sci USA 104:15671–15676
Kabaleeswaran V, Puri N, Walker JE, Leslie AG, Mueller DM (2006) Novel features of the rotary catalytic mechanism revealed in the structure of yeast F1 ATPase. EMBO J 25:5433–5442
Kabaleeswaran V, Shen H, Symersky J, Walker JE, Leslie AG, Mueller DM (2009) Asymmetric structure of the yeast F1 ATPase in the absence of bound nucleotides. J Biol Chem 284:10546–10551
Cingolani G, Duncan TM (2011) Structure of the ATP synthase catalytic complex (F(1)) from Escherichia coli in an autoinhibited conformation. Nat Struct Mol Biol 18:701–707
Shirakihara Y, Shiratori A, Tanikawa H, Nakasako M, Yoshida M, Suzuki T (2015) Structure of a thermophilic F1-ATPase inhibited by an epsilon-subunit: deeper insight into the epsilon-inhibition mechanism. FEBS J 282:2895–2913
Bowler MW, Montgomery MG, Leslie AG, Walker JE (2007) Ground state structure of F1-ATPase from bovine heart mitochondria at 1.9 A resolution. J Biol Chem 282:14238–14242
Noji H, Yasuda R, Yoshida M, Kinosita K Jr (1997) Direct observation of the rotation of F1-ATPase. Nature 386:299–302
Yasuda R, Noji H, Kinosita K Jr, Yoshida M (1998) F1-ATPase is a highly efficient molecular motor that rotates with discrete 120 degree steps. Cell 93:1117–1124
Yasuda R, Noji H, Yoshida M, Kinosita K Jr, Itoh H (2001) Resolution of distinct rotational substeps by submillisecond kinetic analysis of F1-ATPase. Nature 410:898–904
Shimabukuro K, Yasuda R, Muneyuki E, Hara KY, Kinosita K Jr, Yoshida M (2003) Catalysis and rotation of F1 motor: cleavage of ATP at the catalytic site occurs in 1 ms before 40 degree substep rotation. Proc Natl Acad Sci USA 100:14731–14736
Ariga T, Muneyuki E, Yoshida M (2007) F1-ATPase rotates by an asymmetric, sequential mechanism using all three catalytic subunits. Nat Struct Mol Biol 14:841–846
Adachi K, Oiwa K, Nishizaka T, Furuike S, Noji H, Itoh H, Yoshida M, Kinosita K Jr (2007) Coupling of rotation and catalysis in F(1)-ATPase revealed by single-molecule imaging and manipulation. Cell 130:309–321
Nishizaka T, Oiwa K, Noji H, Kimura S, Muneyuki E, Yoshida M, Kinosita K Jr (2004) Chemomechanical coupling in F1-ATPase revealed by simultaneous observation of nucleotide kinetics and rotation. Nat Struct Mol Biol 11:142–148
Masaike T, Koyama-Horibe F, Oiwa K, Yoshida M, Nishizaka T (2008) Cooperative three-step motions in catalytic subunits of F(1)-ATPase correlate with 80 degrees and 40 degrees substep rotations. Nat Struct Mol Biol 15:1326–1333
Watanabe R, Iino R, Noji H (2010) Phosphate release in F1-ATPase catalytic cycle follows ADP release. Nat Chem Biol 6:814–820
Martin JL, Ishmukhametov R, Hornung T, Ahmad Z, Frasch WD (2014) Anatomy of F1-ATPase powered rotation. Proc Natl Acad Sci USA 111:3715–3720
Bilyard T, Nakanishi-Matsui M, Steel BC, Pilizota T, Nord AL, Hosokawa H, Futai M, Berry RM (2013) High-resolution single-molecule characterization of the enzymatic states in Escherichia coli F1-ATPase. Philos Trans R Soc Lond B Biol Sci 368:20120023
Suzuki T, Tanaka K, Wakabayashi C, Saita E, Yoshida M (2014) Chemomechanical coupling of human mitochondrial F1-ATPase motor. Nat Chem Biol 10:930–936
Maher MJ et al (2009) Crystal structure of A3B3 complex of V-ATPase from Thermus thermophilus. EMBO J 28:3771–3779
Numoto N, Hasegawa Y, Takeda K, Miki K (2009) Inter-subunit interaction and quaternary rearrangement defined by the central stalk of prokaryotic V1-ATPase. EMBO Rep 10:1228–1234
Nagamatsu Y, Takeda K, Kuranaga T, Numoto N, Miki K (2013) Origin of asymmetry at the intersubunit interfaces of V1-ATPase from Thermus thermophilus. J Mol Biol 425:2699–2708
Arai S et al (2013) Rotation mechanism of Enterococcus hirae V1-ATPase based on asymmetric crystal structures. Nature 493:703–707
Suzuki K et al (2016) Crystal structures of the ATP-binding and ADP-release dwells of the V1 rotary motor. Nat Commun 7:13235
Minagawa Y et al (2013) Basic properties of rotary dynamics of the molecular motor Enterococcus hirae V1-ATPase. J Biol Chem 288:32700–32707
Ueno H et al (2014) Torque generation of Enterococcus hirae V-ATPase. J Biol Chem 289:31212–31223
Iino R, Minagawa Y, Ueno H, Hara M, Murata T (2014) Molecular structure and rotary dynamics of Enterococcus hirae V(1)-ATPase. IUBMB Life 66:624–630
Iino R, Ueno H, Minagawa Y, Suzuki K, Murata T (2015) Rotational mechanism of Enterococcus hirae V1-ATPase by crystal-structure and single-molecule analyses. Curr Opin Struct Biol 31:49–56
Shirakihara Y et al (1997) The crystal structure of the nucleotide-free alpha 3 beta 3 subcomplex of F1-ATPase from the thermophilic Bacillus PS3 is a symmetric trimer. Structure 5:825–836
Uchihashi T, Iino R, Ando T, Noji H (2011) High-speed atomic force microscopy reveals rotary catalysis of rotorless F-ATPase. Science 333:755–758
Liu Q, Leng XH, Newman PR, Vasilyeva E, Kane PM, Forgac M (1997) Site-directed mutagenesis of the yeast V-ATPase A subunit. J Biol Chem 272:11750–11756
Hayashi S et al (2012) Molecular mechanism of ATP hydrolysis in F1-ATPase revealed by molecular simulations and single-molecule observations. J Am Chem Soc 134:8447–8454
Noji H, Hasler K, Junge W, Kinosita K Jr, Yoshida M, Engelbrecht S (1999) Rotation of Escherichia coli F(1)-ATPase. Biochem Biophys Res Commun 260:597–599
Imamura H, Takeda M, Funamoto S, Shimabukuro K, Yoshida M, Yokoyama K (2005) Rotation scheme of V1-motor is different from that of F1-motor. Proc Natl Acad Sci USA 102:17929–17933
McMillan DGG, Watanabe R, Ueno H, Cook GM, Noji H (2016) Biophysical characterization of a thermoalkaliphilic molecular motor with a high stepping torque gives insight into evolutionary ATP synthase adaptation. J Biol Chem 291:23965–23977
Hirata T, Iwamoto-Kihara A, Sun-Wada GH, Okajima T, Wada Y, Futai M (2003) Subunit rotation of vacuolar-type proton pumping ATPase: relative rotation of the G and C subunits. J Biol Chem 278:23714–23719
Imamura H, Nakano M, Noji H, Muneyuki E, Ohkuma S, Yoshida M, Yokoyama K (2003) Evidence for rotation of V1-ATPase. Proc Natl Acad Sci USA 100:2312–2315
Furuike S, Nakano M, Adachi K, Noji H, Kinosita K Jr, Yokoyama K (2011) Resolving stepping rotation in Thermus thermophilus H(+)-ATPase/synthase with an essentially drag-free probe. Nat Commun 2:233
Isaka Y, Ekimoto T, Kokabu Y, Yamato I, Murata T, Ikeguchi M (2017) Rotation mechanism of molecular motor V1-ATPase studied by multiscale molecular dynamics simulation. Biophys J 112:911–920
Li CB, Ueno H, Watanabe R, Noji H, Komatsuzaki T (2015) ATP hydrolysis assists phosphate release and promotes reaction ordering in F1-ATPase. Nat Commun 6:10223
Schwille P, Diez S (2009) Synthetic biology of minimal systems. Crit Rev Biochem Mol Biol 44:223–242
Kay ER, Leigh DA, Zerbetto F (2007) Synthetic molecular motors and mechanical machines. Angew Chem Int Ed Engl 46:72–191
Chen L, Nakamura M, Schindler TD, Parker D, Bryant Z (2012) Engineering controllable bidirectional molecular motors based on myosin. Nat Nanotechnol 7:252–256
Nakamura M, Chen L, Howes SC, Schindler TD, Nogales E, Bryant Z (2014) Remote control of myosin and kinesin motors using light-activated gearshifting. Nat Nanotechnol 9:693–697
Furuta A, Amino M, Yoshio M, Oiwa K, Kojima H, Furuta K (2017) Creating biomolecular motors based on dynein and actin-binding proteins. Nat Nanotechnol 12:233–237
DelRosso NV, Derr ND (2017) Exploiting molecular motors as nanomachines: the mechanisms of de novo and re-engineered cytoskeletal motors. Curr Opin Biotechnol 46:20–26
Ueno H, Nishikawa S, Iino R, Tabata KV, Sakakihara S, Yanagida T, Noji H (2010) Simple dark-field microscopy with nanometer spatial precision and microsecond temporal resolution. Biophys J 98:2014–2023
Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research (25840053 and 16K14706 to H.U.; 26291009 and 17H03638 to T.M.) and Bilateral Joint Research Projects from the Japan Society for the Promotion of Science (to H.U.) and the Research Program of “Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials” in “Network Joint Research Center for Materials and Devices” (to H.U.). We thank all members of our laboratory.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ueno, H., Suzuki, K. & Murata, T. Structure and dynamics of rotary V1 motor. Cell. Mol. Life Sci. 75, 1789–1802 (2018). https://doi.org/10.1007/s00018-018-2758-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-018-2758-3