
Abstract
Adult neurogenesis, the birth of new neurons in the mature brain, has attracted considerable attention in the last decade. One of the earliest identified and most profound factors that affect adult neurogenesis both positively and negatively is stress. Here, we review the complex interplay between stress and adult neurogenesis. In particular, we review the role of the glucocorticoid receptor, the main mediator of the stress response in the proliferation, differentiation, migration, and functional integration of newborn neurons in the hippocampus. We review a multitude of mechanisms regulating glucocorticoid receptor activity in relationship to adult neurogenesis. We postulate a novel concept in which the level of glucocorticoid receptor expression directly regulates the excitation-inhibition balance, which is key for proper neurogenesis. We furthermore argue that an excitation-inhibition dis-balance may underlie aberrant functional integration of newborn neurons that is associated with psychiatric and paroxysmal brain disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
“In the adult centres, the nerve paths are something fixed, ended, and immutable. Everything may die, nothing may be regenerated.” These words from Ramon y Cajal in 1913 [1] highlight what has been one of the central dogma’s in neuroscience for a long time: the birth of new neurons, i.e., neurogenesis, was restricted to prenatal and early postnatal development and that the adult mammalian brain was unable to produce new neurons. However, in the 1960s, Joseph Altman and colleagues showed first evidence for adult neurogenesis in the brain of rodents [2–4]. Although these data were received with skepticism by the scientific community, results were reproduced and revealed the neuronal phenotype derived from dividing cells in the hippocampus [5].
In the 1990s, pioneering studies by Weiss and colleagues identified stem cell-like cells from the adult brain that were able to differentiate into neurons and astrocytes [6]. Important for the acceptance of the concept of adult neurogenesis has been the development of novel techniques and scientific methods. Cell division for example, can be marked using bromodeoxyuridine (BrdU) or [3H]thymidine. These molecules label and incorporate into DNA of dividing cells and can be visualized with electron and confocal microscopy [7]. By varying the paradigm and the examination time points after injection, these techniques allow quantitative analysis of proliferation, differentiation, and survival. During the last decades, it also became clear that developing neurons express distinct markers during their maturation process [8]. BrdU-labeled DNA in combination with immuno-histochemical analysis of the expression of these specific neuronal markers by confocal microscopy unambiguously revealed the existence of neurogenesis in the adult brain [9]. For example, for immature newborn neurons, doublecortin (DCX) is regularly used, while for mature neurons the specific adult neuronal marker of nuclei NeuN is mostly used [10–12]. Analysis of adult born neurons can also be performed using retroviral genetic marking, since retroviruses exclusively enter the target cell during mitosis [13]. Adult neurogenesis has been shown in the brain of different species of rodents [14, 15], primates [16], and even humans [17, 18]. Newborn neurons have been observed to integrate into neuronal hippocampal circuits and have been functionally associated with complex brain processes like cognition, emotion, and pattern separation.
The discovery of environmental factors regulating adult neurogenesis in a bi-directional manner has been of key importance for the acceptance of the concept [19]. In particular, the finding that stress and adrenal steroid hormones down-regulate adult neurogenesis [14, 20] contributed significantly. As aberrant forms of stress and chronic elevation of adrenal hormones are also linked to the psychopathology of depression, possible involvement of adult neurogenesis in psychiatric diseases has been a topic of intense research. Nowadays, changing rates of adult neurogenesis have been linked to aging, environment, hormones, neurochemicals, and behavior as well as to numerous brain diseases ranging from depression to epilepsy.
Overthrowing a dogma always attracts attention. Therefore, an impressive number of papers have appeared in the last decade and as a consequence, numerous reviews have been published covering specific topics of adult neurogenesis such as general mechanisms and signaling cascades [13, 21], cognition and memory formation [22–24], evolution [25], olfaction [26] psychiatric diseases [27], paroxysmal disorders such as epilepsy [28, 29], and neurodegenerative disorders [30]. Also, the concept of stress and its effect on adult neurogenesis have been reviewed extensively [31–35]. Therefore, we will only briefly review the general aspects of the stress system and the interplay between stress and adult neurogenesis in the hippocampus. More extensively, we will highlight novel findings on the function of the glucocorticoid receptor, the main mediator of the stress response, in neuronal progenitor cells and in adult neurogenesis. We will present a novel concept about the role of the glucocorticoid receptor in the positioning and functional integration of newborn neurons and we discuss how this concept may contribute to the chronification of paroxysmal brain diseases.
Neurogenesis in the adult hippocampus
To understand the possible relevance of adult neurogenesis, it is important to understand the anatomy of the hippocampus. The hippocampus can be divided into three main subregions, i.e., CA1, CA3, and dentate gyrus (DG). Neuronal cells derived from these three subregions are connected by so-called trisynaptic pathways (see Fig. 1a). It is generally assumed—but not proven—that information processing by this tri-synaptic circuit is crucially involved in learning and memory formation. Neurogenesis occurs only in the DG and there is no evidence that other hippocampal regions generate new neurons [36].

Overview of the hippocampal formation and adult neurogenesis. a Schematic representation of the tri-synaptic pathway. Axons derived from the entorhinal cortex (EC) layer II projects via the perforant pathway (purple) towards the dendrites of the DG granule cells (red). The perforant pathway projects also towards CA3 were they end in boutons, which contact dendrites of pyramidal cells (blue). Mossy fibers (red) originating in the DG granule cells project to the same pyramidal cells in the CA3. Via the Schaffer collateral pathway (blue) the CA3 projects towards pyramidal neurons (green) in the CA1, which also receive input from the EC layer III. CA1 pyramidal neurons project (green) towards layer IV of the same EC. Only in the DG, new-born granule cells (yellow) integrate into this network. b Adult neurogenesis can be divided in three main stages: the precursor cell stage, the early postmitotic maturation phase and the late mitotic maturation phase. Stem cells proliferate in the sub-granular zone (SGZ) where after NPCs migrate into the granular cell layer (GCL). During the late postmitotic phase, the newborn neurons develop dendritic trees protruding into the molecular layer (ML). Different stages are characterized by expression of specific markers. Note that the GR is expressed in radial glia cells, but not early precursor cells. For further details, see main text
The DG area consists of three layers: the molecular layer (ML), the granule cell layer (GCL), and the hilus or polymorphic layer (PL; see Fig. 1b). The GCL is densely packed and consists of a stack of roughly six granule cell bodies [2, 3, 37]. These cells have an elliptical cell body of about 10–18 μm, are tightly packed, and are not ensheathed by glia cells. Granule cells have a cone-shaped dendritic tree that projects into the ML. At the other site of the cell body, granule cells give rise to unmyelinated axons called mossy fibers. These fibers have large boutons, which not only connect to pyramidal cells of the CA3 but also contact mossy cells in the hilus [2, 3, 38]. The border between GCL and hilus is called the subgranular zone (SGZ) and inhabits neural stem cells (NSCs), which are the source of adult neurogenesis and generate excitatory granule cells [5, 36].
Neurogenesis in the adult DG can be divided into three main stages (see Fig. 1b) [7, 36]. The first stage is called the precursor cell stage and is characterized by cell division [6, 39]. The primary NSC in the DG is a radial glia (RG)-like astrocyte called the type 1 cell [9, 36]. Like RGs, these NSCs have long processes that project into the ML. The second phase is called the early postmitotic phase and is characterized by neuronal differentiation and migration of neuroblasts/immature neurons. Under the influence of GABA, they start to develop dendrites and axons towards, respectively, the ML and CA3 regions. At this stage, the dendrites lack any spines but receive functional GABAergic input [13, 21, 40]. Initially, GABA has an excitatory effect, which induces dendritic growth. When this excitatory signaling is blocked, immature neurons develop abnormally [22–24, 41].
The immature neurons/neuroblasts migrate radially into the DG. Doublecortin, a gene that is causally involved in migration of neuronal precursor cells and neuroblasts [42], is a major marker in this process (see Fig. 1b). The majority of newborn cells end up in the inner third layer of the DG, while 10–20 % reach the mid third layer and only a few cells ends up in the outer third layer [37, 40]. This non-proportional distribution may be explained by the fact that the majority of GCs, which inhabit the middle and outer third layers, are born during early postnatal development while the inner third layer is generated later during development [43].
The third stage is called the late postmitotic maturation stage, wherein surviving cells integrate into the local DG network and further mature into genuine granule cells [40, 44–46]. Although newborn cells lose the expression of immature markers around 4–5 weeks after cell birth, full maturity is reached around 60 days after cell birth [44]. After the first weeks, newborn neurons continue to extend their dendritic and axonal processes and many new connections are formed with glutamatergic synapses from the EC and output to pyramidal cells in the CA3. Not only newborn GCs increase and reshape their synaptic connectivity. Due to their interaction with newborn GCs, mature GCs also change their connectivity. Spines from newborns’ GCs compete at entorhinal boutons with old synapses from mature GCs and—possibly due to local glutamate spillover at the synapse—will replace the old synaptic connection [47–49]. This pattern of competitive synaptic plasticity in the molecular layer also seems to be present in the hilus and CA3 region at the axonal end of newborn neurons. Although boutons actively connect to synapses already 17 days after cell division, the connections are fully mature 2 months post-cell division [47, 50].
The stress system, glucocorticoids, and glucocorticoid receptors
Stress is the response of an organism aiming to maintain a physiological balance called homeostasis. When homeostasis is challenged by a stressor, the organism responds by behavioral and physiological adaptations, resulting in coping and recovery. A stressor can be an environmental challenge, e.g., taking a test, or a physical challenge, e.g., a viral infection. Whatever the nature of the stressor, the body reacts in a stereotypical way [51]. Firstly, neurohormones, in particular corticotropin-releasing hormone (CRH), are released in the hypothalamus that subsequently project to the pituitary [52]. Secondly, activation of CRH receptor expressing neuro-endocrine cells in the pituitary leads to the subsequent release of adrenocorticotropic hormone (ACTH) into the blood circulation, which activates endocrine cells in the adrenal cortex. Thirdly, ACTH stimulates the release of adrenal glucocorticoid hormones, also called stress hormones, which are cortisol in man and corticosterone in rodents, here collectively referred to as CORT. This adrenal steroid hormone affects many organs in the body: it causes release of glucose in the blood circulation, it acts as a repressor of the immune system, and in the brain it facilitates information processing in limbic neuronal networks involved in emotion, cognition, and memory formation [for a review, see 31, 32]. CORT also functions as a feedback mechanism to the same brain structures in the hypothalamus that triggered its release, thereby stabilizing the hypothalamus–pituitary–adrenal (HPA) axis and preventing sensitive organs in the body from CORT overexposure. HPA axis activity is organized in a circadian rhythm with high levels in the morning, enabling individuals to cope with energy demands ahead of them. It is important to note that rapid HPA axis activation by acute stress and the subsequent turn-off of the HPA axis by the negative feedback response of CORT is healthy, as it helps an individual to cope with the stressor. However, dysregulation of the HPA axis by e.g., chronic stress may endanger the immune system, cardiovascular functions, the regulation of fear, cognition, and memory formation, and as such is associated with numerous diseases, in the brain in particularly with depression [33, 53].
Lipophilic CORT passes the blood–brain barrier easily and enters neuronal target cells by penetrating across the cell membrane. At the neuronal level, CORT controls the stress response through binding to two types of steroid receptors in the cytosol: the mineralocorticoid receptor (MR or NR3C2; [54]) and the glucocorticoid receptor (GR or NR3C1; [55–57]). These steroid receptors belong to a superfamily of ligand-inducible, highly conserved nuclear hormone receptors. Upon binding CORT, GR and MR translocate to the nucleus where they affect expression of specific sets of genes in two ways: (1) by binding to specific so-called glucocorticoid-response element DNA motifs located in promoter regions of target genes; a process called transactivation and (2) by protein–protein interaction with other transcription factors and co-factors, such as cAMP-response element binding (CREB) protein and CREB binding protein (CBP), thereby mainly inhibiting the activation of these transcription factors and as such is called transrepression (for review, see [58]). In addition, CORT evokes fast non-genomic neuronal responses by binding to membrane-bound GR and MR [59]. As these membrane-bound receptors have not yet been identified in NPCs, they are outside the scope of this review.
The hippocampus, a brain area crucially involved in cognition and memory formation, expresses both MR and GR at high levels, and is therefore particularly sensitive for fluctuating levels of CORT. Moreover, as neuronal stem cells in the dentate gyrus of the hippocampus are located in the close vicinity of blood vessels [60], stress and stress-induced elevated CORT levels may target NPCs easily and may be very profound (environmental) factors affecting adult hippocampal neurogenesis.
The glucocorticoid receptor and adult neurogenesis
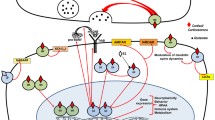
The general view is that stress and stress hormones inhibit adult neurogenesis by inhibiting proliferation of type 2 cells. For example, several chronic stress paradigms, like subordination stress in primates and social defeat in rodents showed diminished cell proliferation in the DG [16, 61–63]. Also, ground-breaking studies in the early 1990s showed that administration of adrenal hormones in rats negatively affect the incorporation of 3H-thymidine while removal of adrenal hormones by adrenalectomy booster the appearance of 3H-thymidine labeled cells [14, 64, 65], suggesting an inhibitory role for stress-induced glucocorticoids in adult neurogenesis. However, the relationship between stress and adult neurogenesis seems far more complex than a simple inhibitory role. For example, the use of the running wheel by mice is well known to booster adult neurogenesis at the proliferation stage and promotes neuronal differentiation [45, 66, 67]. At the same time, physical exercise is a strong activator of the HPA axis, leading to elevated levels of circulating glucocorticoids [68, 69]. Similarly, an enriched environment stimulates neuronal differentiation and survival of newborn cells [66, 70, 71], yet it simultaneously increases glucocorticoid levels [72]. Also, several learning paradigms not only stimulate survival of newborn neurons but also increase HPA axis activity and glucocorticoid levels [73, 74]. Recently, acute stress, induced by 3 h of immobilization stress resulting in elevated plasma CORT levels, was shown to induce (not repress) cell proliferation in the DG [75]. Reversely, hippocampal neurogenesis may also facilitate normalization of glucocorticoid levels after stress [76], suggesting a bi-directional relationship between adult hippocampal neurogenesis and regulation of the HPA axis. Clearly, the effect of stress and stress hormones on adult neurogenesis is complex. These paradoxical findings may be explained by several factors (see also Fig. 2). Firstly, neuronal stem cells in the SGZ of the dentate gyrus are located in a specialized microenvironment, the so-called neurogenic niche, consisting of numerous different cell types, including astrocytes, ependymal cells, blood vessels, interneurons, oligodendrocytes, and myeloid cells, i.e., microglia cells and dendritic cells. All of these cell types may modulate adult neurogenesis. For example, depending on the type of microglia and on the challenge, activated microglia cells release cytokines that may have detrimental or beneficial effects on adult neurogenesis (for review, see [77]). Also, these cell types express GRs indicating that stress-induced glucocorticoid elevation targets these cells as well, and as such these cells may modulate the rate of neurogenesis. Secondly, the nature of the stressor is an important factor. Control or no control over stress may have opposite effects on neuronal plasticity including adult neurogenesis whereby non-controllable stress such as social defeat or learned helplessness has a negative and controllable stress such as voluntary exercise or enriched environment has a positive effect on neurogenesis [72]. The precise neurochemical mechanisms that are differentially activated by controllable and uncontrollable stress are presently unknown but may involve desensitized serotonergic signaling via the dorsal raphe nucleus [78] as serotonin stimulates adult neurogenesis through 5HT1A receptors [79]. Thirdly, the duration of stress, acute stress (e.g., during learning) versus chronic stress, may have opposite effects on adult neurogenesis. Chronic stress has been associated with decreased expression of 5-HT1A receptor expression [80] and thus likely impaired serotonergic signaling in dentate gyrus region of the rodent hippocampus [81, for review see 82] while an enriched environment stimulates 5HT1A expression [83]. Fourthly, early life experiences may also affect the rate of neurogenesis in adult life. For example, exposure to E. coli bacteria in early life not only affects the responsiveness of microglia in the adult brain but also negatively affects the rate of adult neurogenesis after infection compared to non-E. coli-treated pups [84].

Relationship between GR activity and neurogenesis GR activation exerts both positive and negative effects on neurogenesis. We propose an inverted U-shaped model in which there is a relationship between the amount of GR activation and neurogenesis. Low levels of stress seen in animals kept in a poor environment or with a sedentary lifestyle induce low levels of proliferation and maturation. Controllable stress, like enriched environments, physical activity, and learning, coincides with increased levels of GR activation and is associated with increased cell proliferation and correct integration of mature neurons. Too much GR activation as seen during uncontrollable stress negatively affects proliferation and neuronal integration. Neurogenesis-controlling molecular factors, like BDNF, VEGF, and 5-HT signaling, are regulated by low–high GR activity in opposite directions
Besides serotonergic signaling, controllable versus non-controllable stress and acute versus chronic stress may also affect other transmitter systems. Relevant for adult neurogenesis particularly is the interaction with glutamatergic signaling, e.g., acute cortisol exposure may facilitate clustering of AMPA receptors [85], thereby facilitating the pro-plasticity activities of glutamate in the early and post-mitotic maturation phase. Another important group that interacts with stress and stress hormones is neurotrophins, like BDNF, VEGF, and IGF-1 and their Trks. Expression of these neurotrophins is not only regulated by stress and circulating stress hormones (see e.g., [86]) but also by an enriched environment, chronic stress, and by learning, and may be differentially regulated by controllable versus non-controllable stress [87]. The interplay between the stress system on the one hand and neurotrophins and excitatory stimuli on the other may direct the outcome on the proliferation, maturation, and functional integration of newborn cells in the DG. The nature of the stress, e.g., acute versus chronic, and the background of an organism—aging, traumatic early life events, environmental enrichment—further determine the outcome on adult neurogenesis. For more in-depth reviews on this topic, we refer to recent excellent papers highlighting the complex interplay between stress, stress hormones, and adult neurogenesis [27, 34, 39, 88–90].
The glucocorticoid receptor
Corticosterone has profound effects on the excitation-inhibition balance within the hippocampus. This effect is not identical in all hippocampal sub areas. In both CA1 and CA3, high concentrations of corticosterone enhance LTP via short-term MR activation. After this acute effect, GR-mediated inhibition of the LTP normalizes the excitation-inhibition balance in these brain areas. However, within the dentate gyrus, the effect of corticosterone on the excitation-inhibition balance seems reversed. Despite the abundant expression of the GR within the DG, high concentrations of corticosterone do not suppress LTP in the long run but, in contrast to the CA1 and CA3, enhance LTP [91].
The MR and the GR are the main intracellular mediators of the stress response (for review, see [31–33]). In the DG, both receptors are abundantly expressed in all GCLs except in the SGZ. In neuronal precursor cells, MR expression is below detection levels and the GR is expressed in about 50 % of neuronal precursor cells, both in vivo [65, 92] as well as in primary neuronal precursor cell cultures [93]. These data suggest that the GR (and not the MR) is the main mediator of direct stress-suppressive effects on cell proliferation. Indeed, pharmacological blockade of the GR prevents the negative effect of exogenous CORT administration on the proliferation of neuronal progenitor cells [94]. The absence of both MR and GR expression in a subpopulation of neuronal progenitor cells opens up the possibility that the effect of stress hormones is indirect, likely via NMDA receptors. Activation of NMDA receptors inhibits and NMDAR blockade promotes cell proliferation in the dentate gyrus [95–97]. In addition, stress and CORT are known to stimulate glutamate release [85, 98]. Therefore, it may well be possible that the CORT-suppressive effects are mediated by glutamatergic signaling. Indeed, pharmacological blockade of NMDAR by MK801 prevents the inhibitory effect on cell proliferation by CORT administration [99], suggesting that glutamate-induced inhibition acts downstream of CORT signaling.
The presence of GR expression in a number of progenitor cells and in immature neurons [92] also suggests a direct effect of circulating CORT on adult neurogenesis. Such a direct role for the GR is further suggested by the fact that GR activity and GR mRNA and GR protein levels are tightly regulated by a number of factors that also regulate adult neurogenesis (see Table 1). One of these factors is doublecortin-like (DCL), a protein that is specifically expressed in radial glia cells during embryonic neuronal development [100] and that is co-expressed with DCX in progenitor cells in the adult hippocampus [101]. DCL belongs to the DCX gene family [102] and is a microtubule-associated protein regulating retrograde transport of activated GR proteins to the nucleus, indicating that progenitor cells possess specialized mechanisms to quickly translocate activated GRs. Another factor is microRNA(miR)-124, a small non-coding RNA that is able to bind to the 3′-untranslated region of the GR mRNA thereby reducing GR protein levels to 70 % [103]. Interestingly, miR-124 is a neuron-specific microRNA that directs progenitor cells in the brain to differentiate into a neuronal phenotype [104, 105]. To achieve neuronal fate, miR-124 represses the activity of a number of proteins such as REST [106] and Sox9 [105] that are known to antagonize neuronal differentiation. The repression of the GR by miR-124 suggests that reduced GR protein levels are critical for proper neuronal differentiation. Indeed, downregulation of GR proteins by retroviral and lentiviral delivery of GR-targeting small interference (si)RNA molecules specifically in neuronal progenitor cells in the mouse DG [107] accelerates their neuronal differentiation. Moreover, newborn granule cells with reduced GR protein levels exhibit more complex dendritic arbors, have increased numbers of mature dendritic spines, and more mature mossy fiber boutons. In line with this, cells with reduced GR expression exhibit increased basal excitability (see Fig. 3) [108]. A striking finding in this study was the positioning of newborn granule cells in the GCL: a large percentage of the cells with reduced GR levels were located in the middle and outer layer. This position is significantly different from newborn granule cells with normal GR expression that were predominantly present in the inner and middle layer. These data suggest a role for the GR in the accurate migration and functional integration of newborn cells in the GCL, a role which is in line with the effect of glucocorticoids on cortical neuron migration in embryonic development [109] and the changed positioning of DCX-positive newborn granule cells after adrenalectomy [108].

GR knockdown stimulates differentiation of post-mitotic neuronal precursor cells. GR knockdown increases the number of dendritic mature mushroom spines (1), axonal bouton size (2), and number of mini-excitatory postsynaptic currents (mEPSC; 4). GR knockdown also leads to more complex dendritic arbors (2) and ectopically located new-born granule cells (not illustrated). After [108]
The altered morphology and mis-positioning of newborn granule cells with reduced GR expression raise the question about the consequences for hippocampal functioning. Contextual fear conditioning, a task that is often used to monitor functional consequences of altered adult neurogenesis [110, 111], showed impairment of contextual but not of cued freezing behavior in mice with reduced GR expression in newborn, 5-week-old granule cells [108]. This behavioral finding is remarkable, as only a limited number of cells, approximately 20,000, are transduced by the lentivirus [107], which further underscores the importance of adult neurogenesis for proper hippocampal functioning.
Aberrant placement of newborn granule cells has also been found in other disease-related models. Neuronal diseases, characterized by extensive glutamate release, such as epilepsy and stroke, are associated with massive neurogenesis [112, 113], dendritic abnormalities, and ectopic positioning of newborn cells [114]. Remarkably, in the case of status epilepticus-induced changes in neurogenesis, newly formed neurons are found at ectopic locations [115]. Similarly, as after GR reduction, these ectopic adult-born neurons are hyper-excitable, and are believed to be part of the mechanisms underlying increased susceptibility towards seizures (see Fig. 4) [29, 116, 117]. Reduced levels of disrupted-in-schizophrenia-1 (DISC1) in newborn granule cells cause a highly similar phenotype as after reduced GR levels. DISC1 knockdown in NPCs exhibit accelerated neuronal differentiation, functional integration at ectopic locations and impairment in hippocampus-dependent memory consolidation [118, 119]. Collectively, these studies suggest that integration of newborn granule cells at ectopic locations may contribute to the pathogenesis of both neurological and psychiatric diseases and possibly are part of mechanisms underlying chronification of paroxysmal disorders such as stroke, epilepsy, and schizophrenia [117]. As the GR has been implicated in epilepsy and schizophrenia [120–126], aberrant GR signaling might be part of the mechanisms underlying aberrant integration of newborn cells in these diseases.

The excitation–inhibition balance determines the number and final positioning of newborn granule cells. Excessive excitation, as is the case after seizures, stroke, and GR knockdown, leads to an increase and ectopic location of newborn granule cells. Reversely, excessive inhibition leads to reduced numbers of newborn cells. Although suggested in this cartoon, it is unknown whether newborn granule cells are ectopically located in the GCL. For further details, see main text
The studies outlined above suggest that the level of GR expression is important for adequate adult neurogenesis and should be in a narrow window. Too low expression leads to ectopic integration of newborn granule cells and too high expression may block proliferation and differentiation of NPCs. In line with the importance of a narrow window for GR expression is the finding that a 30 % reduction of GR protein levels results in a 70 % reduction of the expression of glucocorticoid-induced leucine zipper (GILZ), a GR-responsive gene [103]. Indeed, intensive research of the last decades has revealed a myriad of mechanisms all aiming at the regulation of GR activity (see Table 1). Alternative splicing of the GR gene results in at least three different GR isoforms. In addition, alternative translation initiation results in the formation of another eight different GR isoforms. Each of these GR isoforms is expressed in a cell-specific way and control the expression of a unique set of genes [127, 128]. As such, these GR isoforms may contribute to mechanisms underlying cell-specific responses to CORT. GR activity can also be modulated by GR phosphorylation [129]. In human neuronal progenitor cells, GR responsiveness towards CORT also depends on GR phosphorylation by serum- and glucocorticoid-inducible kinase 1 phosphorylation, a target gene for activated GRs [130]. A further complicating factor for the outcome of GR action is the interaction with other proteins, in particular co-activators and co-inhibitors that are expressed in a cell-specific manner in the brain (for review, see [131]). Relevant for adult neurogenesis is Ube3a, a co-activator of the GR, which is crucially involved in the Angelman syndrome [132]. Lack of Ube3a expression not only leads to impaired cognition and decreased numbers of NeuN/BrdU-positive cells [133] but may also result in impaired GR signaling [134]. CBP is another important cofactor and integration point, bridging CREB or zif68/egr1 activity with GR signaling [131, 135]. Together with the regulation of GR levels by neurogenesis-related microRNA-124 [103] and the presence of specific retrograde transport mechanisms for the GR in neuronal progenitor cells such as DCL [136], the different mechanisms outlined above clearly indicate that GR expression and activity are under tight control in neuronal progenitor cells.
An important question that emerges is what type of (environmental) factors regulating adult neurogenesis also affect GR levels in neuronal progenitor cells (see Table 1). Interestingly, early life events, such as maternal separation and parental care, are known to reduce GR levels at adult age by epigenetic programming of the GR promoter [137–140] and is also associated with impaired adult neurogenesis [141, 142]. Aging is associated with lower rates of hippocampal neurogenesis [143–145], impaired negative feedback of CORT on the HPA axis, and reduced levels of the GR [146, 147]. Chronic stress is another factor negatively affecting both GR levels and adult neurogenesis [61, 148–150]. It is unknown whether aging, early life events, and chronic stress directly affect GR levels, thereby impairing proliferation and differentiation of neuronal progenitor cells. It is likely, however, that these effects are indirect, as proliferation, differentiation, migration, and survival depend on neurotrophins and excitatory input, first by GABA input and later in development by glutamatergic input. In other words, different phases of adult neurogenesis require excitatory neuronal activity [for review, see 151]. Chronic stress not only reduces GR levels but also impairs glutamate release via BDNF [152] and reduces expression of other neurotrophins as well [86, 153]. Thus, it seems likely that environmental factors negatively affecting adult neurogenesis attenuate multiple excitatory and inhibitory signaling cascades through the GR.
The glucocorticoid receptor and the excitation-inhibition balance in neuronal progenitor cells
The precise mechanisms by which GR levels directly regulate proliferation and neuronal differentiation in neuronal progenitor cells are unknown. As a transcription factor, the GR affects the expression of hundreds of genes in the hippocampus [154, 155] and without a doubt a number of these genes are involved in the differentiation of neuronal progenitor cells. We suggest that the majority of these GR-responsive genes are regulated by a transrepression model (see also Fig. 5). Studies aiming at the identification of GR-responsive genes in the hippocampus showed that 1 h after corticosterone exposure all responsive genes are downregulated, suggesting a GR-mediated transrepression effect of fast glucocorticoid action [156]. Also, a GR-mediated transactivation mechanism seems to underlie retarded migration of immature neurons in the developing rat brain [109]. This finding strongly suggests that proliferation and neuronal differentiation of neuronal progenitor cells require reduced GR activation, which seems more in line with a transrepression model. Also, this finding is in line with the repression of GR translation of the proneurogenic microRNA-124 [103]. However, recent in vitro studies indicate involvement of a GR-mediated transactivation effect in the pro-neurogenic action of antidepressants [157], which suggest that the GR function in neuronal progenitor cells is context dependent, for example depending on serotonin signaling.

Hypothetical model illustrating possible molecular mechanisms underlying the action of the glucocorticoid receptor in the excitation-inhibition balance in NPCs. Activation of NMDA and AMPA receptors lead to an intracellular signaling cascade leading to the activation of the pro-neurogenic transcription factors CREB and zif68/EGR via CaMK. Likewise, binding of neurotrophins (e.g., BDNF) to Trks leads to activation of CREB and zif68/egr1 via the MAPK pathway. CORT-activated GR is retrograde transported by doublecortin-like (DCL) along the microtubules towards the nucleus. GR may interact with p-CREB by interaction with co-factors such as CREB binding protein (CPB). The net result of this GR-CPB-CREB interaction is the decreased CREB activity and subsequent inhibition of neurogenesis. Pro-neurogenic miR-124 represses GR activity, which may facilitate neuronal differentiation via NMDAR and/or Trk activation
Proper proliferation and neuronal differentiation of neuronal progenitor cells require excitatory input. As outlined before, in early stages this is excitatory GABA [41] and later this is glutamate that act on AMPA receptors and NMDA receptors [158, 159]. Another group of molecules that stimulate neurogenesis are neurotrophins, such as BDNF and VEGF, which are induced and released by environmental enrichment and learning, thereby promoting neurogenesis. Neurotrophins bind to tyrosine kinase receptors (Trk), membrane-bound receptors that are necessary for adult neurogenesis [160]. Activated AMPA receptors, NMDA receptors, and Trk receptors are all known to activate an intracellular signaling cascade that results in the activation of transcription factors that subsequently stimulate proliferation of neuronal precursor cells, their differentiation, and functional integration in hippocampal circuits (for review, see [161]). Examples of such transcription factors are Zif68/egr1 [162] and CREB [163–165], which are key transcription factors in the development and survival of newborn granule cells. Activated GR may interact with these pathways either by binding directly to these transcription factors and/or by interaction with co-factors such as CBP [166] (for review, see [131]). The net result of this GR–CPB–CREB interaction is the decreased CREB activity. The emerging picture in this concept is that CORT and the activated GR dampen excitatory input in neuronal progenitor cells thereby preventing the neurogenesis process from overshoot. Reduction of GR levels, e.g., by miR-124 (see Fig. 5) in NPCs or removal of CORT by adrenalectomy will relief this functional brake on excitatory signaling leading to increased proliferation, differentiation and (aberrant) integration of newborn granule cells in hippocampal circuits. Extrapolation of this model may explain ectopic newborn granule cells in the hilus after seizures, which are characterized by excessive glutamate and neurotrophin release, in particular BDNF. Other paroxysmal disorders like migraine are often also explained by a disturbed excitation-inhibition balance [167] with high levels of cortical glutamate release, which may reach limbic structures during cortical spreading depression [168] and increases proliferative activity in the dentate gyrus [112]. Reversely, chronic high levels of CORT and diminished neuronal activity, i.e., diminished glutamate signaling and neurotrophin release—often associated with psychiatric diseases like depression—may lead to reduced proliferation, differentiation and survival of newborn granule cells (see Fig. 4) [27, 34, 89]. However, it is presently unknown if this high CORT-low glutamate signaling will lead to diminished neuronal migration and functional integration of newborn granule cells mainly in the inner layer of the GCL.
Perspectives
During the last decade, tremendous progress has been made in understanding adult neurogenesis. The process of morphological changes of neuronal stem cells leading to mature excitatory granule cells, the synaptic connectivity, functional integration and, to a lesser extent, the functional implications for learning and cognition, has been described in considerable detail. Similarly, many genes that are orchestrating the different stages of adult neurogenesis have been identified and numerous intrinsic and extrinsic factors influencing the rate of proliferation and fate of resulting newborn granule cells have been documented. Strangely, despite the fact that stress and stress hormones were one of the earliest discovered factors that manipulate adult neurogenesis [14, 64], their action on neurogenesis is still not fully understood. Likely, this is due to the complexity of the stress system that is controlled by several brain areas and peripheral tissues, i.e., the HPA axis; to different physiological responses after acute stress versus chronic stress and as outlined above, to the complex control of the GR, the main mediator of the stress response in relation to adult neurogenesis. A number of issues remain to be resolved. For example, which GR isoforms are expressed in neuronal progenitor cells. Perhaps more importantly, can reduced GR levels, as observed after chronic stress, aging, and traumatic early life events, also be found in NPCs? Does epigenetic programming of the GR promoter by early life stress also take place in NPCs? If so, what will be the consequences for adult neurogenesis with respect to the positioning of newborn granule cells?
Both chronic stress, a dysfunctional HPA axis, and aberrant neurogenesis have been implicated in psychiatric diseases [27, 33]. A proper excitation-inhibition balance seems key for the control of both the stress system and adult neurogenesis. Both the MR and GR are important mediators for the stress response and interact with proteins that are activated by neuronal activity. Therefore, the question emerges: are these receptors suitable therapeutic targets for restoring excitation-inhibition balances in not only psychiatric diseases [53] but also in paroxysmal disorders like epilepsy [169]? With availability of numerous potent GR and MR antagonists and agonists, which have been developed by the pharmaceutical companies during the last 50 years, and modern genetic technologies like in vivo viral delivery of RNA-interference molecules, these questions can now be addressed.
References
Cajal Y, Ramón S (1928) Degeneration and regeneration of the nervous system. Oxford University Press, London
Altman J, Das GD (1965) Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol 124:319–335
Altman J (1963) Autoradiographic investigation of cell proliferation in the brains of rats and cats. Anat Rec 145:573–591
Altman J, Das GD (1965) Post-natal origin of microneurones in the rat brain. Nature 207:953–956
Kaplan MS, Hinds JW (1977) Neurogenesis in the adult rat: electron microscopic analysis of light radioautographs. Science 197:1092–1094
Reynolds BA, Weiss S (1992) Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 255:1707–1710
Nottebohm F (1989) From bird song to neurogenesis. Sci Am 260:74–79
Kempermann G, Jessberger S, Steiner B, Kronenberg G (2004) Milestones of neuronal development in the adult hippocampus. Trends Neurosci 27:447–452. doi:10.1016/j.tins.2004.05.013
Encinas JM, Enikolopov G (2008) Identifying and Quantitating Neural Stem and Progenitor Cells in the Adult Brain. In: Methods in Cell Biology. Elsevier, pp 243–272
Couillard-Despres S, Winner B, Schaubeck S et al (2005) Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci 21:1–14. doi:10.1111/j.1460-9568.2004.03813.x
Brown JP, Couillard-Despres S, Cooper-Kuhn CM et al (2003) Transient expression of doublecortin during adult neurogenesis. J Comp Neurol 467:1–10. doi:10.1002/cne.10874
Rao MS, Shetty AK (2004) Efficacy of doublecortin as a marker to analyse the absolute number and dendritic growth of newly generated neurons in the adult dentate gyrus. Eur J Neurosci 19:234–246
Ming G-L, Song H (2005) Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci 28:223–250. doi:10.1146/annurev.neuro.28.051804.101459
Gould E, Cameron HA, Daniels DC et al (1992) Adrenal hormones suppress cell division in the adult rat dentate gyrus. J Neurosci 12:3642–3650
Cameron HA, Woolley CS, McEwen BS, Gould E (1993) Differentiation of newly born neurons and glia in the dentate gyrus of the adult rat. NSC 56:337–344
Gould E, Reeves AJ, Fallah M et al (1999) Hippocampal neurogenesis in adult Old World primates. Proc Natl Acad Sci USA 96:5263–5267
Curtis MA, Kam M, Faull RLM (2011) Neurogenesis in humans. Eur J Neurosci 33:1170–1174. doi:10.1111/j.1460-9568.2011.07616.x
Eriksson PS, Perfilieva E, Björk-Eriksson T et al (1998) Neurogenesis in the adult human hippocampus. Nat Med 4:1313–1317. doi:10.1038/3305
Gross CG (2000) Neurogenesis in the adult brain: death of a dogma. Nat Rev Neurosci 1:67–73. doi:10.1038/35036235
Gould E, Woolley CS, McEwen BS (1991) Adrenal steroids regulate postnatal development of the rat dentate gyrus: I. Effects of glucocorticoids on cell death. J Comp Neurol 313:479–485. doi:10.1002/cne.903130308
Faigle R, Song H (2013) Biochimica et Biophysica Acta. BBA Gen Subj 1830:2435–2448. doi:10.1016/j.bbagen.2012.09.002
Sahay A, Wilson DA, Hen R (2011) Pattern separation: a common function for new neurons in hippocampus and olfactory bulb. Neuron 70:582–588. doi:10.1016/j.neuron.2011.05.012
Aimone JB, Deng W, Gage FH (2011) Resolving new memories: a critical look at the dentate gyrus, adult neurogenesis, and pattern separation. Neuron 70:589–596. doi:10.1016/j.neuron.2011.05.010
Aimone JB, Deng W, Gage FH (2010) Adult neurogenesis: integrating theories and separating functions. Trends Cogn Sci (Regul Ed) 14:325–337. doi:10.1016/j.tics.2010.04.003
Kempermann G (2012) New neurons for ‘survival of the fittest’. Nat Rev Neurosci 13:727–736. doi:10.1038/nrn3319
Lazarini F, Lledo P-M (2011) Is adult neurogenesis essential for olfaction? Trends Neurosci 34:20–30. doi:10.1016/j.tins.2010.09.006
Eisch AJ, Petrik D (2012) Depression and hippocampal neurogenesis: a road to remission? Science 338:72–75. doi:10.1126/science.1222941
Parent JM (2008) Persistent hippocampal neurogenesis and epilepsy. Epilepsia 49(Suppl 5):1–2. doi:10.1111/j.1528-1167.2008.01631.x
Scharfman HE, McCloskey DP (2009) Postnatal neurogenesis as a therapeutic target in temporal lobe epilepsy. Epilepsy Res 85:150–161. doi:10.1016/j.eplepsyres.2009.03.006
Winner B, Kohl Z, Gage FH (2011) Neurodegenerative disease and adult neurogenesis. Eur J Neurosci 33:1139–1151. doi:10.1111/j.1460-9568.2011.07613.x
de Kloet ER, Vreugdenhil E, Oitzl MS, Joëls M (1998) Brain corticosteroid receptor balance in health and disease. Endocr Rev 19:269–301
de Kloet ER, Fitzsimons CP, Datson NA et al (2009) Glucocorticoid signaling and stress-related limbic susceptibility pathway: about receptors, transcription machinery and microRNA. Brain Res 1293:129–141. doi:10.1016/j.brainres.2009.03.039
de Kloet ER, Joëls M, Holsboer F (2005) Stress and the brain: from adaptation to disease. Nat Rev Neurosci 6:463–475. doi:10.1038/nrn1683
Schoenfeld TJ, Gould E (2013) Differential effects of stress and glucocorticoids on adult neurogenesis. Curr Top Behav Neurosci 15:139–164. doi:10.1007/7854_2012_233
Karten YJG, Olariu A, Cameron HA (2005) Stress in early life inhibits neurogenesis in adulthood. Trends Neurosci 28:171–172. doi:10.1016/j.tins.2005.01.009
Kempermann G, Song H, Gage FH (2008) Neurogenesis in the hippocampus. In: Gage FH, Kempermann G, Song H (eds) Adult neurogenesis. Cold Spring Harbor Laboratory Press, New York, pp 159–174
Kempermann G, Gast D, Kronenberg G et al (2003) Early determination and long-term persistence of adult-generated new neurons in the hippocampus of mice. Development 130:391–399
Amaral DG, Scharfman HE, Lavenex P (2007) The dentate gyrus: fundamental neuroanatomical organization (dentate gyrus for dummies). Prog Brain Res 163:3–22. doi:10.1016/S0079-6123(07)63001-5
Schoenfeld TJ, Gould E (2011) Stress, stress hormones, and adult neurogenesis. Exp Neurol 233:12–21. doi:10.1016/j.expneurol.2011.01.008
Espósito MS, Piatti VC, Laplagne DA et al (2005) Neuronal differentiation in the adult hippocampus recapitulates embryonic development. J Neurosci 25:10074–10086. doi:10.1523/JNEUROSCI.3114-05.2005
Ge S, Goh ELK, Sailor KA et al (2006) GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature 439:589–593. doi:10.1038/nature04404
Francis F, Koulakoff A, Boucher D et al (1999) Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron 23:247–256
Muramatsu R, Ikegaya Y, Matsuki N, Koyama R (2007) Neonatally born granule cells numerically dominate adult mice dentate gyrus. Neuroscience 148:593–598. doi:10.1016/j.neuroscience.2007.06.040
Laplagne DA, Espósito MS, Piatti VC et al (2006) Functional convergence of neurons generated in the developing and adult hippocampus. PLoS Biol 4:e409. doi:10.1371/journal.pbio.0040409
van Praag H, Schinder AF, Christie BR et al (2002) Functional neurogenesis in the adult hippocampus. Nature 415:1030–1034. doi:10.1038/4151030a
Zhao C, Teng EM, Summers RG et al (2006) Distinct morphological stages of dentate granule neuron maturation in the adult mouse hippocampus. J Neurosci 26:3–11. doi:10.1523/JNEUROSCI.3648-05.2006
Toni N, Laplagne DA, Zhao C et al (2008) Neurons born in the adult dentate gyrus form functional synapses with target cells. Nat Neurosci 11:901–907. doi:10.1038/nn.2156
Toni N, Sultan S (2011) Synapse formation on adult-born hippocampal neurons. Eur J Neurosci 33:1062–1068. doi:10.1111/j.1460-9568.2011.07604.x
Toni N, Teng EM, Bushong EA et al (2007) Synapse formation on neurons born in the adult hippocampus. Nat Neurosci 10:727–734. doi:10.1038/nn1908
Faulkner RL, Jang M-H, Liu X-B et al (2008) Development of hippocampal mossy fiber synaptic outputs by new neurons in the adult brain. Proc Natl Acad Sci USA 105:14157–14162. doi:10.1073/pnas.0806658105
Selye H (1998) A syndrome produced by diverse nocuous agents. 1936. J Neuropsychiatry Clin Neurosci 10:230–231
Vale W, Spiess J, Rivier C, Rivier J (1981) Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science 213:1394–1397
Holsboer F (2000) The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 23:477–501. doi:10.1016/S0893-133X(00)00159-7
Arriza JL, Weinberger C, Cerelli G et al (1987) Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science 237:268–275
Reul JM, de Kloet ER (1985) Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology 117:2505–2511
McEwen BS, de Kloet ER, Rostene W (1986) Adrenal steroid receptors and actions in the nervous system. Physiol Rev 66:1121–1188
Hollenberg SM, Weinberger C, Ong ES et al (1985) Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 318:635–641
Bamberger CM, Schulte HM, Chrousos GP (1996) Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev 17:245–261
Groeneweg FL, Karst H, de Kloet ER, Joëls M (2012) Mineralocorticoid and glucocorticoid receptors at the neuronal membrane, regulators of nongenomic corticosteroid signalling. Mol Cell Endocrinol 350:299–309. doi:10.1016/j.mce.2011.06.020
Wurmser AE, Palmer TD, Gage FH (2004) Neuroscience. Cellular interactions in the stem cell niche. Science 304:1253–1255. doi:10.1126/science.1099344
Gould E, McEwen BS, Tanapat P et al (1997) Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J Neurosci 17:2492–2498
Gould E, Tanapat P, McEwen BS et al (1998) Proliferation of granule cell precursors in the dentate gyrus of adult monkeys is diminished by stress. Proc Natl Acad Sci USA 95:3168–3171
Lagace DC, Donovan MH, DeCarolis NA et al (2010) Adult hippocampal neurogenesis is functionally important for stress-induced social avoidance. Proc Natl Acad Sci USA 107:4436–4441. doi:10.1073/pnas.0910072107
Cameron HA, Gould E (1994) Adult neurogenesis is regulated by adrenal steroids in the dentate gyrus. NSC 61:203–209
Cameron HA, Woolley CS, Gould E (1993) Adrenal steroid receptor immunoreactivity in cells born in the adult rat dentate gyrus. Brain Res 611:342–346
van Praag H, Kempermann G, Gage FH (1999) Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci 2:266–270. doi:10.1038/6368
Snyder JS, Glover LR, Sanzone KM et al (2009) The effects of exercise and stress on the survival and maturation of adult-generated granule cells. Hippocampus 19:898–906. doi:10.1002/hipo.20552
Droste SK, Gesing A, Ulbricht S et al (2003) Effects of long-term voluntary exercise on the mouse hypothalamic–pituitary–adrenocortical axis. Endocrinology 144:3012–3023
Stranahan AM, Khalil D, Gould E (2006) Social isolation delays the positive effects of running on adult neurogenesis. Nat Neurosci 9:526–533. doi:10.1038/nn1668
Rizzi S, Bianchi P, Guidi S et al (2011) Impact of environmental enrichment on neurogenesis in the dentate gyrus during the early postnatal period. Brain Res 1415:23–33. doi:10.1016/j.brainres.2011.08.007
Veena J, Srikumar BN, Raju TR, Shankaranarayana Rao BS (2009) Exposure to enriched environment restores the survival and differentiation of new born cells in the hippocampus and ameliorates depressive symptoms in chronically stressed rats. Neurosci Lett 455:178–182. doi:10.1016/j.neulet.2009.03.059
Lehmann ML, Brachman RA, Martinowich K et al (2013) Glucocorticoids orchestrate divergent effects on mood through adult neurogenesis. J Neurosci 33:2961–2972. doi:10.1523/JNEUROSCI.3878-12.2013
Leuner B, Mendolia-Loffredo S, Kozorovitskiy Y et al (2004) Learning enhances the survival of new neurons beyond the time when the hippocampus is required for memory. J Neurosci 24:7477–7481. doi:10.1523/JNEUROSCI.0204-04.2004
Leuner B, Gould E, Shors TJ (2006) Is there a link between adult neurogenesis and learning? Hippocampus 16:216–224. doi:10.1002/hipo.20153
Kirby ED, Muroy SE, Sun WG et al (2013) Acute stress enhances adult rat hippocampal neurogenesis and activation of newborn neurons via secreted astrocytic FGF2. Elife 2:e00362. doi:10.7554/eLife.00362
Snyder JS, Soumier A, Brewer M et al (2011) Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature 476:1–5. doi:10.1038/nature10287
Ekdahl CT (2012) Microglial activation—tuning and pruning adult neurogenesis. Front Pharmacol 3:41. doi:10.3389/fphar.2012.00041
Maier SF, Watkins LR (1998) Stressor controllability, anxiety, and serotonin. Cogn Ther Res 22:595–613
Banasr M, Hery M, Printemps R, Daszuta A (2004) Serotonin-induced increases in adult cell proliferation and neurogenesis are mediated through different and common 5-HT receptor subtypes in the dentate gyrus and the subventricular zone. Neuropsychopharmacology 29:450–460. doi:10.1038/sj.npp.1300320
Flügge G, Kramer M, Rensing S, Fuchs E (1998) 5HT1A-receptors and behaviour under chronic stress: selective counteraction by testosterone. Eur J Neurosci 10:2685–2693
Meijer OC, de Kloet ER (1994) Corticosterone suppresses the expression of 5-HT1A receptor mRNA in rat dentate gyrus. Eur J Pharmacol 266:255–261
Meijer OC, de Kloet ER (1998) Corticosterone and serotonergic neurotransmission in the hippocampus: functional implications of central corticosteroid receptor diversity. Crit Rev Neurobiol 12:1–20
Rasmuson S, Olsson T, Henriksson BG et al (1998) Environmental enrichment selectively increases 5-HT1A receptor mRNA expression and binding in the rat hippocampus. Brain Res Mol Brain Res 53:285–290
Bland ST, Beckley JT, Young S et al (2010) Enduring consequences of early-life infection on glial and neural cell genesis within cognitive regions of the brain. Brain Behav Immun 24:329–338. doi:10.1016/j.bbi.2009.09.012
Martin S, Henley JM, Holman D et al (2009) Corticosterone alters AMPAR mobility and facilitates bidirectional synaptic plasticity. PLoS ONE 4:e4714. doi:10.1371/journal.pone.0004714
Schaaf MJ, de Kloet ER, Vreugdenhil E (2000) Corticosterone effects on BDNF expression in the hippocampus. Implications for memory formation. Stress (Amsterdam, Netherlands) 3:201–208
Barrientos RM, Frank MG, Crysdale NY et al (2011) Little exercise, big effects: reversing aging and infection-induced memory deficits, and underlying processes. J Neurosci 31:11578–11586. doi:10.1523/JNEUROSCI.2266-11.2011
Kheirbek MA, Klemenhagen KC, Sahay A, Hen R (2012) Neurogenesis and generalization: a new approach to stratify and treat anxiety disorders. Nat Neurosci 15:1613–1620. doi:10.1038/nn.3262
Petrik D, Lagace DC, Eisch AJ (2012) The neurogenesis hypothesis of affective and anxiety disorders: are we mistaking the scaffolding for the building? Neuropharmacology 62:21–34. doi:10.1016/j.neuropharm.2011.09.003
Mirescu C, Gould E (2006) Stress and adult neurogenesis. Hippocampus 16:233–238. doi:10.1002/hipo.20155
Joëls M, Baram TZ (2009) The neuro-symphony of stress. Nat Rev Neurosci 10:459–466. doi:10.1038/nrn2632
Garcia A, Steiner B, Kronenberg G et al (2004) Age-dependent expression of glucocorticoid- and mineralocorticoid receptors on neural precursor cell populations in the adult murine hippocampus. Aging Cell 3:363–371. doi:10.1111/j.1474-9728.2004.00130.x
Boku S, Nakagawa S, Masuda T et al (2009) Glucocorticoids and lithium reciprocally regulate the proliferation of adult dentate gyrus-derived neural precursor cells through GSK-3beta and beta-catenin/TCF pathway. Neuropsychopharmacology 34:805–815. doi:10.1038/npp.2008.198
Wong EYH, Herbert J (2005) Roles of mineralocorticoid and glucocorticoid receptors in the regulation of progenitor proliferation in the adult hippocampus. Eur J Neurosci 22:785–792. doi:10.1111/j.1460-9568.2005.04277.x
Cameron HA, McEwen BS, Gould E (1995) Regulation of adult neurogenesis by excitatory input and NMDA receptor activation in the dentate gyrus. J Neurosci 15:4687–4692
Okuyama N, Takagi N, Kawai T et al (2004) Phosphorylation of extracellular-regulating kinase in NMDA receptor antagonist-induced newly generated neurons in the adult rat dentate gyrus. J Neurochem 88:717–725
Bernabeu R, Sharp FR (2000) NMDA and AMPA/kainate glutamate receptors modulate dentate neurogenesis and CA3 synapsin-I in normal and ischemic hippocampus. J Cereb Blood Flow Metab 20:1669–1680. doi:10.1097/00004647-200012000-00006
Abrahám I, Juhasz G, Kékesi KA, Kovács KJ (1998) Corticosterone peak is responsible for stress-induced elevation of glutamate in the hippocampus. Stress (Amsterdam, Netherlands) 2:171–181
Cameron HA, Tanapat P, Gould E (1998) Adrenal steroids and N-methyl-d-aspartate receptor activation regulate neurogenesis in the dentate gyrus of adult rats through a common pathway. NSC 82:349–354
Vreugdenhil E, Kolk SM, Boekhoorn K, et al (2007) Doublecortin-like, a microtubule-associated protein expressed in radial glia, is crucial for neuronal precursor division and radial process stability. Eur J Neurosci 25:635–648. doi:10.1111/j.1460-9568.2007.05318.x
Saaltink D-J, Håvik B, Verissimo CS et al (2012) Doublecortin and doublecortin-like are expressed in overlapping and non-overlapping neuronal cell population: implications for neurogenesis. J Comp Neurol 520:2805–2823. doi:10.1002/cne.23144
Dijkmans TF, van Hooijdonk LWA, Fitzsimons CP, Vreugdenhil E (2010) The doublecortin gene family and disorders of neuronal structure. Cent Nerv Syst Agents Med Chem 10:32–46
Vreugdenhil E, Verissimo CSL, Mariman R et al (2009) MicroRNA 18 and 124a down-regulate the glucocorticoid receptor: implications for glucocorticoid responsiveness in the brain. Endocrinology 150:2220–2228. doi:10.1210/en.2008-1335
Yoo AS, Sun AX, Li L et al (2011) MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 476:228–231. doi:10.1038/nature10323
Cheng L-C, Pastrana E, Tavazoie M, Doetsch F (2009) miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat Neurosci 12:399–408. doi:10.1038/nn.2294
Conaco C, Otto S, Han J–J, Mandel G (2006) Reciprocal actions of REST and a microRNA promote neuronal identity. Proc Natl Acad Sci USA 103:2422–2427. doi:10.1073/pnas.0511041103
van Hooijdonk LWA, Ichwan M, Dijkmans TF et al (2009) Lentivirus-mediated transgene delivery to the hippocampus reveals sub-field specific differences in expression. BMC Neurosci 10:2. doi:10.1186/1471-2202-10-2
Fitzsimons CP, van Hooijdonk LWA, Schouten M et al (2013) Knockdown of the glucocorticoid receptor alters functional integration of newborn neurons in the adult hippocampus and impairs fear-motivated behavior. Mol Psychiatry 18:396–407. doi:10.1038/mp.2012.123
Fukumoto K, Morita T, Mayanagi T et al (2009) Detrimental effects of glucocorticoids on neuronal migration during brain development. Mol Psychiatry 14:1119–1131. doi:10.1038/mp.2009.60
Deng W, Aimone JB, Gage FH (2010) New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci 11:339–350. doi:10.1038/nrn2822
Kirby ED, Friedman AR, Covarrubias D et al (2011) Basolateral amygdala regulation of adult hippocampal neurogenesis and fear-related activation of newborn neurons. Mol Psychiatry 17:10. doi:10.1038/mp.2011.71
Urbach A, Redecker C, Witte OW (2008) Induction of neurogenesis in the adult dentate gyrus by cortical spreading depression. Stroke 39:3064–3072. doi:10.1161/STROKEAHA.108.518076
Parent JM, Yu TW, Leibowitz RT et al (1997) Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci 17:3727–3738
Niv F, Keiner S, Krishna K et al (2012) Aberrant neurogenesis after stroke: a retroviral cell labeling study. Stroke 43:2468–2475. doi:10.1161/STROKEAHA.112.660977
Jessberger S, Nakashima K, Clemenson GD et al (2007) Epigenetic modulation of seizure-induced neurogenesis and cognitive decline. J Neurosci 27:5967–5975. doi:10.1523/JNEUROSCI.0110-07.2007
Scharfman HE, Hen R (2007) Neuroscience. Is more neurogenesis always better? Science 315:336–338. doi:10.1126/science.1138711
Parent JM (2007) Adult neurogenesis in the intact and epileptic dentate gyrus. Prog Brain Res 163:529–540. doi:10.1016/S0079-6123(07)63028-3
Duan X, Chang JH, Ge S et al (2007) Disrupted-in-schizophrenia 1 regulates integration of newly generated neurons in the adult brain. Cell 130:1146–1158. doi:10.1016/j.cell.2007.07.010
Mao Y, Ge X, Frank CL et al (2007) Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3β/β-catenin signaling. Cell 136:1017–1031. doi:10.1016/j.cell.2008.12.044
Gallagher BB, Murvin A, Flanigin HF et al (1984) Pituitary and adrenal function in epileptic patients. Epilepsia 25:683–689
Kumar G, Couper A, O’Brien TJ et al (2007) The acceleration of amygdala kindling epileptogenesis by chronic low-dose corticosterone involves both mineralocorticoid and glucocorticoid receptors. Psychoneuroendocrinology 32:834–842. doi:10.1016/j.psyneuen.2007.05.011
Sinclair D, Tsai SY, Woon HG, Weickert CS (2011) Abnormal glucocorticoid receptor mRNA and protein isoform expression in the prefrontal cortex in psychiatric illness. Neuropsychopharmacology 36:2698–2709. doi:10.1038/npp.2011.160
Sinclair D, Webster MJ, Wong J, Weickert CS (2011) Dynamic molecular and anatomical changes in the glucocorticoid receptor in human cortical development. Mol Psychiatry 16:504–515. doi:10.1038/mp.2010.28
Sinclair D, Fullerton JM, Webster MJ, Shannon Weickert C (2012) Glucocorticoid receptor 1B and 1C mRNA transcript alterations in schizophrenia and bipolar disorder, and their possible regulation by GR gene variants. PLoS ONE 7:e31720. doi:10.1371/journal.pone.0031720
Webster MJ, Knable MB, O’Grady J et al (2002) Regional specificity of brain glucocorticoid receptor mRNA alterations in subjects with schizophrenia and mood disorders. Mol Psychiatry 7:985–994. doi:10.1038/sj.mp.4001139
Perlman WR, Webster MJ, Kleinman JE, Weickert CS (2004) Reduced glucocorticoid and estrogen receptor alpha messenger ribonucleic acid levels in the amygdala of patients with major mental illness. Biol Psychiatry 56:844–852. doi:10.1016/j.biopsych.2004.09.006
Lu NZ, Cidlowski JA (2005) Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell 18:331–342. doi:10.1016/j.molcel.2005.03.025
Oakley RH, Cidlowski JA (2011) Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. J Biol Chem 286:3177–3184. doi:10.1074/jbc.R110.179325
Ismaili N, Garabedian MJ (2004) Modulation of glucocorticoid receptor function via phosphorylation. Ann N Y Acad Sci 1024:86–101. doi:10.1196/annals.1321.007
Anacker C, Cattaneo A, Musaelyan K et al (2013) Role for the kinase SGK1 in stress, depression, and glucocorticoid effects on hippocampal neurogenesis. Proc Natl Acad Sci USA 110:8708–8713. doi:10.1073/pnas.1300886110
Zalachoras I, Houtman R, Meijer OC (2013) Understanding stress-effects in the brain via transcriptional signal transduction pathways. Neuroscience 242:97–109. doi:10.1016/j.neuroscience.2013.03.038
Sutcliffe JS, Jiang YH, Galijaard RJ et al (1997) The E6-Ap ubiquitin-protein ligase (UBE3A) gene is localized within a narrowed Angelman syndrome critical region. Genome Res 7:368–377
Mardirossian S, Rampon C, Salvert D et al (2009) Impaired hippocampal plasticity and altered neurogenesis in adult Ube3a maternal deficient mouse model for Angelman syndrome. Exp Neurol 220:341–348. doi:10.1016/j.expneurol.2009.08.035
Godavarthi SK, Dey P, Maheshwari M, Jana NR (2012) Defective glucocorticoid hormone receptor signaling leads to increased stress and anxiety in a mouse model of Angelman syndrome. Hum Mol Genet 21:1824–1834. doi:10.1093/hmg/ddr614
Yu J, de Belle I, Liang H, Adamson ED (2004) Coactivating factors p300 and CBP are transcriptionally crossregulated by Egr1 in prostate cells, leading to divergent responses. Mol Cell 15:83–94. doi:10.1016/j.molcel.2004.06.030
Fitzsimons CP, Ahmed S, Wittevrongel CFW et al (2008) The microtubule-associated protein doublecortin-like regulates the transport of the glucocorticoid receptor in neuronal progenitor cells. Mol Endocrinol 22:248–262. doi:10.1210/me.2007-0233
Meaney MJ, Szyf M, Seckl JR (2007) Epigenetic mechanisms of perinatal programming of hypothalamic–pituitary–adrenal function and health. Trends Mol Med 13:269–277. doi:10.1016/j.molmed.2007.05.003
Zhang TY, Labonté B, Wen XL et al (2013) Epigenetic mechanisms for the early environmental regulation of hippocampal glucocorticoid receptor gene expression in rodents and humans. Neuropsychopharmacology 38:111–123. doi:10.1038/npp.2012.149
Weaver ICG, Cervoni N, Champagne FA et al (2004) Epigenetic programming by maternal behavior. Nat Neurosci 7:847–854. doi:10.1038/nn1276
Szyf M, Weaver ICG, Champagne FA et al (2005) Maternal programming of steroid receptor expression and phenotype through DNA methylation in the rat. Front Neuroendocrinol 26:139–162. doi:10.1016/j.yfrne.2005.10.002
Mirescu C, Peters JD, Gould E (2004) Early life experience alters response of adult neurogenesis to stress. Nat Neurosci 7:841–846. doi:10.1038/nn1290
Mak GK, Antle MC, Dyck RH, Weiss S (2013) Bi-parental care contributes to sexually dimorphic neural cell genesis in the adult mammalian brain. PLoS ONE 8:e62701. doi:10.1371/journal.pone.0062701
Kuhn HG, Dickinson-Anson H, Gage FH (1996) Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci 16:2027–2033
Montaron MF, Drapeau E, Dupret D et al (2006) Lifelong corticosterone level determines age-related decline in neurogenesis and memory. Neurobiol Aging 27:645–654. doi:10.1016/j.neurobiolaging.2005.02.014
Leuner B, Kozorovitskiy Y, Gross CG, Gould E (2007) Diminished adult neurogenesis in the marmoset brain precedes old age. Proc Natl Acad Sci USA 104:17169–17173. doi:10.1073/pnas.0708228104
Mizoguchi K, Ikeda R, Shoji H et al (2009) Aging attenuates glucocorticoid negative feedback in rat brain. NSC 159:259–270. doi:10.1016/j.neuroscience.2008.12.020
Murphy EK, Spencer RL, Sipe KJ, Herman JP (2002) Decrements in nuclear glucocorticoid receptor (GR) protein levels and DNA binding in AgedRat hippocampus. Endocrinology 143:1362–1370
Tanapat P, Galea LA, Gould E (1998) Stress inhibits the proliferation of granule cell precursors in the developing dentate gyrus. Int J Dev Neurosci 16:235–239
Gould E, Tanapat P (1999) Stress and hippocampal neurogenesis. Biol Psychiatry 46:1472–1479
Avishai-Eliner S, Gilles EE, Eghbal-Ahmadi M et al (2001) Altered regulation of gene and protein expression of hypothalamic-pituitary-adrenal axis components in an immature rat model of chronic stress. J Neuroendocrinol 13:799–807
Ma DK, Kim WR, Ming G-L, Song H (2009) Activity-dependent extrinsic regulation of adult olfactory bulb and hippocampal neurogenesis. Ann N Y Acad Sci 1170:664–673. doi:10.1111/j.1749-6632.2009.04373.x
Chiba S, Numakawa T, Ninomiya M et al (2012) Chronic restraint stress causes anxiety- and depression-like behaviors, downregulates glucocorticoid receptor expression, and attenuates glutamate release induced by brain-derived neurotrophic factor in the prefrontal cortex. Prog Neuropsychopharmacol Biol Psychiatry 39:112–119. doi:10.1016/j.pnpbp.2012.05.018
McEwen BS (2012) The ever-changing brain: cellular and molecular mechanisms for the effects of stressful experiences. Devel Neurobiol 72:878–890. doi:10.1002/dneu.20968
Datson NA, van der Perk J, de Kloet ER, Vreugdenhil E (2001) Expression profile of 30,000 genes in rat hippocampus using SAGE. Hippocampus 11:430–444. doi:10.1002/hipo.1058
Datson NA, Morsink MC, Meijer OC, de Kloet ER (2008) Central corticosteroid actions: search for gene targets. Eur J Pharmacol 583:272–289. doi:10.1016/j.ejphar.2007.11.070
Morsink MC, Steenbergen PJ, Vos JB et al (2006) Acute activation of hippocampal glucocorticoid receptors results in different waves of gene expression throughout time. J Neuroendocrinol 18:239–252. doi:10.1111/j.1365-2826.2006.01413.x
Anacker C, Zunszain PA, Cattaneo A et al (2011) Antidepressants increase human hippocampal neurogenesis by activating the glucocorticoid receptor. Mol Psychiatry 16:750. doi:10.1038/mp.2011.26
Tashiro A, Sandler VM, Toni N et al (2006) NMDA-receptor-mediated, cell-specific integration of new neurons in adult dentate gyrus. Nature 442:929–933. doi:10.1038/nature05028
Ge S, Yang C-H, Hsu K-S et al (2007) A critical period for enhanced synaptic plasticity in newly generated neurons of the adult brain. Neuron 54:559–566. doi:10.1016/j.neuron.2007.05.002
Li Y, Luikart BW, Birnbaum S et al (2008) TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 59:399–412. doi:10.1016/j.neuron.2008.06.023
Mu Y, Lee SW, Gage FH (2010) Signaling in adult neurogenesis. Curr Opin Neurobiol 20:416–423. doi:10.1016/j.conb.2010.04.010
Veyrac A, Gros A, Bruel-Jungerman E et al (2013) Zif268/egr1 gene controls the selection, maturation and functional integration of adult hippocampal newborn neurons by learning. Proc Natl Acad Sci USA 110:7062–7067. doi:10.1073/pnas.1220558110
Merz K, Herold S, Lie DC (2011) CREB in adult neurogenesis—master and partner in the development of adult-born neurons? Eur J Neurosci 33:1078–1086. doi:10.1111/j.1460-9568.2011.07606.x
Herold S, Jagasia R, Merz K et al (2011) CREB signalling regulates early survival, neuronal gene expression and morphological development in adult subventricular zone neurogenesis. Mol Cell Neurosci 46:79–88. doi:10.1016/j.mcn.2010.08.008
Jagasia R, Steib K, Englberger E et al (2009) GABA-cAMP response element-binding protein signaling regulates maturation and survival of newly generated neurons in the adult hippocampus. J Neurosci 29:7966–7977. doi:10.1523/JNEUROSCI.1054-09.2009
Conway-Campbell BL, George CL, Pooley JR et al (2011) The HSP90 molecular chaperone cycle regulates cyclical transcriptional dynamics of the glucocorticoid receptor and its coregulatory molecules CBP/p300 during ultradian ligand treatment. Mol Endocrinol 25:944–954. doi:10.1210/me.2010-0073
Vecchia D, Pietrobon D (2012) Migraine: a disorder of brain excitatory–inhibitory balance? Trends Neurosci 35:507–520. doi:10.1016/j.tins.2012.04.007
Eikermann-Haerter K, Yuzawa I, Qin T et al (2011) Enhanced subcortical spreading depression in familial hemiplegic migraine Type 1 mutant mice. J Neurosci 31:5755–5763. doi:10.1523/JNEUROSCI.5346-10.2011
Joëls M (2009) Stress, the hippocampus, and epilepsy. Epilepsia 50:586–597. doi:10.1111/j.1528-1167.2008.01902.x
Szyf M (2009) The early life environment and the epigenome. Biochim Biophys Acta 1790:878–885. doi:10.1016/j.bbagen.2009.01.009
Ridder S, Chourbaji S, Hellweg R et al (2005) Mice with genetically altered glucocorticoid receptor expression show altered sensitivity for stress-induced depressive reactions. J Neurosci 25:6243–6250. doi:10.1523/JNEUROSCI.0736-05.2005
Kronenberg G, Kirste I, Inta D et al (2009) Reduced hippocampal neurogenesis in the GR(+/−) genetic mouse model of depression. Eur Arch Psychiatry Clin Neurosci 259:499–504. doi:10.1007/s00406-009-0036-y
Vreugdenhil E, Berezikov E (2010) Fine-tuning the brain: MicroRNAs. Front Neuroendocrinol 31:128–133. doi:10.1016/j.yfrne.2009.08.001
Wang J, Barak LS, Mook RA, Chen W (2011) Glucocorticoid hedgehog agonists in neurogenesis. Vitam Horm 87:207–215. doi:10.1016/B978-0-12-386015-6.00030-5
Moors M, Bose R, Johansson-Haque K et al (2012) Dickkopf 1 mediates glucocorticoid-induced changes in human neural progenitor cell proliferation and differentiation. Toxicol Sci 125:488–495. doi:10.1093/toxsci/kfr304
Furukawa-Hibi Y, Yun J, Nagai T, Yamada K (2012) Transcriptional suppression of the neuronal PAS domain 4 (Npas4) gene by stress via the binding of agonist-bound glucocorticoid receptor to its promoter. J Neurochem 123:866–875. doi:10.1111/jnc.12034
Ibi D, Takuma K, Koike H et al (2008) Social isolation rearing-induced impairment of the hippocampal neurogenesis is associated with deficits in spatial memory and emotion-related behaviors in juvenile mice. J Neurochem 105:921–932. doi:10.1111/j.1471-4159.2007.05207.x
Murray F, Smith DW, Hutson PH (2008) Chronic low dose corticosterone exposure decreased hippocampal cell proliferation, volume and induced anxiety and depression like behaviours in mice. Eur J Pharmacol 583:115–127. doi:10.1016/j.ejphar.2008.01.014
Nacher J, Gomez-Climent MA, Mcewen B (2004) Chronic non-invasive glucocorticoid administration decreases polysialylated neural cell adhesion molecule expression in the adult rat dentate gyrus. Neurosci Lett 370:40–44. doi:10.1016/j.neulet.2004.07.062
Liu D, Diorio J, Tannenbaum B et al (1997) Maternal care, hippocampal glucocorticoid receptors, and hypothalamic-pituitary-adrenal responses to stress. Science 277:1659–1662
Coe CL, Kramer M, Czéh B et al (2003) Prenatal stress diminishes neurogenesis in the dentate gyrus of juvenile rhesus monkeys. Biological psychiatry 54:1025–1034
Fujioka A, Fujioka T, Ishida Y et al (2006) Differential effects of prenatal stress on the morphological maturation of hippocampal neurons. Neuroscience 141:907–915. doi:10.1016/j.neuroscience.2006.04.046
Lemaire V, Koehl M, Le Moal M, Abrous DN (2000) Prenatal stress produces learning deficits associated with an inhibition of neurogenesis in the hippocampus. Proc Natl Acad Sci USA 97:11032–11037
Bredy TW, Grant RJ, Champagne DL, Meaney MJ (2003) Maternal care influences neuronal survival in the hippocampus of the rat. Eur J Neurosci 18:2903–2909
Cameron HA, McKay RD (1999) Restoring production of hippocampal neurons in old age. Nat Neurosci 2:894–897. doi:10.1038/13197
Simon M, Czéh B, Fuchs E (2005) Age-dependent susceptibility of adult hippocampal cell proliferation to chronic psychosocial stress. Brain Res 1049:244–248. doi:10.1016/j.brainres.2005.05.006
Seki T, Arai Y (1995) Age-related production of new granule cells in the adult dentate gyrus. Neuroreport 6:2479–2482
Dunlap KD, Chung M, Castellano JF (2013) Influence of long-term social interaction on chirping behavior, steroid levels and neurogenesis in weakly electric fish. J Exp Biol 216:2434–2441. doi:10.1242/jeb.082875
Magariños AM, McEwen BS, Flügge G, Fuchs E (1996) Chronic psychosocial stress causes apical dendritic atrophy of hippocampal CA3 pyramidal neurons in subordinate tree shrews. J Neurosci 16:3534–3540
Dong H, Goico B, Martin M et al (2004) Modulation of hippocampal cell proliferation, memory, and amyloid plaque deposition in APPsw (Tg2576) mutant mice by isolation stress. Neuroscience 127:601–609. doi:10.1016/j.neuroscience.2004.05.040
Yap JJ, Takase LF, Kochman LJ et al (2006) Repeated brief social defeat episodes in mice: effects on cell proliferation in the dentate gyrus. Behav Brain Res 172:344–350. doi:10.1016/j.bbr.2006.05.027
Ferragud A, Haro A, Sylvain A et al (2010) Enhanced habit-based learning and decreased neurogenesis in the adult hippocampus in a murine model of chronic social stress. Behav Brain Res 210:134–139. doi:10.1016/j.bbr.2010.02.013
Branchi I, D'Andrea I, Sietzema J et al (2006) Early social enrichment augments adult hippocampal BDNF levels and survival of BrdU-positive cells while increasing anxiety- and “depression-”like behavior. J Neurosci Res 83:965–973. doi:10.1002/jnr.20789
Kannangara TS, Webber A, Gil-Mohapel J, Christie BR (2009) Stress differentially regulates the effects of voluntary exercise on cell proliferation in the dentate gyrus of mice. Hippocampus 19:889–897. doi:10.1002/hipo.20514
Bednarczyk MR, Hacker LC, Fortin-Nunez S et al (2011) Distinct stages of adult hippocampal neurogenesis are regulated by running and the running environment. Hippocampus 21:1334–1347. doi:10.1002/hipo.20831
Oomen CA, Mayer JL, de Kloet ER et al (2007) Brief treatment with the glucocorticoid receptor antagonist mifepristone normalizes the reduction in neurogenesis after chronic stress. Eur J Neurosci 26:3395–3401. doi:10.1111/j.1460-9568.2007.05972.x
Falconer EM, Galea LAM (2003) Sex differences in cell proliferation, cell death and defensive behavior following acute predator odor stress in adult rats. Brain Res 975:22–36
Tanapat P, Hastings NB, Rydel TA et al (2001) Exposure to fox odor inhibits cell proliferation in the hippocampus of adult rats via an adrenal hormone-dependent mechanism. J Comp Neurol 437:496–504
Heine VM, Maslam S, Zareno J et al (2004) Suppressed proliferation and apoptotic changes in the rat dentate gyrus after acute and chronic stress are reversible. Eur J Neurosci 19:131–144
Pham K, Nacher J, Hof PR, McEwen BS (2003) Repeated restraint stress suppresses neurogenesis and induces biphasic PSA-NCAM expression in the adult rat dentate gyrus. Eur J Neurosci 17:879–886
Rosenbrock H, Koros E, Bloching A et al (2005) Effect of chronic intermittent restraint stress on hippocampal expression of marker proteins for synaptic plasticity and progenitor cell proliferation in rats. Brain Res 1040:55–63. doi:10.1016/j.brainres.2005.01.065
Acknowledgments
We would like to thank Dr. Jan van Minnen for a critical evaluation of our manuscript. Erno Vreugdenhil and Dirk-Jan Saaltink were supported by grants from The Netherlands Technology Foundation (STW), the Applied Science Division of the Netherlands Scientific Organization, the Technology Program of the Netherlands Ministry of Economic affairs (LFA6332) and The Center for Medical System Biology, Leiden, The Netherlands.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Saaltink, DJ., Vreugdenhil, E. Stress, glucocorticoid receptors, and adult neurogenesis: a balance between excitation and inhibition?. Cell. Mol. Life Sci. 71, 2499–2515 (2014). https://doi.org/10.1007/s00018-014-1568-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-014-1568-5