
Abstract
Proteins routed to the secretory pathway start their journey by being transported across biological membranes, such as the endoplasmic reticulum. The essential nature of this protein translocation process has led to the evolution of several factors that specifically target the translocon and block translocation. In this review, various translocation pathways are discussed together with known inhibitors of translocation. Properties of signal peptide-specific systems are highlighted for the development of new therapeutic and antimicrobial applications, as compounds can target signal peptides from either host cells or pathogens and thereby selectively prevent translocation of those specific proteins. Broad inhibition of translocation is also an interesting target for the development of new anticancer drugs because cancer cells heavily depend on efficient protein translocation into the endoplasmic reticulum to support their fast growth.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Different signal peptide-dependent translocation pathways
More than 30% of all human genes encode proteins destined for the extracellular environment, cell membrane or components of the secretory pathway [1]. Since protein synthesis occurs in the cytosol, translocation of proteins across biological membranes is essential for cellular function. Multiple pathways of protein translocation have been proposed. In general, translocation from the cytosol requires three key steps: (1) substrate recognition and targeting to the destination membrane, while maintaining the substrate in a translocation-competent state (2) translocation across or integration into that membrane, which usually requires energy expenditure in the form of GTP, ATP or proton motive force and (3) release, folding and maturation of the protein substrate.
Targeting signals contain the principal information that drives protein translocation. They direct newly synthesized proteins to their target membrane for translocation or membrane integration [2]. Signals present at the N-terminus of the synthesized protein are termed signal peptides (SP) or signal sequences. SPs are cleaved from the mature protein after translocation by the signal peptidase complex (SPC) and are characterized by a short (8–12 amino acids) hydrophobic segment, but their length and amino acid composition are highly divergent [3, 4]. Alternatively, targeting is facilitated by uncleaved amino-terminal signals termed signal anchors (SA). Signal anchors can act as a transmembrane segment and usually contain about 20 (or more) hydrophobic residues, a length required to physically span the approximately 3 nm wide hydrophobic interior of the phospholipid bilayer in an α-helical fold [5]. A third distinction is made for a class of proteins called tail-anchored membrane proteins (TA proteins), which have a single hydrophobic transmembrane region at their C-terminus that acts both as a targeting signal and membrane anchor [6].
The conserved Sec-dependent pathway is used for the translocation of most eukaryotic proteins [7]. The central component of this system is the heterotrimeric Sec61 translocon complex, also known as the SecY complex in bacteria and archaea [8]. It forms an aqueous channel in the endoplasmic reticulum (ER) membrane which allows protein transport across the membrane and it facilitates insertion of hydrophobic protein segments into the lipid bilayer. The Sec pathway operates in two major modes of translocation: (1) co-translational translocation couples the ribosomal protein synthesis directly to translocation through the channel, which efficiently uses the energy from mRNA translation in ribosomes to drive protein translocation across the membrane, (2) while post-translational translocation delivers completely synthesized polypeptide chains to the membrane which is best understood in fungi and bacteria. Mitochondrial, chloroplast and peroxisomal protein import, as well as specialized bacterial secretion systems (suggested reviews: [9,10,11,12]) are not discussed here, but the general concepts of signal peptide-dependent translocation (use of targeting signals and specialized protein-conducting channels) are conserved in these systems.
Co-translational translocation
The process of co-translational translocation is a multistep sequence that depends on dynamic interactions between many factors. For most of these steps, natural and synthetic inhibitors have been discovered that usually affect translocation of a broad range of co-translational substrates. However, some compounds are able to operate in a signal peptide-selective way.
Synthesis of proteins destined for the co-translational pathway starts with mRNA translation in cytosolic ribosomes (Fig. 1). Once the hydrophobic targeting signal of the nascent chain (NC) emerges from the ribosomal exit tunnel, it is recognized by the signal recognition particle (SRP), a universally conserved ribonucleoprotein complex [13]. The S-domain contains the evolutionarily conserved SRP54 subunit that binds signal peptides, functions as a GTPase and interacts with the membrane-associated SRP receptor (SR). Eukaryotic SRP has an additional Alu domain that forms an elongated, kinked structure. After recognition of the signal by the S-domain, the Alu domain reaches into the elongation factor binding site of the ribosome [13] and slows down elongation of the polypeptide chain. The SRP and ribosome-nascent chain complex (RNC) is then targeted to the ER membrane where SRP binds the SRP receptor (SR). The current model of SRP function suggests that this mechanism maintains the RNC in a translocation-competent state during the targeting step and GTP-dependent transfer of the RNC to the translocon [14, 15]. The Sec61/SecY translocon forms a passive pore in the ER membrane (or plasma membrane in bacteria) where the targeting signal needs to be recognized a second time. After successful recognition and opening of the pore, the polypeptide is finally translocated across or embedded into the membrane.

Co-translational translocation in eukaryotes relies on SRP for targeting to the Sec61 translocon. Ribosomal protein synthesis is coupled to translocation, which protects the peptide and effectively uses the energy from chain elongation as a driving force. The channel also facilitates membrane integration of hydrophobic transmembrane domains (TMDs, dark blue box) through a lateral gate. Multiple accessory factors reside in the local membrane environment and are dynamically recruited to assist the function of the translocon. The lumenal signal peptidase complex (SPC) cleaves signal peptides (orange box) from the mature protein, while uncleaved signal anchors (yellow box) function as transmembrane domains in integral membrane proteins. Inhibitors of co-translational translocation are indicated in blue text and the targets are explained in more detail in Table 1
The Sec translocon is a dynamic protein complex
The Sec61 complex consists of a central Sec61α subunit (referred to as SecY in bacteria and archaea) which forms the channel, and two smaller peripheral subunits Sec61β and Sec61γ [8]. Sec61γ is homologous to SecE in bacteria and archaea, but Sec61β shows little homology to the bacterial SecG subunit. Sec61α contains ten transmembrane helices divided into two halves of the channel (TM 1-5 and TM 6-10) with a hinge point between TM helix 5 and 6, often referred to as a ‘clam shell’ design. The ‘lateral gate’ of Sec61α, formed by the interface of transmembrane helices 2 and 7, allows opening of the channel towards the lipid bilayer for lipid insertion of transmembrane domains [16]. It also serves as the recognition site of signal peptides [17] and allows hydrophobic peptide region access to the lipid layer [18]. The inside of the channel is hourglass-shaped, with a ring at the center consisting of six bulky hydrophobic amino acid residues (the pore ring) which position their side chains to the center of the pore. This ring prevents leakage of ions through the inactive channel and during translocation of a protein substrate. The lumenal side of the closed channel is occupied by a short helix (TM2a) called the plug domain.
The translocon provides a dynamic interface between the water filled inside of the channel and the lipid environment. Hence, most eukaryotic membrane proteins with a (trans)membrane domain (such as a hydrophobic α-helix) are inserted into the ER membrane during co-translational translocation. In the current understanding of membrane integration, individual TM segments insert sequentially in the membrane layer through the lateral gate of Sec61 [19]. Furthermore, the channel can accommodate several TM helices at the same time, and facilitates early folding of these segments before release into the membrane [20]. Multiple accessory factors are dynamically recruited to the translocon and assist the Sec channel (Fig. 1), e.g., through the chaperoning functions of translocon-associated protein (TRAP) and translocating chain-associated membrane protein (TRAM). Other factors associate with the translocon to perform post-translational modification of the peptide substrates: oligosaccharyl–transferase (OST) complex facilitates N-linked glycosylation, while the SPC cleaves signal peptides from the mature protein.
Non-selective inhibition of RNC transfer to the translocon
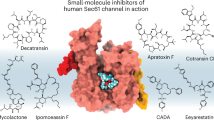
The chemical compound eeyarestatin I (ESI) (Fig. 2) was discovered as an inhibitor of endoplasmic reticulum-associated protein degradation (ERAD) that induces an ER stress response in cells which leads to cytotoxicity [21]. ERAD removes misfolded proteins (and certain folded proteins) from the ER and plays a key role in ER homeostasis [22]. After recognition for ERAD, target proteins are modified by E3 ubiquitin ligases, retrotranslocated towards the cytosol and finally degraded by the proteasome. ESI inhibits the action of p97/VCP, a cytosolic ATPase that extracts polyubiquitinated ERAD substrates from the ER membrane [23]. ESI also interferes with co-translational protein import into the ER, as it blocks the transfer of a SP from the RNC-SRP complex into the Sec61 channel’s acceptor site [24]. This ESI-induced general block of protein translocation results in cytosolic accumulation of polyubiquitinated proteins and induces the unfolded protein response (UPR) [25]. The UPR normally protects the cell during ER stress, and is often upregulated in cancer cells. However, prolonged activation of UPR can induce cell death through apoptosis [26]. Consequently, ESI can be considered as an anticancer agent. In addition, Aletrari et al. showed that ESI also interferes with vesicular trafficking of Shiga-like toxin, which uses endosomal vesicles to reach the ER lumen and then exploits the ERAD machinery to enter the cytosol through ER retrotranslocation [27].

Chemical structure of several natural and synthetic inhibitors of co-translational translocation. Eeyarestatin I blocks the transfer of a SP from the RNC–SRP complex to Sec61. Mycolactone induces a conformational change in the Sec61 channel. HUN-7293 and its derivatives cotransin and CAM741 interfere with signal peptide insertion at the translocon. Decatransin and apratoxin A inhibit translocation into the ER lumen and can prevent growth of tumor cells. CADA prevents co-translational translocation of hCD4 and sortilin
Inhibitors of translocon gating
Mycolactone
The polyketide macrolide mycolactone (Fig. 2) is a virulence factor produced by the human pathogen Mycobacterium ulcerans and causes necrotizing lesions of the skin without acute inflammation [28]. Hall et al. showed that mycolactone is a non-selective inhibitor of Sec61-dependent translocation across the ER [29]. Additionally, the inhibitory effect on cells was irreversible, indicative of a high affinity binding. The lack of immune response to this molecule is thus due to its suppression of inflammatory cytokine and receptor production in immune cells, and due to an indirect inhibition of antigen cross-presentation [30]. The eukaryotic Sec61 channel was recently identified as the target of mycolactone. Chemical crosslinking data suggest that the compound induces a conformational change of the channel that significantly disturbs co-translational translocation efficiency, but has less impact on post-translational translocation substrates [31].
Exotoxin A
The Pseudomonas aeruginosa protein exotoxin A is a cytotoxic ADP-ribosyltransferase that enters the eukaryotic cytosol trough retrograde transport and inhibits retrograde export of immunogenic peptides from the ER towards the cytosol. It binds to Sec61α and prevents both co- and post-translational translocation [32, 33]. Exotoxin A also competes with cytosolic protein calmodulin (CaM) for binding to an N-terminal IQ motif on Sec61α and prevents Ca2+ leakage through the channel in human cells [34]. These observations suggest that the protein keeps the Sec61 channel in a closed state.
Cotransins
A group of cyclic heptadepsipeptides are derived from the fungal macrocycle HUN-7293. The latter inhibits expression of three endothelial cell adhesion molecules: intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule (VCAM-1) and E-selectin [35]. One derivative called cotransin (Fig. 2) was shown to inhibit the co-translational translocation of several proteins into the ER, in a signal peptide-selective way [36]. These initial studies reported inhibition of VCAM-1, P-selectin, angiotensinogen, β-lactamase, and corticotropin-releasing factor receptor 1 (CRF-R-1). Later studies also identified endothelin B receptor [37], human epidermal growth factor receptor 3 [38] and tumor necrosis factor alpha (TNFα) [39], a type II integral membrane protein with uncleaved signal anchor, as targets of cotransin.
Cotransin does not affect SRP recognition or targeting, but prevents access of NCs to the ER lumen, suggesting that the compound inhibits signal peptide-dependent gating of the Sec61 channel (Fig. 1). Accessory translocon factors such as TRAP, TRAM, Sec62/63 and binding immunoglobulin protein (BiP) are not required for cotransin activity, as the compound was able to selectively prevent translocation of VCAM-1 in minimal proteoliposomes (containing only Sec61 and SR) [36]. Garrison et al. suggested that cotransin either stabilizes the channel in a closed conformation or that it allosterically alters the signal peptide binding site of Sec61. These hypotheses, respectively, restrict productive interaction of low-affinity SPs or decrease the SP binding site flexibility, which both result in substrate selection at the translocon.
It must be noted that the reported compound concentrations used in the different translocation assays varies widely, which is important for the interpretation of the selectivity concept. For example, cotransin operates selectively at low nanomolar concentrations [36]. In contrast, Klein et al. have recently shown that a saturating concentration of cotransin (30 µM) actually inhibits translocation of a broad range of secreted proteins, while integral membrane proteins are mostly unaffected [40].
Decatransin
Decatransin is a fungal cyclic decadepsipeptide (Fig. 2) that prevents growth of human carcinoma cells [41]. It is synthesized by a non-ribosomal peptide synthetase. Such very large modular enzymes are often used by microorganisms to produce complex secondary metabolites [42]. Decatransin prevents Sec61/SecY-dependent co- and post-translational translocation into the ER lumen but does not affect SRP recognition or SR targeting [41].
Apratoxin A
Apratoxins are natural secondary metabolites isolated from a marine cyanobacterium. They are also produced by a non-ribosomal peptide synthetase [43]. The cyclic depsipeptide apratoxin A (Fig. 2) was discovered as a cytotoxic antitumor drug and prevents growth of various cancer cell lines by inducing G1 cell cycle arrest and apoptosis [43, 44]. Proteomic data showed that expression of a subset of secreted and membrane proteins are downregulated by apratoxin A, and this effect was due to an inhibition of their co-translational translocation [45].
Gating inhibitors likely operate through a common mechanism
Mutations in Sec61 provide cross-resistance to gating inhibitors
Photo-crosslinking experiments showed that HUN-7293 derivatives bind to the Sec61α subunit [46], as predicted earlier. A more recent study used genetic selection in the DNA repair-defective HCT-116 tumor cell line to identify mutations that confer cotransin resistance [47]. These mutations, located in a region near the lateral gate and plug domain of Sec61α, stabilize the plug domain in the closed state and allosterically prevent the translocation substrate from opening the channel (Fig. 3). Extensive crosslinking experiments showed that the arrested transmembrane domain (TMD) of TNFα is positioned at the interface of Sec61alpha helices 2 and 7, perpendicular to the channel helices. However, TMDs with increased hydrophobicity or helical propensity are able to escape the cotransin-induced block [47].

Location of the mutations in mammalian Sec61 that confer resistance to different gating inhibitors. Cryo-electron microscopy model of mammalian Sec61 from Protein Data Bank accession code 3J7Q [52]. The lateral gate (TM 2 and 7) is shown in blue and the plug helix in orange. The loop that connects TM 3 and 4 contains M136 but was not resolved in the model and is inferred from the Archaeal SecY structure (PDB 1RHZ) [8]
Accordingly, resistance mutations for apratoxin A were all located near the Sec61 plug domain and several of these mutations confer cross-resistance against cotransin [48]. Apratoxin A also competes with cotransin for translocon binding, suggesting a mutually exclusive binding site near the lumenal plug region. Compared to cotransin, apratoxin A does not arrest the TNFα TMD in a preferred orientation on the cytosolic side of Sec61. This suggests that the compound blocks TNFα translocation in an earlier step, before TMD insertion occurs [48].
Decatransin might act similar to cotransin, as several (but not all) mutations in Sec61 that provide resistance to cotransin also confer decatransin resistance. Additionally, both decatransin and CP2, a compound very similar to HUN-7293/cotransin, inhibit translocation in yeast Sec61α and bacterial SecY [41], suggesting that the translocon might contain a universal binding site for natural translocation inhibitors. Junne et al. hypothesized that these hydrophobic, peptide-like molecules mimic signal peptides and can bind to the Sec translocon, where they block incoming peptides and stabilize the closed translocon conformation [41].
Mycolactone showed a broad effect not only on co-translational translocation but also dose-dependently competed with a cotransin variant (CT7) for translocon binding suggesting that they likely share a binding site [49]. When tested for cross-resistance in the cotransin-resistant cell lines, several mutations near the lumenal plug indeed provided mycolactone resistance (Fig. 3).
Gating inhibitors stabilize the translocon in a closed state
The prl (protein localization) phenotype of bacteria and yeast, characterized by the ability to translocate defective signal peptides, has historically been associated with mutations in the translocon plug and pore ring, as these mutations destabilize the closed translocon conformation [50, 51]. Interestingly, the observed resistance mutations for HUN-7293 derivatives, decatransin, apratoxin and mycolactone also map to residues located on the lumenal side of the translocon lateral gate, near the plug domain and thus resemble the prl phenotype. The resistance mutations are believed to increase the flexibility of the translocon, which overrules the action of the inhibitors, i.e., keeping the translocon in a closed state [41]. Paatero et al. suggested that these natural and synthetic compounds all target a similar binding site on the Sec61α translocon to regulate translocation (Fig. 3). Due to significant differences in their chemical structure and potency though, each of these natural compounds produces a distinct inhibitory profile [48].
Since the translocon machinery evolved to accept a broad range of targeting signals, it is surprising to see a selective inhibition of only a small subset of translocation substrates for some of the inhibitors. Nevertheless, such a selective suppression of cell surface receptor expression can have many novel therapeutic and antiviral applications. Hegde and Kang proposed that, in absence of accessory translocon components, the interaction between Sec61 and most signal peptides is intrinsically unstable [53]. Only a limited set of ‘strong’ sequences are able to engage the channel on their own, for a sufficiently long duration to allow for complete insertion of the elongating chain, and subsequent translocation of the protein. All other targeting signals (‘weak’ sequences) are assumed to have a very low basal translocation efficiency. In this model, the occurrence of many Sec61-associated protein complexes (e.g., TRAP, TRAM, Sec62/63, BiP and OST) that dynamically assist the translocon, is the key concept that regulates substrate-specific translocation efficiency. Selective inhibition of translocation is possible too, either through (allosteric) destabilization of channel/signal peptide interactions or through stabilization of the translocon channel in a closed state. Additional regulatory factors likely exist, as the current knowledge is mostly obtained from a very limited set of model translocation systems (e.g., dog pancreas cells, model yeast and bacterial systems). Properties of Sec-dependent signal peptides are thus linked to the observed variation in selectivity (the inhibitory profiles) of these gating inhibitors.
Selective inhibition of signal peptide topology inversion
The small molecule cyclotriazadisulfonamide (CADA) is a synthetic macrocycle (Fig. 2) that showed antiviral activity against a broad range of human immunodeficiency virus (HIV) strains and human herpes virus-7 (HHV-7). Analysis of receptor expression showed that CADA treatment induced a downregulation of the cell surface- and intracellular CD4 levels. Since both HIV and HHV-7 use CD4 as the primary receptor for cell entry, this down-modulation of CD4 is responsible for CADA’s inhibition of viral entry [54].
CADA selectively inhibits the cell surface expression of human CD4 (hCD4) on a post-transcriptional level. The compound interacts with the signal peptide of hCD4 and it prevents co-translational translocation of the pre-protein chain across the ER membrane [55]. Instead, the affected precursor protein chains end up in the cytosol where they are degraded by the proteasome. Targeting of RNCs to the ER translocon is not affected by the compound. The hCD4 signal peptide initially inserts head-on (Nexo/Ccyt) into the translocon, and inverts to a looped topology (Ncyt/Cexo) upon chain elongation (transitioning, Fig. 1). Early models assumed that signal peptides generally insert into the translocon with a looped topology, but head-on insertion and a dynamic topology inversion has been described for signal anchor proteins [56, 57]. Vermeire et al. suggest that CADA interferes with the mandatory inversion (transitioning) of hCD4’s signal peptide, and thereby prevents translocation of the chain.
Furthermore, the down-modulating effect of CADA seems to be selective for hCD4 and the membrane glycoprotein sortilin. As previously shown for hCD4, CADA also inhibits the co-translational translocation of sortilin in a signal peptide-dependent way [58]. The effects of CADA on sortilin were less pronounced as compared to hCD4 though, and sortilin appears to be a secondary substrate of CADA. Importantly, expression of the homologous mouse CD4 protein is not affected by CADA. Mutagenesis of the CD4 signal peptide identified the central hydrophobic signal peptide region as crucial for CADA sensitivity, with a lesser contribution from the C-terminal SP region [55].
The binding site of CADA is not known, but quantitative structure–activity relationship studies of CADA analogues suggest a two-site binding model for these compounds [59, 60], and surface plasmon resonance experiments showed a selective, non-covalent interaction between CADA and the human CD4 signal peptide [55].
Another HUN-7293 derivative, CAM741 (Fig. 2), also interferes with the co-translational translocation of only a limited set of substrates in a signal peptide-selective way. VCAM-1 [61, 62] and VEGF [63] are reported CAM741 targets. Blocked polypeptide chains are directed towards the cytosol and degraded by the proteasome [36, 61]. Their cytosolic accumulation also induces the unfolded protein response [25]. The compound CAM741 prevents correct insertion of the VCAM-1 SP into the translocon [62]. Using a diagnostic amino-terminal glycosylation tag, the topology of the VCAM-1 SP was determined during the early post-targeting phase: it initially inserts head-on (Nexo/Ccyt) into the translocon channel and reorients upon polypeptide chain elongation. However, the amino-terminus does not enter the ER lumen in the presence of CAM741. Systematic analysis of VCAM-1 signal peptide mutants identified residues in the SP C-region, h-region and the first residue of the VCAM-1 mature domain as crucial elements for full CAM741 sensitivity [62]. VEGF-1 mutagenesis revealed a different pattern, as mutagenesis of leucines in the N-terminal SP region and hydrophobic residues in the h-region resulted in a loss of compound sensitivity [63].
Signal peptides with increased hydrophobicity are able to escape CAM741 activity, while reducing the hydrophobicity of VCAM-1 and VEGF-1 SP’s h-region increases the inhibitory effect of CAM741 and vice versa [62, 63]. Hydrophobicity is a major determinant of TM segment recognition at the translocon [64], and signal peptide recognition at the translocon is also dependent on hydrophobic interactions. Targeting signals were shown to interact with a specific hydrophobic patch on the cytosolic side of Sec61, after which they intercalate between the channel helices [18]. Mutations in the signal peptide that increase its hydrophobicity are thus better at opening the channel (they are “stronger” signal peptides according to Hegde’s theorem of translational regulation [53]) and this should allow them to overcome the translocational block imposed by CAM741 more easily.
SRP-independent translocation
After more than 30 years of study, it is clear that SRP-dependent co-translational translocation is highly efficient. However, it is now also evident that a significant fraction of all ER-targeted proteins do not utilize the SRP pathway for targeting, and/or do not even rely on the Sec61 translocon for translocation [65,66,67]. Membrane proteins with large translocated domains or multiple TM domains are usually constrained to the SRP-dependent co-translational pathway, but other physical limitations can restrict SRP recognition. In bacteria and yeast, SRP recognition requires targeting signals with sufficiently high hydrophobicity. SPs with low or moderate hydrophobicity are targeted towards the Sec61-dependent post-translational pathway [68]. Mammalian SRP does not differentiate between SPs of different hydrophobicity, while microorganisms likely favor post-translational translocation to support higher rates of protein secretion, as this pathway does not use up the limited pool of ribosomes [69].
These SRP-independent polypeptides must often be kept in a translocation-competent state during the targeting towards the translocon. The translocon pore is quite narrow and only allows passage of proteins with limited secondary structure. Therefore, excessive folding or aggregation of pre-proteins must be prevented (Fig. 4). Multiple cytosolic proteins, e.g., heat shock protein 70 (Hsp70) and Hsp40 chaperones, assist this process after completion of translation [70]. Hsp70 s bind to exposed hydrophobic protein regions in an ATP-dependent way, while their ATPase activity is regulated by Hsp40 co-chaperones. It is still unclear which factors target these chaperones towards the ER [66].

Sec-dependent post-translational translocation of pre-proteins involves several chaperone proteins. a Calmodulin binds signal peptides (orange box) and is targeted to Sec61, as Sec61 contains a cytosolic signal peptide-binding site. Hsp chaperones retain cytosolic proteins in a translocation-competent state and are targeted to Sec62/63. Other chaperone-independent targeting pathways are possible too, e.g., with intrinsically disordered proteins. The J domain of Sec63 converts BiP to a state with high affinity for protein binding. Sequential binding of BiP molecules works as a ratcheting mechanism that drives post-translational translocation. b Targeting of bacterial pre-proteins can occur either in a chaperone-dependent way through the help of SecA, SecB or trigger factor (TF), or in a chaperone-independent way. SecY-dependent post-translational translocation in bacteria relies on the essential ATPase motor protein SecA and the proton motive force (PMF) to drive translocation of pre-proteins through the SecY pore and across the plasma membrane. Inhibitors of translocation are indicated in blue text
Another one of these cytosolic factors is the calcium-binding protein calmodulin, that was shown to maintain the small protein preprocecropin A (64 amino acids) in a translocation-competent state inside the cytosol, by selective binding to the signal peptide [71]. CaM also binds to the cytosolic N terminus of Sec61α, which is proposed as the targeting mechanism for these peptides.
Chaperone inhibitors
NSC 630668-R/1
In a screen for heat shock cognate 70 (Hsc70, a member of the Hsp70 chaperone family)-interacting compounds, NSC 630668-R/1 (referred to as R/1) was identified as an inhibitor of Hsc70 ATPase activity (Fig. 4). R/1 almost completely inhibits in vitro post-translational translocation of pre-pro-α-factor (ppαF) in yeast microsomes at a concentration of 300 µM, with an IC50 of 6 µM [72].
Calmodulin inhibitors
Ophiobolin A and E6 Berbamine are specific antagonists of CaM, and thus disrupt the translocation competence of small-secreted proteins (SSPs) that depend on CaM during the targeting phase towards the ER [71].
Driving forces in post-translational translocation
The polymerization of peptides in the ribosome provides a directional driving force during co-translational translation. GTP is hydrolyzed during the ribosomal chain elongation step, which pushes the preprotein through the ribosomal exit tunnel and translocon pore [73] (Fig. 1). However, roughly 70 residues still remain inside the tunnel and channel after completion of translation [74] and this chain can move freely up and down through the channel. Cells employ a secondary mechanism to complete the translocation of this free-moving substrate: after sufficient downwards diffusion (due to Brownian motion) of the chain through Sec61 and towards the lumen, the chaperone BiP can bind to the exposed chain segment. Once bound to BiP, the chain segment cannot diffuse back to the cytosolic side. This starts a cycle, where stepwise binding of additional BiP molecules acts as a ‘molecular ratchet’ that pulls nascent chains towards the lumen [75, 76] (Fig. 1).
Sec61-dependent post-translational translocation (Fig. 4) requires the Sec62/63 complex and the lumenal BiP chaperone for directional protein movement through the translocon channel towards the lumen [77], similar to the function of BiP in co-translational translocation. Sec62 is an integral membrane protein that forms a stable complex with Sec63 and associates with ER-bound ribosomes, where it binds near the ribosomal exit tunnel [78] (Fig. 1). These proteins are highly abundant in the mammalian ER [79]. A study by Reithinger et al. used the yeast model system to show that Sec62 is required in addition to SRP for the targeting and translocation of uncleaved signal anchor sequences with moderate hydrophobicity [80]. Sec62 is also required for post-translational translocation of SSPs, an important class of SRP-independent proteins [77]. Due to their small size (< 160 aa), the signal peptide of these SSPs remains (partially) buried inside the ribosomal exit tunnel after completion of translation, which prevents recognition by SRP [81]. Additionally, Sec62 is suggested to function as a receptor that detects Ca2+ leakage through Sec61 and facilitates CaM recruitment [82], which in turn mediates Ca2+-dependent closure of the channel [83]. Interestingly, the different functional studies and the location of Sec62 near the ribosome suggests that both (co- and post-) translocational systems overlap at this site.
The BiP chaperone has multiple functions during both co- and post-translational translocation
ER-resident Hsp40/DnaJ-like proteins ERdj1 and Sec63 (ERdj2) are integral membrane proteins that recruit the lumenal Hsp70 member BiP (termed Kar2p in yeast) to the translocon (Figs. 1, 4a) [77, 84,85,86]. BiP consists of a substrate binding domain and a nucleotide-binding domain and can perform several functions during protein translocation: (1) BiP is proposed to drive translocon gating from the closed to the open state during early stages of translocation, as it was found to assist with insertion of precursor peptides into the channel [87]. The chaperone can bind to loop 7 of Sec61α, which forms the hinge region between both halves of the channel, and this binding energy facilitates insertion of ‘weak’ nascent peptides which are otherwise unable to open the channel by themselves [7, 88]. (2) It closes off the lumenal side of Sec61 and returns the channel to a closed state to prevent Ca2+ efflux after translocation has ended [88, 89]. (3) BiP acts as a molecular ratchet to complete translocation of pre-proteins in both co- and post-translational Sec61-dependent translocation (Figs. 1, 4a) [76, 90]. The substrate-binding domain of BiP has affinity for unfolded hydrophobic oligopeptides [91] and this affinity is regulated by its nucleotide-binding domain. The ATP-bound state of BiP has low substrate affinity while the ADP-bound state has high affinity [92]. Recruitment of BiP to the translocon complex allows Sec63 and ERdj1 to activate BiP’s ATPase activity with their lumenal J domain. This converts the bound ATP to ADP and thus increases the substrate binding affinity, which allows BiP to bind peptides as they emerge from the translocon. After completion of translocation, release of bound proteins from BiP requires exchange of ADP for ATP, which is performed by two lumenal nucleotide exchange factors: Grp170 and Sil1 [92].
SecA-dependent post-translational translocation
Bacteria (and chloroplasts in plants) uniquely contain SecA as part of their translocation systems. SecA recognizes targeting signals in cytoplasmic pre-proteins and functions as an essential motor protein that mechanically drives post-translational translocation, as it uses sequential cycles of substrate clamping and ATP-dependent domain movement to push the preprotein through the SecYEG channel [14, 93]. Interestingly, mature protein regions are also bound to a (currently unknown) site on SecA and can target the preprotein independent of their signal peptide, but this only occurs after allosteric activation of SecA by a bound signal peptide [94, 95].
Inhibition of the translocational driving force
Mycolactone
In addition to the previously described actions, mycolactone also depletes BiP and this may affect the translocation of other proteins indirectly [49]. Co-translational translocation of BiP itself could be inhibited by mycolactone too, resulting in lower luminal BiP levels, and further amplifying the inhibition of translocation for BiP-dependent substrates.
Valinomycin
The macrocycle valinomycin, isolated from an Actinomycete culture, is a known potassium anionophore [96]. Interestingly, it was identified as a down-regulator of BiP expression and induces cell death in cancer cells with glucose starvation [97]. The chaperone function of BiP protects cells during ER stress and supports the unfolded protein response. Reduced BiP levels in valinomycin-treated cancer cells can thus result in cell death under ER stress conditions, which are typical for solid tumor environments where nutrient supply is limited due to the poor vascularization. Valinomycin is also a signal peptide-specific inhibitor of hamster prion protein (PrP) translocation [98]. Inefficient translocation of PrP leads to cytosolic accumulation and misfolding of the protein, resulting in cytotoxic protein aggregates. Interestingly, human PrP was not affected by valinomycin and this selectivity was due to small differences in the signal peptides of these two homologous proteins. The downregulation of BiP is suggested as a mechanism for the selectivity of the compound, as BiP dependency is linked to targeting properties of the signal peptide: ‘weak’ signal peptides require assistance from accessory factors for translocon gating, which is one of the proposed functions of BiP.
R/1 derivatives
Two derivatives of R/1, MAL3-39 and MAL3-101, were shown to inhibit the in vitro post-translational translocation of ppαF substrates, but these derivatives are less active and only inhibit, respectively, 45 and 30% of translocation at 300 µM [99]. While R/1 affects both the innate and Hsp40-stimulated ATPase activity of Hsp70 chaperones, MAL3-39 and MAL3-101 only inhibit the J domain-mediated stimulation of Hsp70 ATPase activity. It is suggested that these two derivatives enter the ER lumen, where they modulate the functions of BiP/Kar2p and Sec63, and therefore, affect post-translational translocation.
Bactericidal ATPase inhibitors
In bacteria, most secretory and membrane proteins (~ 95% in Escherichia coli) rely on the highly conserved Sec pathway for their membrane insertion or translocation across the plasma membrane [100]. SecA is essential for SecY-dependent post-translational translocation in bacteria (Fig. 4b), but it is absent in eukaryotes, which makes this an attractive target for the development of new antibacterial drugs (Table 1).
CJ-21058, a derivative of the fungal antibiotic equisetin was discovered as the first inhibitor of E. coli SecA ATPase activity [101]. It also showed antibacterial activity against multidrug-resistant Staphylococcus aureus and Enterococcus faecalis. The fluorescein analogs rose Bengal and erythrosin B are able to inhibit the in vitro translocation of proOmpA through inhibition of the SecA ATPase activity. These compounds are predicted to occupy the ATP binding site in SecA [102]. Inhibition of ATPase activity was indeed competitive at low ATP concentrations, however, more recent data shows that rose bengal non-competitively inhibits the translocation activity of SecA at high ATP concentrations [103]. They have both bacteriostatic and bactericidal effects. Five compounds (termed P97-A9 family in Fig. 4) were identified in a small molecule screening as inhibitors of SecA translocase activity [104]. These compounds target the signal peptide binding site in SecA, a conserved and essential feature in SecA from both Gram positive and Gram negative bacterial species, and also showed weak antimicrobial activity. Recently discovered analogues of bisthiouracil [105, 106] and the bistriazole compound SCA-21 [107] are more potent inhibitors of the ATPase activity and SecA-dependent protein translocation. Moreover, they are effective against methicillin-resistant S. aureus strains. For most of these SecA inhibitors, permeability of the outer membrane in Gram negative bacteria was required for the antimicrobial activity [108]. These novel antibacterial mechanisms are promising solutions for the urgent problem of multidrug-resistant pathogens.
Alternative targeting pathways
Tail-anchored proteins
Due to their structure, TA proteins should not be able to use the SRP-driven targeting system, as their C-terminal targeting signal remains hidden inside the ribosome during protein synthesis (Fig. 5). Instead, they rely on the Bag6/SGTA complex (or Sgt2 in yeast) and a completely different set of proteins termed transmembrane domain recognition complexes (TRCs) for recognition and targeting to the ER membrane receptors WRB and CAML. These protein complexes then facilitate membrane insertion independent of Sec61 [109]. The yeast ortholog of TRC is GET; “guided entry of tail-anchored proteins”.

Alternative targeting pathways for translocation. a Tail-anchored proteins have a single transmembrane domain at their C terminus that also acts as a targeting signal (purple box). A pretargeting complex captures these C-terminal signals after release from the ribosome and transfers them to TRC40 (or Get3 in yeast) for targeting to the membrane receptor WRB/CAML (Get1/Get2 in yeast). The targeting factor TRC40 operates as a dimer and requires ATP to transition between open and closed states. Inhibitors of translocation are indicated in blue text. b The SRP-independent (SND) pathway serves as a backup system for the classical Sec61 and TRC40 targeting pathways. The membrane proteins Snd2 and Snd3 function as targeting receptors for Snd1
SRP-independent targeting
A route that functions in parallel with the SRP and TA pathways, termed SND (SRP-independent targeting), was recently discovered in Saccharomyces cerevisiae [110, 111]. Three proteins were identified (Fig. 5): Snd1 is located in the cytosol and predicted to interact with the ribosome, where it may act as the receptor for hydrophobic targeting signals. Snd2 and Snd3 form a complex in the ER membrane, together with the translocon, and could act as targeting receptors. Though only described in yeast, a human ortholog of Snd2 exists (hSnd2) and its function as a membrane receptor was recently confirmed [112]. The SND pathway was originally shown to serve as a backup targeting system for both SRP-dependent and TRC40-dependent pathways in yeast [110]. Recent evidence also showed that the TRC40 targeting pathway is not essential for integration of TA proteins in human cells, as membrane integration of TA proteins in TRC40 knockouts can be complemented by both the SND and SRP pathways [113]. This supports the idea that SRP is likely able to recognize some targeting signals inside the ribosomal exit tunnel, near the end of TA protein translation, instead of outside the ribosome [114,115,116].
Inhibitors of the TRC40 pathway
Calmodulin inhibits ER membrane insertion of mammalian TA proteins in multiple translocation pathways, likely due to binding of CaM to the targeting signals as in Sec-dependent post-translational translocation, masking them for recognition. This function of CaM has been suggested as a regulatory mechanism for the TRC40 pathway [117].
TRC40 is also involved in ER targeting of SSPs [118] and glycosylphosphatidylinositol (GPI)-anchored proteins [66]. Targeting of these proteins is thus apparently facilitated by multiple SRP-independent pathways. Mycolactone is an interesting inhibitor in this regard because it only partially affects translocation of SSPs. Some SSP pre-proteins that normally use the Sec61 pathway are able to escape the translocational block; in the presence of mycolactone, they are redirected to alternative pathways such as the TRC40 pathway. Importantly, hydrophobicity of the signal peptide and properties of the mature domain were shown to affect mycolactone sensitivity of SSPs [31].
Species-specific signal peptides
The GPI-anchored surface protein VSG_117 protein from Trypanosoma brucei, a protozoan parasite responsible for human African trypanosomiasis (sleeping sickness), is used for immune evasion in the bloodstream. Interestingly, post-translational translocation of VSG_117 is inhibited by MAL3-101, equisetin and CJ-21058 [119], three ATPase-related inhibitors (Table 1).
Trypanosomatida diverged early from other eukaryotes and they developed a surprisingly different SRP complex [120]. Importantly, T. brucei signal peptides are incompatible with the eukaryotic post-translational translocation pathway [121]. Properties of the hydrophobic region of the targeting signals appear to determine compatibility with the mammalian translocation system, likely due to their different interactions with the unique trypanosomal SRP. Most trypanosomal signal peptides are also able to use multiple translocation pathways, but they depend heavily on the SRP-independent post-translational translocation pathway for the production of their GPI-anchored proteins. Sec71, a non-essential post-translational translocon component in yeast which is not present in mammalian cells, is also present in T. brucei and essential for their survival [122].
Compounds like MAL3-101 might, therefore, offer a novel method for the treatment of trypanosomal infection. Selective inhibition of only trypanosomal post-translational translocation is possible due to the differences in the composition of host and parasite signal peptides and the corresponding translocon complexes. This key therapeutic concept of signal peptide-selective translocation inhibition was previously demonstrated in mammalian cells for the HUN-7293/cotransin family and CADA compounds (Table 1).
Conclusions
Since the discovery of the ubiquitous Sec-dependent protein translocation pathway, various translocation inhibitors have been discovered (Table 1). Furthermore, recent reports have uncovered a significant redundancy between the different protein translocation pathways, in which properties of the targeting signals determine the preference for the selected translocation system. In this review, we highlighted how multiple stages in the different translocation pathways can be modulated or even inhibited, leading to either selective or broad inhibition of protein translocation. In general, most of the translocation inhibitors described here affect the recognition, chaperoning or function of the signal peptides.
Based on the characteristic inhibitory profile of each translocation inhibitor, there is some optimism that modulation of protein translocation can be exploited for the development of new therapeutic and antimicrobial applications. Recent CRISPR/Cas9-based screenings of host factors involved in viral replication revealed an important role of translocon-associated components [123, 124]. For cotransin, it has already been shown that it limits proteostasis of enveloped viruses such as influenza virus, human immunodeficiency virus and dengue virus [125]. In addition for mycolactone, one might expect a similar broad antiviral effect as it influences translocation of a broad range of proteins. Screening for translocation inhibitors has resulted in the discovery of eeyarestatin I and apratoxin A with anticancer properties, and a new class of broad-spectrum antibacterial compounds is being developed based on inhibition of SecA. Notwithstanding the long road to go for translocation inhibitors to become therapeutics, in the mean time they are valuable as research tools to decipher the remaining mysteries of protein translocation across membranes.
Abbreviations
- BiP:
-
Binding immunoglobulin protein
- CADA:
-
Cyclotriazadisulfonamide
- CaM:
-
Calmodulin
- CRF-R-1:
-
Corticotropin-releasing factor receptor 1
- ER:
-
Endoplasmic reticulum
- ERAD:
-
Endoplasmic reticulum-associated protein degradation
- ESI:
-
Eeyarestatin I
- GET:
-
Guided entry of tail-anchored proteins
- GPI:
-
Glycosylphosphatidylinositol
- hCD4:
-
Human CD4
- HHV-7:
-
Human herpes virus-7
- HIV:
-
Human immunodeficiency virus
- Hsc70:
-
Heat shock cognate 70
- Hsp70:
-
Heat shock protein 70
- ICAM-1:
-
Intercellular adhesion molecule 1
- NC:
-
Nascent chain
- OST:
-
Oligosaccharyl-transferase complex
- PMF:
-
Proton motive force
- ppαF:
-
Pre-pro-α-factor
- prl :
-
Protein localization
- PrP:
-
Prion protein
- RNC:
-
Ribosome-nascent chain complex
- SA:
-
Signal anchor
- SND:
-
SRP-independent targeting
- SP:
-
Signal peptide
- SPC:
-
Signal peptidase complex
- SR:
-
SRP receptor
- SRP:
-
Signal recognition particle
- SSP:
-
Small secreted protein
- TA:
-
Tail-anchored membrane protein
- TMD:
-
Transmembrane domain
- TNFα:
-
Tumor necrosis factor alpha
- TRAM:
-
Translocating chain-associated membrane protein
- TRAP:
-
Translocon-associated protein
- TRC:
-
Transmembrane domain recognition complex
- UPR:
-
Unfolded protein response
- VCAM:
-
Vascular cell adhesion molecule
References
Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F (2015) Proteomics. Tissue-based map of the human proteome. Science 347(6220):1260419. https://doi.org/10.1126/science.1260419
Blobel G, Dobberstein B (1975) Transfer of proteins across membranes. II. Reconstitution of functional rough microsomes from heterologous components. J Cell Biol 67(3):852–862
Zheng N, Gierasch LM (1996) Signal sequences: the same yet different. Cell 86(6):849–852
von Heijne G (1985) Signal sequences. The limits of variation. J Mol Biol 184(1):99–105
Osborne AR, Rapoport TA, van den Berg B (2005) Protein translocation by the Sec61/SecY channel. Annu Rev Cell Dev Biol 21:529–550. https://doi.org/10.1146/annurev.cellbio.21.012704.133214
Kutay U, Hartmann E, Rapoport TA (1993) A class of membrane proteins with a C-terminal anchor. Trends Cell Biol 3(3):72–75
Dudek J, Pfeffer S, Lee PH, Jung M, Cavalie A, Helms V, Forster F, Zimmermann R (2015) Protein transport into the human endoplasmic reticulum. J Mol Biol 427(6):1159–1175. https://doi.org/10.1016/j.jmb.2014.06.011
Van den Berg B, Clemons WM Jr, Collinson I, Modis Y, Hartmann E, Harrison SC, Rapoport TA (2004) X-ray structure of a protein-conducting channel. Nature 427(6969):36–44. https://doi.org/10.1038/nature02218
Kang Y, Fielden LF, Stojanovski D (2017) Mitochondrial protein transport in health and disease. Semin Cell Dev Biol. https://doi.org/10.1016/j.semcdb.2017.07.028
Chotewutmontri P, Holbrook K, Bruce BD (2017) Plastid protein targeting: preprotein recognition and translocation. Int Rev Cell Mol Biol 330:227–294. https://doi.org/10.1016/bs.ircmb.2016.09.006
Francisco T, Rodrigues TA, Dias AF, Barros-Barbosa A, Bicho D, Azevedo JE (2017) Protein transport into peroxisomes: knowns and unknowns. BioEssays. https://doi.org/10.1002/bies.201700047
Green ER, Mecsas J (2016) Bacterial secretion systems: an overview. Microbiol Spectr. https://doi.org/10.1128/microbiolspec.VMBF-0012-2015
Halic M, Becker T, Pool MR, Spahn CM, Grassucci RA, Frank J, Beckmann R (2004) Structure of the signal recognition particle interacting with the elongation-arrested ribosome. Nature 427(6977):808–814. https://doi.org/10.1038/nature02342
Park E, Rapoport TA (2012) Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Annu Rev Biophys 41:21–40. https://doi.org/10.1146/annurev-biophys-050511-102312
Saraogi I, Shan SO (2011) Molecular mechanism of co-translational protein targeting by the signal recognition particle. Traffic 12(5):535–542. https://doi.org/10.1111/j.1600-0854.2011.01171.x
Gogala M, Becker T, Beatrix B, Armache JP, Barrio-Garcia C, Berninghausen O, Beckmann R (2014) Structures of the Sec61 complex engaged in nascent peptide translocation or membrane insertion. Nature 506(7486):107–110. https://doi.org/10.1038/nature12950
Plath K, Mothes W, Wilkinson BM, Stirling CJ, Rapoport TA (1998) Signal sequence recognition in posttranslational protein transport across the yeast ER membrane. Cell 94(6):795–807
Voorhees RM, Hegde RS (2016) Structure of the Sec61 channel opened by a signal sequence. Science 351(6268):88–91. https://doi.org/10.1126/science.aad4992
Rapoport TA, Goder V, Heinrich SU, Matlack KE (2004) Membrane-protein integration and the role of the translocation channel. Trends Cell Biol 14(10):568–575. https://doi.org/10.1016/j.tcb.2004.09.002
Sadlish H, Pitonzo D, Johnson AE, Skach WR (2005) Sequential triage of transmembrane segments by Sec61α during biogenesis of a native multispanning membrane protein. Nat Struct Mol Biol 12(10):870–878. https://doi.org/10.1038/nsmb994
Wang Q, Mora-Jensen H, Weniger MA, Perez-Galan P, Wolford C, Hai T, Ron D, Chen W, Trenkle W, Wiestner A, Ye Y (2009) ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc Natl Acad Sci USA 106(7):2200–2205. https://doi.org/10.1073/pnas.0807611106
Ruggiano A, Foresti O, Carvalho P (2014) Quality control: ER-associated degradation: protein quality control and beyond. J Cell Biol 204(6):869–879. https://doi.org/10.1083/jcb.201312042
Wang Q, Li L, Ye Y (2008) Inhibition of p97-dependent protein degradation by Eeyarestatin I. J Biol Chem 283(12):7445–7454. https://doi.org/10.1074/jbc.M708347200
Cross BC, McKibbin C, Callan AC, Roboti P, Piacenti M, Rabu C, Wilson CM, Whitehead R, Flitsch SL, Pool MR, High S, Swanton E (2009) Eeyarestatin I inhibits Sec61-mediated protein translocation at the endoplasmic reticulum. J Cell Sci 122(Pt 23):4393–4400. https://doi.org/10.1242/jcs.054494
McKibbin C, Mares A, Piacenti M, Williams H, Roboti P, Puumalainen M, Callan AC, Lesiak-Mieczkowska K, Linder S, Harant H, High S, Flitsch SL, Whitehead RC, Swanton E (2012) Inhibition of protein translocation at the endoplasmic reticulum promotes activation of the unfolded protein response. Biochem J 442(3):639–648. https://doi.org/10.1042/BJ20111220
Hetz C, Papa FR (2017) The unfolded protein response and cell fate control. Mol Cell. https://doi.org/10.1016/j.molcel.2017.06.017
Aletrari MO, McKibbin C, Williams H, Pawar V, Pietroni P, Lord JM, Flitsch SL, Whitehead R, Swanton E, High S, Spooner RA (2011) Eeyarestatin 1 interferes with both retrograde and anterograde intracellular trafficking pathways. PLoS ONE 6(7):e22713. https://doi.org/10.1371/journal.pone.0022713
George KM, Chatterjee D, Gunawardana G, Welty D, Hayman J, Lee R, Small PL (1999) Mycolactone: a polyketide toxin from Mycobacterium ulcerans required for virulence. Science 283(5403):854–857
Hall BS, Hill K, McKenna M, Ogbechi J, High S, Willis AE, Simmonds RE (2014) The pathogenic mechanism of the Mycobacterium ulcerans virulence factor, mycolactone, depends on blockade of protein translocation into the ER. PLoS Pathog 10(4):e1004061. https://doi.org/10.1371/journal.ppat.1004061
Grotzke JE, Kozik P, Morel JD, Impens F, Pietrosemoli N, Cresswell P, Amigorena S, Demangel C (2017) Sec61 blockade by mycolactone inhibits antigen cross-presentation independently of endosome-to-cytosol export. Proc Natl Acad Sci USA 114(29):E5910–E5919. https://doi.org/10.1073/pnas.1705242114
McKenna M, Simmonds RE, High S (2016) Mechanistic insights into the inhibition of Sec61-dependent co- and post-translational translocation by mycolactone. J Cell Sci 129(7):1404–1415. https://doi.org/10.1242/jcs.182352
Koopmann JO, Albring J, Huter E, Bulbuc N, Spee P, Neefjes J, Hammerling GJ, Momburg F (2000) Export of antigenic peptides from the endoplasmic reticulum intersects with retrograde protein translocation through the Sec61p channel. Immunity 13(1):117–127
Wirth A, Jung M, Bies C, Frien M, Tyedmers J, Zimmermann R, Wagner R (2003) The Sec61p complex is a dynamic precursor activated channel. Mol Cell 12(1):261–268
Schauble N, Cavalie A, Zimmermann R, Jung M (2014) Interaction of Pseudomonas aeruginosa Exotoxin A with the human Sec61 complex suppresses passive calcium efflux from the endoplasmic reticulum. Channels (Austin) 8(1):76–83. https://doi.org/10.4161/chan.26526
Foster CA, Dreyfuss M, Mandak B, Meingassner JG, Naegeli HU, Nussbaumer A, Oberer L, Scheel G, Swoboda EM (1994) Pharmacological modulation of endothelial cell-associated adhesion molecule expression: implications for future treatment of dermatological diseases. J Dermatol 21(11):847–854
Garrison JL, Kunkel EJ, Hegde RS, Taunton J (2005) A substrate-specific inhibitor of protein translocation into the endoplasmic reticulum. Nature 436(7048):285–289. https://doi.org/10.1038/nature03821
Westendorf C, Schmidt A, Coin I, Furkert J, Ridelis I, Zampatis D, Rutz C, Wiesner B, Rosenthal W, Beyermann M, Schulein R (2011) Inhibition of biosynthesis of human endothelin B receptor by the cyclodepsipeptide cotransin. J Biol Chem 286(41):35588–35600. https://doi.org/10.1074/jbc.M111.239244
Ruiz-Saenz A, Sandhu M, Carrasco Y, Maglathlin RL, Taunton J, Moasser MM (2015) Targeting HER3 by interfering with its Sec61-mediated cotranslational insertion into the endoplasmic reticulum. Oncogene 34(41):5288–5294. https://doi.org/10.1038/onc.2014.455
Maifeld SV, MacKinnon AL, Garrison JL, Sharma A, Kunkel EJ, Hegde RS, Taunton J (2011) Secretory protein profiling reveals TNF-alpha inactivation by selective and promiscuous Sec61 modulators. Chem Biol 18(9):1082–1088. https://doi.org/10.1016/j.chembiol.2011.06.015
Klein W, Westendorf C, Schmidt A, Conill-Cortes M, Rutz C, Blohs M, Beyermann M, Protze J, Krause G, Krause E, Schulein R (2015) Defining a conformational consensus motif in cotransin-sensitive signal sequences: a proteomic and site-directed mutagenesis study. PLoS ONE 10(3):e0120886. https://doi.org/10.1371/journal.pone.0120886
Junne T, Wong J, Studer C, Aust T, Bauer BW, Beibel M, Bhullar B, Bruccoleri R, Eichenberger J, Estoppey D, Hartmann N, Knapp B, Krastel P, Melin N, Oakeley EJ, Oberer L, Riedl R, Roma G, Schuierer S, Petersen F, Tallarico JA, Rapoport TA, Spiess M, Hoepfner D (2015) Decatransin, a new natural product inhibiting protein translocation at the Sec61/SecYEG translocon. J Cell Sci 128(6):1217–1229. https://doi.org/10.1242/jcs.165746
Winn M, Fyans JK, Zhuo Y, Micklefield J (2016) Recent advances in engineering nonribosomal peptide assembly lines. Nat Prod Rep 33(2):317–347. https://doi.org/10.1039/c5np00099h
Luesch H, Yoshida WY, Moore RE, Paul VJ, Corbett TH (2001) Total structure determination of apratoxin A, a potent novel cytotoxin from the marine cyanobacterium Lyngbya majuscula. J Am Chem Soc 123(23):5418–5423
Luesch H, Chanda SK, Raya RM, DeJesus PD, Orth AP, Walker JR, Izpisua Belmonte JC, Schultz PG (2006) A functional genomics approach to the mode of action of apratoxin A. Nat Chem Biol 2(3):158–167. https://doi.org/10.1038/nchembio769
Liu Y, Law BK, Luesch H (2009) Apratoxin a reversibly inhibits the secretory pathway by preventing cotranslational translocation. Mol Pharmacol 76(1):91–104. https://doi.org/10.1124/mol.109.056085
MacKinnon AL, Garrison JL, Hegde RS, Taunton J (2007) Photo-leucine incorporation reveals the target of a cyclodepsipeptide inhibitor of cotranslational translocation. J Am Chem Soc 129(47):14560–14561. https://doi.org/10.1021/ja076250y
Mackinnon AL, Paavilainen VO, Sharma A, Hegde RS, Taunton J (2014) An allosteric Sec61 inhibitor traps nascent transmembrane helices at the lateral gate. Elife 3:e01483. https://doi.org/10.7554/eLife.01483
Paatero AO, Kellosalo J, Dunyak BM, Almaliti J, Gestwicki JE, Gerwick WH, Taunton J, Paavilainen VO (2016) Apratoxin kills cells by direct blockade of the Sec61 protein translocation channel. Cell Chem Biol 23(5):561–566. https://doi.org/10.1016/j.chembiol.2016.04.008
Baron L, Paatero AO, Morel JD, Impens F, Guenin-Mace L, Saint-Auret S, Blanchard N, Dillmann R, Niang F, Pellegrini S, Taunton J, Paavilainen VO, Demangel C (2016) Mycolactone subverts immunity by selectively blocking the Sec61 translocon. J Exp Med 213(13):2885–2896. https://doi.org/10.1084/jem.20160662
Emr SD, Hanley-Way S, Silhavy TJ (1981) Suppressor mutations that restore export of a protein with a defective signal sequence. Cell 23(1):79–88
Junne T, Schwede T, Goder V, Spiess M (2007) Mutations in the Sec61p channel affecting signal sequence recognition and membrane protein topology. J Biol Chem 282(45):33201–33209. https://doi.org/10.1074/jbc.M707219200
Voorhees RM, Fernandez IS, Scheres SH, Hegde RS (2014) Structure of the mammalian ribosome-Sec61 complex to 3.4 A resolution. Cell 157(7):1632–1643. https://doi.org/10.1016/j.cell.2014.05.024
Hegde RS, Kang SW (2008) The concept of translocational regulation. J Cell Biol 182(2):225–232. https://doi.org/10.1083/jcb.200804157
Vermeire K, Zhang Y, Princen K, Hatse S, Samala MF, Dey K, Choi HJ, Ahn Y, Sodoma A, Snoeck R, Andrei G, De Clercq E, Bell TW, Schols D (2002) CADA inhibits human immunodeficiency virus and human herpesvirus 7 replication by down-modulation of the cellular CD4 receptor. Virology 302(2):342–353
Vermeire K, Bell TW, Van Puyenbroeck V, Giraut A, Noppen S, Liekens S, Schols D, Hartmann E, Kalies KU, Marsh M (2014) Signal peptide-binding drug as a selective inhibitor of co-translational protein translocation. PLoS Biol 12(12):e1002011. https://doi.org/10.1371/journal.pbio.1002011
Goder V, Spiess M (2003) Molecular mechanism of signal sequence orientation in the endoplasmic reticulum. EMBO J 22(14):3645–3653. https://doi.org/10.1093/emboj/cdg361
Devaraneni PK, Conti B, Matsumura Y, Yang Z, Johnson AE, Skach WR (2011) Stepwise insertion and inversion of a type II signal anchor sequence in the ribosome-Sec61 translocon complex. Cell 146(1):134–147. https://doi.org/10.1016/j.cell.2011.06.004
Van Puyenbroeck V, Claeys E, Schols D, Bell TW, Vermeire K (2017) A proteomic survey indicates sortilin as a secondary substrate of the ER translocation inhibitor cyclotriazadisulfonamide (CADA). Mol Cell Proteomics 16(2):157–167. https://doi.org/10.1074/mcp.M116.061051
Demillo VG, Goulinet-Mateo F, Kim J, Schols D, Vermeire K, Bell TW (2011) Unsymmetrical cyclotriazadisulfonamide (CADA) compounds as human CD4 receptor down-modulating agents. J Med Chem 54(16):5712–5721. https://doi.org/10.1021/jm2002603
Chawla R, Van Puyenbroeck V, Pflug NC, Sama A, Ali R, Schols D, Vermeire K, Bell TW (2016) Tuning side arm electronics in unsymmetrical cyclotriazadisulfonamide (CADA) endoplasmic reticulum (ER) translocation inhibitors to improve their human cluster of differentiation 4 (CD4) receptor down-modulating potencies. J Med Chem 59(6):2633–2647. https://doi.org/10.1021/acs.jmedchem.5b01832
Besemer J, Harant H, Wang S, Oberhauser B, Marquardt K, Foster CA, Schreiner EP, de Vries JE, Dascher-Nadel C, Lindley IJ (2005) Selective inhibition of cotranslational translocation of vascular cell adhesion molecule 1. Nature 436(7048):290–293. https://doi.org/10.1038/nature03670
Harant H, Lettner N, Hofer L, Oberhauser B, de Vries JE, Lindley IJ (2006) The translocation inhibitor CAM741 interferes with vascular cell adhesion molecule 1 signal peptide insertion at the translocon. J Biol Chem 281(41):30492–30502. https://doi.org/10.1074/jbc.M607243200
Harant H, Wolff B, Schreiner EP, Oberhauser B, Hofer L, Lettner N, Maier S, de Vries JE, Lindley IJ (2007) Inhibition of vascular endothelial growth factor cotranslational translocation by the cyclopeptolide CAM741. Mol Pharmacol 71(6):1657–1665. https://doi.org/10.1124/mol.107.034249
Hessa T, Meindl-Beinker NM, Bernsel A, Kim H, Sato Y, Lerch-Bader M, Nilsson I, White SH, von Heijne G (2007) Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature 450(7172):1026–1030. https://doi.org/10.1038/nature06387
Johnson N, Powis K, High S (2013) Post-translational translocation into the endoplasmic reticulum. Biochim Biophys Acta 11:2403–2409. https://doi.org/10.1016/j.bbamcr.2012.12.008
Ast T, Schuldiner M (2013) All roads lead to Rome (but some may be harder to travel): SRP-independent translocation into the endoplasmic reticulum. Crit Rev Biochem Mol Biol 48(3):273–288. https://doi.org/10.3109/10409238.2013.782999
Aviram N, Schuldiner M (2014) Embracing the void—how much do we really know about targeting and translocation to the endoplasmic reticulum? Curr Opin Cell Biol 29:8–17. https://doi.org/10.1016/j.ceb.2014.02.004
Ng DT, Brown JD, Walter P (1996) Signal sequences specify the targeting route to the endoplasmic reticulum membrane. J Cell Biol 134(2):269–278
Hegde RS, Bernstein HD (2006) The surprising complexity of signal sequences. Trends Biochem Sci 31(10):563–571. https://doi.org/10.1016/j.tibs.2006.08.004
Cross BC, Sinning I, Luirink J, High S (2009) Delivering proteins for export from the cytosol. Nat Rev Mol Cell Biol 10(4):255–264. https://doi.org/10.1038/nrm2657
Shao S, Hegde RS (2011) A calmodulin-dependent translocation pathway for small secretory proteins. Cell 147(7):1576–1588. https://doi.org/10.1016/j.cell.2011.11.048
Fewell SW, Day BW, Brodsky JL (2001) Identification of an inhibitor of hsc70-mediated protein translocation and ATP hydrolysis. J Biol Chem 276(2):910–914. https://doi.org/10.1074/jbc.M008535200
Connolly T, Gilmore R (1986) Formation of a functional ribosome-membrane junction during translocation requires the participation of a GTP-binding protein. J Cell Biol 103(6 Pt 1):2253–2261
Zimmermann R, Eyrisch S, Ahmad M, Helms V (2011) Protein translocation across the ER membrane. Biochim Biophys Acta 3:912–924. https://doi.org/10.1016/j.bbamem.2010.06.015
Matlack KES, Misselwitz B, Plath K, Rapoport TA (1999) BiP acts as a molecular ratchet during posttranslational transport of prepro-alpha factor across the ER membrane. Cell 97(5):553–564. https://doi.org/10.1016/S0092-8674(00)80767-9
Tyedmers J, Lerner M, Wiedmann M, Volkmer J, Zimmermann R (2003) Polypeptide-binding proteins mediate completion of co-translational protein translocation into the mammalian endoplasmic reticulum. EMBO Rep 4(5):505–510. https://doi.org/10.1038/sj.embor.embor826
Lang S, Benedix J, Fedeles SV, Schorr S, Schirra C, Schauble N, Jalal C, Greiner M, Hassdenteufel S, Tatzelt J, Kreutzer B, Edelmann L, Krause E, Rettig J, Somlo S, Zimmermann R, Dudek J (2012) Different effects of Sec61alpha, Sec62 and Sec63 depletion on transport of polypeptides into the endoplasmic reticulum of mammalian cells. J Cell Sci 125(Pt 8):1958–1969. https://doi.org/10.1242/jcs.096727
Muller L, de Escauriaza MD, Lajoie P, Theis M, Jung M, Muller A, Burgard C, Greiner M, Snapp EL, Dudek J, Zimmermann R (2010) Evolutionary gain of function for the ER membrane protein Sec62 from yeast to humans. Mol Biol Cell 21(5):691–703. https://doi.org/10.1091/mbc.E09-08-0730
Tyedmers J, Lerner M, Bies C, Dudek J, Skowronek MH, Haas IG, Heim N, Nastainczyk W, Volkmer J, Zimmermann R (2000) Homologs of the yeast Sec complex subunits Sec62p and Sec63p are abundant proteins in dog pancreas microsomes. Proc Natl Acad Sci USA 97(13):7214–7219
Reithinger JH, Kim JE, Kim H (2013) Sec62 protein mediates membrane insertion and orientation of moderately hydrophobic signal anchor proteins in the endoplasmic reticulum (ER). J Biol Chem 288(25):18058–18067. https://doi.org/10.1074/jbc.M113.473009
Lakkaraju AK, Thankappan R, Mary C, Garrison JL, Taunton J, Strub K (2012) Efficient secretion of small proteins in mammalian cells relies on Sec62-dependent posttranslational translocation. Mol Biol Cell 23(14):2712–2722. https://doi.org/10.1091/mbc.E12-03-0228
Linxweiler M, Schorr S, Schauble N, Jung M, Linxweiler J, Langer F, Schafers HJ, Cavalie A, Zimmermann R, Greiner M (2013) Targeting cell migration and the endoplasmic reticulum stress response with calmodulin antagonists: a clinically tested small molecule phenocopy of SEC62 gene silencing in human tumor cells. BMC Cancer 13:574. https://doi.org/10.1186/1471-2407-13-574
Erdmann F, Schauble N, Lang S, Jung M, Honigmann A, Ahmad M, Dudek J, Benedix J, Harsman A, Kopp A, Helms V, Cavalie A, Wagner R, Zimmermann R (2011) Interaction of calmodulin with Sec61alpha limits Ca2+ leakage from the endoplasmic reticulum. EMBO J 30(1):17–31. https://doi.org/10.1038/emboj.2010.284
Dudek J, Greiner M, Muller A, Hendershot LM, Kopsch K, Nastainczyk W, Zimmermann R (2005) ERj1p has a basic role in protein biogenesis at the endoplasmic reticulum. Nat Struct Mol Biol 12(11):1008–1014. https://doi.org/10.1038/nsmb1007
Blau M, Mullapudi S, Becker T, Dudek J, Zimmermann R, Penczek PA, Beckmann R (2005) ERj1p uses a universal ribosomal adaptor site to coordinate the 80S ribosome at the membrane. Nat Struct Mol Biol 12(11):1015–1016. https://doi.org/10.1038/nsmb998
Otero JH, Lizak B, Hendershot LM (2010) Life and death of a BiP substrate. Semin Cell Dev Biol 21(5):472–478. https://doi.org/10.1016/j.semcdb.2009.12.008
Dierks T, Volkmer J, Schlenstedt G, Jung C, Sandholzer U, Zachmann K, Schlotterhose P, Neifer K, Schmidt B, Zimmermann R (1996) A microsomal ATP-binding protein involved in efficient protein transport into the mammalian endoplasmic reticulum. EMBO J 15(24):6931–6942
Schauble N, Lang S, Jung M, Cappel S, Schorr S, Ulucan O, Linxweiler J, Dudek J, Blum R, Helms V, Paton AW, Paton JC, Cavalie A, Zimmermann R (2012) BiP-mediated closing of the Sec61 channel limits Ca2+ leakage from the ER. EMBO J 31(15):3282–3296. https://doi.org/10.1038/emboj.2012.189
Hamman BD, Hendershot LM, Johnson AE (1998) BiP maintains the permeability barrier of the ER membrane by sealing the lumenal end of the translocon pore before and early in translocation. Cell 92(6):747–758. https://doi.org/10.1016/S0092-8674(00)81403-8
Nicchitta CV, Blobel G (1993) Lumenal proteins of the mammalian endoplasmic reticulum are required to complete protein translocation. Cell 73(5):989–998
Flynn GC, Pohl J, Flocco MT, Rothman JE (1991) Peptide-binding specificity of the molecular chaperone BiP. Nature 353(6346):726–730. https://doi.org/10.1038/353726a0
Dudek J, Benedix J, Cappel S, Greiner M, Jalal C, Muller L, Zimmermann R (2009) Functions and pathologies of BiP and its interaction partners. Cell Mol Life Sci 66(9):1556–1569. https://doi.org/10.1007/s00018-009-8745-y
Chatzi KE, Sardis MF, Economou A, Karamanou S (2014) SecA-mediated targeting and translocation of secretory proteins. Biochim Biophys Acta 8:1466–1474. https://doi.org/10.1016/j.bbamcr.2014.02.014
Gouridis G, Karamanou S, Gelis I, Kalodimos CG, Economou A (2009) Signal peptides are allosteric activators of the protein translocase. Nature 462(7271):363–367. https://doi.org/10.1038/nature08559
Tsirigotaki A, De Geyter J, Sostaric N, Economou A, Karamanou S (2017) Protein export through the bacterial Sec pathway. Nat Rev Microbiol 15(1):21–36. https://doi.org/10.1038/nrmicro.2016.161
Berezin SK (2015) Valinomycin as a classical anionophore: mechanism and ion selectivity. J Membr Biol 248(4):713–726. https://doi.org/10.1007/s00232-015-9784-y
Ryoo IJ, Park HR, Choo SJ, Hwang JH, Park YM, Bae KH, Shin-Ya K, Yoo ID (2006) Selective cytotoxic activity of valinomycin against HT-29 Human colon carcinoma cells via down-regulation of GRP78. Biol Pharm Bull 29(4):817–820
Kim J, Choi I, Park JY, Kang SW (2013) Specific inhibition of hamster prion protein translocation by the dodecadepsipeptide valinomycin. Exp Cell Res 319(13):2049–2057. https://doi.org/10.1016/j.yexcr.2013.04.012
Fewell SW, Smith CM, Lyon MA, Dumitrescu TP, Wipf P, Day BW, Brodsky JL (2004) Small molecule modulators of endogenous and co-chaperone-stimulated Hsp70 ATPase activity. J Biol Chem 279(49):51131–51140. https://doi.org/10.1074/jbc.M404857200
Orfanoudaki G, Economou A (2014) Proteome-wide subcellular topologies of E. coli polypeptides database (STEPdb). Mol Cell Proteomics 13(12):3674–3687. https://doi.org/10.1074/mcp.O114.041137
Sugie Y, Inagaki S, Kato Y, Nishida H, Pang CH, Saito T, Sakemi S, Dib-Hajj F, Mueller JP, Sutcliffe J, Kojima Y (2002) CJ-21,058, a new SecA inhibitor isolated from a fungus. J Antibiot (Tokyo) 55(1):25–29
Huang YJ, Wang H, Gao FB, Li M, Yang H, Wang B, Tai PC (2012) Fluorescein analogues inhibit SecA ATPase: the first sub-micromolar inhibitor of bacterial protein translocation. ChemMedChem 7(4):571–577. https://doi.org/10.1002/cmdc.201100594
Hsieh YH, Huang YJ, Jin JS, Yu L, Yang H, Jiang C, Wang B, Tai PC (2014) Mechanisms of Rose Bengal inhibition on SecA ATPase and ion channel activities. Biochem Biophys Res Commun 454(2):308–312. https://doi.org/10.1016/j.bbrc.2014.10.070
De Waelheyns E, Segers K, Sardis MF, Anne J, Nicolaes GA, Economou A (2015) Identification of small-molecule inhibitors against SecA by structure-based virtual ligand screening. J Antibiot (Tokyo) 68(11):666–673. https://doi.org/10.1038/ja.2015.53
Cui P, Li X, Zhu M, Wang B, Liu J, Chen H (2017) Design, synthesis and antimicrobial activities of thiouracil derivatives containing triazolo-thiadiazole as SecA inhibitors. Eur J Med Chem 127:159–165. https://doi.org/10.1016/j.ejmech.2016.12.053
Cui P, Li X, Zhu M, Wang B, Liu J, Chen H (2017) Design, synthesis and antibacterial activities of thiouracil derivatives containing acyl thiourea as SecA inhibitors. Bioorg Med Chem Lett 27(10):2234–2237. https://doi.org/10.1016/j.bmcl.2016.11.060
Cui J, Jin J, Chaudhary AS, Hsieh YH, Zhang H, Dai C, Damera K, Chen W, Tai PC, Wang B (2016) Design, synthesis and evaluation of triazole-pyrimidine analogues as SecA inhibitors. ChemMedChem 11(1):43–56. https://doi.org/10.1002/cmdc.201500447
Jin J, Hsieh YH, Cui J, Damera K, Dai C, Chaudhary AS, Zhang H, Yang H, Cao N, Jiang C, Vaara M, Wang B, Tai PC (2016) Using chemical probes to assess the feasibility of targeting SecA for developing antimicrobial agents against Gram-negative bacteria. ChemMedChem 11(22):2511–2521. https://doi.org/10.1002/cmdc.201600421
Hegde RS, Keenan RJ (2011) Tail-anchored membrane protein insertion into the endoplasmic reticulum. Nat Rev Mol Cell Biol 12(12):787–798. https://doi.org/10.1038/nrm3226
Aviram N, Ast T, Costa EA, Arakel EC, Chuartzman SG, Jan CH, Hassdenteufel S, Dudek J, Jung M, Schorr S, Zimmermann R, Schwappach B, Weissman JS, Schuldiner M (2016) The SND proteins constitute an alternative targeting route to the endoplasmic reticulum. Nature 540(7631):134–138. https://doi.org/10.1038/nature20169
Ast T, Cohen G, Schuldiner M (2013) A network of cytosolic factors targets SRP-independent proteins to the endoplasmic reticulum. Cell 152(5):1134–1145. https://doi.org/10.1016/j.cell.2013.02.003
Hassdenteufel S, Sicking M, Schorr S, Aviram N, Fecher-Trost C, Schuldiner M, Jung M, Zimmermann R, Lang S (2017) hSnd2 protein represents an alternative targeting factor to the endoplasmic reticulum in human cells. FEBS Lett. https://doi.org/10.1002/1873-3468.12831
Casson J, McKenna M, Hassdenteufel S, Aviram N, Zimmerman R, High S (2017) Multiple pathways facilitate the biogenesis of mammalian tail-anchored proteins. J Cell Sci. https://doi.org/10.1242/jcs.207829
Abell BM, Pool MR, Schlenker O, Sinning I, High S (2004) Signal recognition particle mediates post-translational targeting in eukaryotes. EMBO J 23(14):2755–2764. https://doi.org/10.1038/sj.emboj.7600281
Berndt U, Oellerer S, Zhang Y, Johnson AE, Rospert S (2009) A signal-anchor sequence stimulates signal recognition particle binding to ribosomes from inside the exit tunnel. Proc Natl Acad Sci USA 106(5):1398–1403. https://doi.org/10.1073/pnas.0808584106
Voorhees RM, Hegde RS (2015) Structures of the scanning and engaged states of the mammalian SRP-ribosome complex. Elife. https://doi.org/10.7554/eLife.07975
Hassdenteufel S, Schauble N, Cassella P, Leznicki P, Muller A, High S, Jung M, Zimmermann R (2011) Ca2+ -calmodulin inhibits tail-anchored protein insertion into the mammalian endoplasmic reticulum membrane. FEBS Lett 585(21):3485–3490. https://doi.org/10.1016/j.febslet.2011.10.008
Johnson N, Vilardi F, Lang S, Leznicki P, Zimmermann R, High S (2012) TRC40 can deliver short secretory proteins to the Sec61 translocon. J Cell Sci 125(Pt 15):3612–3620. https://doi.org/10.1242/jcs.102608
Patham B, Duffy J, Lane A, Davis RC, Wipf P, Fewell SW, Brodsky JL, Mensa-Wilmot K (2009) Post-translational import of protein into the endoplasmic reticulum of a trypanosome: an in vitro system for discovery of anti-trypanosomal chemical entities. Biochem J 419(2):507–517. https://doi.org/10.1042/BJ20081787
Liu L, Ben-Shlomo H, Xu YX, Stern MZ, Goncharov I, Zhang Y, Michaeli S (2003) The trypanosomatid signal recognition particle consists of two RNA molecules, a 7SL RNA homologue and a novel tRNA-like molecule. J Biol Chem 278(20):18271–18280. https://doi.org/10.1074/jbc.M209215200
Al-Qahtani A, Teilhet M, Mensa-Wilmot K (1998) Species-specificity in endoplasmic reticulum signal peptide utilization revealed by proteins from Trypanosoma brucei and Leishmania. Biochem J 331(Pt 2):521–529
Goldshmidt H, Sheiner L, Butikofer P, Roditi I, Uliel S, Gunzel M, Engstler M, Michaeli S (2008) Role of protein translocation pathways across the endoplasmic reticulum in Trypanosoma brucei. J Biol Chem 283(46):32085–32098. https://doi.org/10.1074/jbc.M801499200
Marceau CD, Puschnik AS, Majzoub K, Ooi YS, Brewer SM, Fuchs G, Swaminathan K, Mata MA, Elias JE, Sarnow P, Carette JE (2016) Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 535(7610):159–163. https://doi.org/10.1038/nature18631
Zhang R, Miner JJ, Gorman MJ, Rausch K, Ramage H, White JP, Zuiani A, Zhang P, Fernandez E, Zhang Q, Dowd KA, Pierson TC, Cherry S, Diamond MS (2016) A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature 535(7610):164–168. https://doi.org/10.1038/nature18625
Heaton NS, Moshkina N, Fenouil R, Gardner TJ, Aguirre S, Shah PS, Zhao N, Manganaro L, Hultquist JF, Noel J, Sachs D, Hamilton J, Leon PE, Chawdury A, Tripathi S, Melegari C, Campisi L, Hai R, Metreveli G, Gamarnik AV, Garcia-Sastre A, Greenbaum B, Simon V, Fernandez-Sesma A, Krogan NJ, Mulder LCF, van Bakel H, Tortorella D, Taunton J, Palese P, Marazzi I (2016) Targeting viral proteostasis limits influenza virus, HIV, and dengue virus infection. Immunity 44(1):46–58. https://doi.org/10.1016/j.immuni.2015.12.017
Wang Q, Shinkre BA, Lee JG, Weniger MA, Liu Y, Chen W, Wiestner A, Trenkle WC, Ye Y (2010) The ERAD inhibitor Eeyarestatin I is a bifunctional compound with a membrane-binding domain and a p97/VCP inhibitory group. PLoS ONE 5(11):e15479. https://doi.org/10.1371/journal.pone.0015479
Fiebiger E, Hirsch C, Vyas JM, Gordon E, Ploegh HL, Tortorella D (2004) Dissection of the dislocation pathway for type I membrane proteins with a new small molecule inhibitor, eeyarestatin. Mol Biol Cell 15(4):1635–1646. https://doi.org/10.1091/mbc.E03-07-0506
Hu ZY, Gong YS, Huang WL (1992) Interaction of berbamine compound E6 and calmodulin-dependent myosin light chain kinase. Biochem Pharmacol 44(8):1543–1547
Au TK, Chick WS, Leung PC (2000) The biology of ophiobolins. Life Sci 67(7):733–742
Acknowledgements
This work was supported by the KU Leuven (GOA no. 10/014 and PF/10/018) and the FWO (No. G.485.08).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Van Puyenbroeck, V., Vermeire, K. Inhibitors of protein translocation across membranes of the secretory pathway: novel antimicrobial and anticancer agents. Cell. Mol. Life Sci. 75, 1541–1558 (2018). https://doi.org/10.1007/s00018-017-2743-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-017-2743-2