
Abstract
The human genome contains more than 1,000 microRNA (miRNA) genes, which are transcribed mainly by RNA polymerase II. The canonical pathway of miRNA biogenesis includes the nuclear processing of primary transcripts (pri-miRNAs) by the ribonuclease Drosha and further cytoplasmic processing of pre-miRNAs by the ribonuclease Dicer. This review discusses the issue of miRNA end heterogeneity generated primarily by Drosha and Dicer cleavage and focuses on the structural aspects of the Dicer step of miRNA biogenesis. We examine the structures of miRNA precursors, both predicted and experimentally determined, as well as the influence of various motifs that disturb the regularity of pre-miRNA structure on Dicer cleavage specificity. We evaluate the structural determinants of the length diversity of miRNA generated by Dicer from different precursors and highlight the importance of asymmetrical motifs. Finally, we discuss the impact of Dicer protein partners on cleavage efficiency and specificity and propose the contribution of pre-miRNA structural plasticity to the dynamics of the dicing complex.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Biogenesis and activity of human microRNAs
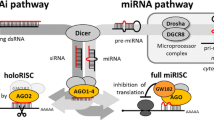
MicroRNAs (miRNAs) are small endogenous post-transcriptional regulators of gene expression found in a wide range of eukaryotes [1]. These 20- to 24-nucleotide (nt)-long RNAs regulate numerous physiological processes, including cell proliferation, differentiation, apoptosis and development [2–5]. Deregulation of miRNA expression has been linked to many disorders, including cancer [6–8] and neurodegeneration [9, 10]. Most of the miRNA genes, which are located predominantly in introns of protein coding genes and intergenic regions, are transcribed by RNA polymerase II (Pol II) [11, 12], and some are transcribed by RNA polymerase III (Pol III) [13] (Fig. 1). The primary transcripts (pri-miRNAs), which are typically capped and polyadenylated, contain one or more long hairpin structures. The structural features of these hairpins are unique to pri-miRNAs, thus distinguishing them from the various RNA stem-loop-like structures present in the nucleus. The pri-miRNA hairpin typically contains a long imperfect stem of approximately 30 bp with flanking single-stranded RNA segments at its base (termed the single-stranded–double-stranded RNA junction) [14, 15]. This structure is recognized and cleaved by the Microprocessor complex containing the ribonuclease Drosha (RNase III enzyme), the RNA binding protein DGCR8 (DiGeorge syndrome critical region gene 8) [16–18] and other proteins [17, 19]. Drosha cleaves the pri-miRNA hairpin at a distance of approximately 11 bp from the single-stranded RNA–dsRNA junction, which is recognized by DGCR8 [15]. The Drosha cleavage of intronic miRNAs occurs co-transcriptionally before splicing of the host RNA [20, 21]. As a result, a long truncated hairpin of approximately 60 nt (pre-miRNA) is generated, which usually has a 2-nt 3′ overhang, the hallmark of RNase III products [22]. The 3′ overhang and the pre-miRNA double-stranded stem with a minimal length of 16 bp [23] are essential structural elements that are recognized by Exportin-5 (Exp-5) [24, 25]. In cooperation with the guanine triphosphatase (GTPase) Ran, Exp-5 transports the pre-miRNA from the nucleus to the cytoplasm irrespective of its nucleotide sequence and the presence of various structural motifs. The high-resolution X-ray structure of the pre-miRNA nuclear export machinery has recently been reported and the crucial role of the structural features of the precursor for Exp-5 recognition confirmed [26]. However, the exact mechanism by which the pre-miRNA is transferred from the Microprocessor to Exp-5 in the nucleus and from Exp-5 to the RISC loading complex (RLC) in the cytoplasm remains to be elucidated. It is thought that Exp-5 passes pre-miRNA to Dicer directly or via additional components [27]. The pre-miRNA cutting machine of the RLC is the ribonuclease Dicer (another RNase III enzyme), which converts the pre-miRNA to mature miRNA. Dicer recognizes the 3′-ends generated by Drosha and cleaves the pre-miRNA two helical turns (approx. 22 nt) away, near the terminal loop, to produce a miRNA–miRNA* duplex having 2-nt 3′ overhangs at both ends [28, 29]. The human Dicer is an approximately 200-kDa multidomain protein that contains an N-terminal DEAD-box helicase domain, a domain originally named the domain of unknown function (DUF283), a PAZ domain, two conserved catalytic RNase III domains (RIIIA and RIIIB) and a C-terminal dsRNA-binding domain (dsRBD) [30]. The two RNase III domains of Dicer form a single processing center for pre-miRNA cleavage, and the 5′ and 3′ arms of the precursor are cleaved by domains RIIIB and RIIIA, respectively [31, 32]. Dicer does not function alone; it cooperates within the RLC with other proteins [33, 34]. Its protein partners are members of the argonaute (AGO) family [35, 36], HIV-1 transactivation response (TAR) RNA-binding protein (TRBP) [37, 38] and possibly other proteins (see the section Dicer partners in pre-miRNA cleavage). Once Dicer has cleaved the pre-miRNA, only one miRNA strand (guide strand) of the duplex is loaded onto AGO to form the programmed RISC (referred to as the miRISC); the other strand (passenger strand or miRNA*) is released and degraded [37, 39, 40]. The thermodynamic stability of the ends of the miRNA duplexes is thought to play a crucial role in miRNA strand selection [41–43]. Other factors contributing to strand selection are structural features of the miRNA/miRNA* duplex (e.g. positions of base mismatches) [44–46] and sequence composition [27, 47]. These features may be disregarded, since there are cases where both miRNA and miRNA* strands may be involved in RISC-mediated gene silencing [46, 48]. The finding of the cognate target is thought to be a diffusion-driven process [49], and the recognition of mRNA by miRISC is based on the partial complementarity of miRNA to mRNA. The “seed” region, i.e. nt 2–8 of the miRNAs, typically forms perfect matches with their target sequences located in the 3′ untranslated region (UTR) of mRNAs, but these interactions fall into several categories [50–52]. MiRNAs downregulate gene expression in several ways; in animal cells, the downregulation is usually achieved by translational arrest, mRNA deadenylation and degradation, or less frequently by mRNA cleavage (reviewed in [1]). More recently, mRNA destabilization and degradation have been proposed to constitute the predominant mechanism of gene expression regulation by miRNAs [53]. Moreover, the existence of “non-canonical” pathways of miRNA biogenesis has also been reported [54]. Pre-miRNA-like introns (mirtrons) are spliced out of mRNA transcripts [55–57]. Mirtrons bypass the Drosha requirements, but other steps of their biogenesis, such as nuclear export and cleavage by Dicer, follow the canonical pathway [58]. Instead, an erythropoetic miR-451 circumvents the Dicer step; miR-451 is cleaved by Ago2 within the 3′ arm, and the exact miRNA 3′-end is generated by trimming, which is mediated by other 3′-5′ exonucleases [59–61]. Like all RNAs, miRNAs also have inherent half-lives. Their turnover has been evaluated in various experiments with Pol II inhibitors and/or by the RNAi-mediated depletion of miRNA processing enzymes. Most miRNAs are highly stable, and their half-lives range from hours to even days, but several miRNAs with an accelerated turnover have also been reported [62–66]. The different aspects of miRNA biogenesis, function and regulation at multiple levels have been extensively reviewed elsewhere [67–71].

Canonical pathway of microRNA (miRNA) biogenesis and activity. Sequential processing reactions of the primary transcript by Drosha in the nucleus and of pre-miRNA by Dicer in the cytoplasm are presented schematically (details are given in the text). The structural requirements for Drosha, Exportin-5 and Dicer are highlighted in the yellow boxes
Here we review recent progress in understanding the mechanism of Drosha and Dicer cleavages from the perspective of their end-products. We describe the specificity of these cleavages and analyze miRNA length variety in terms of both diversity and heterogeneity, searching for their sources and consequences. Finally, we discuss the composition of the core RLC complex, the role of each Dicer protein partner and the structure of this complex as a whole interacting with pre-miRNA.
Relaxed specificity of Drosha and Dicer cleavage and its consequences
It was predicted 6 years ago that as many as 1,000 miRNAs may regulate the expression of most human protein coding genes [72]. The latest release of the miRNA repository (miRBase, release 16) [73] indeed matches that prediction, and the phenomenon of miRNA end and length heterogeneity substantially increases this number. This kind of miRNA heterogeneity, i.e. the existence of several length variants and shifted sequence variants of the same miRNA, has been known since early miRNA studies [74] and has recently been termed miRNA end polymorphism or iso-miR formation [75–77]. Since the routine application of deep sequencing technology in miRNA discovery studies [75, 78–82], the scale of miRNA heterogeneity has been found to be greater than anticipated (Fig. 2a). At the same time, the proper interpretation of deep sequencing results and the separation of real biological effects from various deep sequencing artifacts remain ambiguous. Different deep sequencing platforms generate platform-specific biases [83, 84] that stem from the methods of miRNA library construction [85]. Among the biological effects involved, Drosha- and Dicer-induced cuts with relaxed specificity are one of the most likely explanations for the phenomenon of miRNA heterogeneity [75, 78, 81, 86].

Sources of miRNA end heterogeneity. a Example of deep sequencing data for an miRNA generated from both precursor arms. The number of sequence reads and fraction of miRNA variants are indicated on the right. Variants represented by only one sequence read are not shown. The major contributor (miRNA-MAIN) is shown in boldface, and the miRNA weighted average length (miRNA-WAL) is also indicated. The underlined letters indicate nontemplated nucleotides. The reference precursor and annotated miRNAs (red type), along with the dot-bracket pattern of structure of the precursor, are shown below the aligned sequences. Note that only the miRNA–MAIN generated from the 5′ arm corresponds to the miRNA annotated in miRBase. The figure was prepared from deep sequencing data deposited in miRBase [73, 149]. b–d Different proposed hypotheses to explain the observed miRNA end heterogeneity. b Drosha and Dicer cleavages are the primary sources of miRNA heterogeneity, and the ends generated by Drosha are less heterogeneous than those generated by Dicer. c No matter which arm the miRNA is generated from, the 5′-end is always less heterogeneous than the 3′-end. d Shifted Drosha cleavages result in shifted Dicer cleavages (leading to miRNA with two heterogeneous ends). e AGO2 loading is the selection step for binding miRNAs with U and A at their 5′-ends. f Nontemplated heterogeneity results from modifications of the 3′-end occurring after Drosha/Dicer cleavages
Based on deep sequencing results, it has been proposed that Drosha-induced cleavages generate much less miRNA end heterogeneity than Dicer cleavages [87] (Fig. 2b). Other authors have concluded that the 5′-ends of miRNAs are always less heterogeneous than the 3′-ends, regardless of the precursor arm from which the functional miRNAs were generated [27, 75, 78, 81, 88, 89] (Fig. 2c). This conclusion is in agreement with the notion that evolutionary pressure favors homogenous miRNA 5′-end formation, which is important for specific target recognition [90]. However, the above propositions are mutually exclusive as Drosha defines the 5′-ends of miRNAs from the pre-miRNA 5′ arm and the 3′-ends of miRNAs from the 3′ arm, whereas Dicer defines the 3′ ends of miRNAs from the pre-miRNA 5′ arm and the 5′-ends of miRNAs from the pre-miRNA 3′ arm (see Fig. 2). Drosha and Dicer belong to the same RNase III class; both possess a catalytic center formed by two RNase III domains. The difference in cleavage specificity for Drosha and Dicer may arise from their different strategies of substrate recognition. Dicer combines precursor recognition and cleavage activities, as it contains the PAZ and RNase III domains within one protein. This enzyme design makes the structure flexible and adaptable to various structures of pre-miRNA substrates. Drosha requires another protein (DGCR8) to recognize and bind to pri-miRNAs. It may be these differences between Drosha and Dicer which influence their cleavage specificities (Fig. 2b). It would appear that the discriminative power of the Drosha/DGCR8 complex between substrates and the multitude of nonsubstrate hairpins need to be very high; also, the use of two specialized proteins to perform this task offers more precision in the cleavage. However, it seems unlikely that Drosha and Dicer can sense which precursor arm will generate the functional miRNA and that arm will be cleaved more precisely (Fig. 2c). Therefore, the hypothesis suggesting that miRNA end heterogeneity is the result of different Drosha and Dicer cleavage specificities seems more appealing. Another model of miRNA heterogeneity that was proposed is a parallel shift of Drosha and Dicer cuts to generate several miRNAs from the same precursor [86] (Fig. 2d). This model simply reflects the fact that nonspecific Drosha cleavages will have consequences for nonspecific Dicer cleavages. Apart from the imprecise cleavages by Drosha and Dicer being the primary source of miRNA heterogeneity, the AGO binding step may also introduce a bias toward the U and A residues at the 5′ nucleotides of the miRNA [91] (Fig. 2e).
Starting from the release 16, the deep sequencing data are deposited in miRBase. Most miRNAs are represented by numerous length/sequence variants; in the majority of cases, the miRNAs are generated from one precursor arm (5′ or 3′). Such miRNAs are often represented by approximately 99% of all sequence reads. There are, however, examples of substantial amounts of miRNAs generated from both arms of one precursor (Fig. 2a). The frequency of individual miRNA variants varies strongly, with the most frequent approaching 100% and the rarest being represented by very small fractions (considerably less than 1% of all the reads). Only the most frequent variants (miRNA-MAIN and those that contribute to more than 5% of total reads) can probably be considered functional. If many miRNAs are derived from one precursor arm, the parameter miRNA-WAL (weighted average length) would be appropriate to describe the lengths of miRNAs obtained from deep sequencing and derived from single genes.
In addition to deep sequencing, conventional techniques of miRNA detection have also been used to demonstrate and characterize miRNA heterogeneity. Northern blotting, a “gold standard” in molecular biology, has been frequently used to detect newly identified miRNAs [74, 92–95]. Recent improvements in northern blotting protocols have allowed us to distinguish not only miRNAs but also pre-miRNAs differing in length by 1 nt; the northern blot method has also been used for the quantitative analysis of miRNA and pre-miRNA heterogeneity [96–98]. High-resolution northern blotting used in conjunction with primer extension (which detects 5′-end heterogeneity) has made it possible to distinguish shares held by Drosha and Dicer in generating miRNA heterogeneity. Both Drosha and Dicer generate substantial miRNA end heterogeneity, but the contribution of Dicer is slightly greater [96].
The observed heterogeneity in miRNA ends and lengths may have important functional implications. Different nucleotides at the miRNA 5′-end may change the relative thermodynamic stability of the miRNA/miRNA* duplex ends and cause preferential RISC activation by a different strand. Furthermore, the miRNA 5′-end, particularly its seed sequence, is responsible for the recognition of a complementary sequence and the binding to mRNA. MiRNAs with shifted 5′-ends have different seed sequences and may regulate different targets [77, 78, 96, 99, 100]. It has recently been shown that different miRNA length variants (iso-miRs) may be loaded to different AGO proteins [101]. Thus, the generation of 5′-end heterogeneity may be another mechanism of miRNA activity regulation that functions either by increasing the number of targets regulated by one miRNA or by decreasing the fraction and functional impact of a dominant, canonical miRNA. The role of miRNA 3′-end heterogeneity is also gaining the attention of researchers. In addition to the frequently detected templated heterogeneity (miRNA end nucleotides match the genomic sequence), the presence of nontemplated nucleotides (Fig. 2f) has also been observed in some miRNAs. The nucleotides that most often differ from genomic DNA are typically 3′-end A and U [75, 79, 89, 102]. It is thought that these nucleotides are added to either miRNA or pre-miRNA ends by specific enzymes [102–105] following Drosha or Dicer cleavage. In addition to contributing to the miRNA–mRNA interaction (compensatory site effect) [50, 51], the 3′-end of the miRNA may influence its localization [63]. Extra nucleotides added at the 3′-ends of some miRNAs may also influence their stabilities [103–106], regulate the Dicer step of biogenesis by blocking cleavage [107] or modulate miRNA uptake by RISC bound with different AGO proteins [102]. The 3′-end modification has also been linked to a reduction in the efficiency of mRNA targeting [102, 104].
The 5′- and 3′-end heterogeneity of the Drosha and Dicer cleavage products, as well as the nontemplated effects, may also have implications for the RNA interference (RNAi) and miRNA technologies. The end heterogeneity of biologically processed short RNAi triggers or exogenous miRNAs may create problems in reproducing the silencing effects achieved with synthetic small interfering RNAs (siRNAs) or miRNAs having defined ends [98]. The nonspecific Dicer and Drosha cleavages generate a population of products, only a fraction of which may have the desired sequence and exert the expected effects.
Structural aspects of pre-miRNA cleavage by recombinant Dicer
Another phenomenon related to the variation in the length of miRNAs is their length diversity, i.e. the formation of miRNAs differing in length from different miRNA genes [96]. miRNA length diversity is generated by Dicer from Drosha cleavage products. This effect originates from the structural features of the pre-miRNA hairpins, which differ from each other in the number and localization of various types of structural motifs. The range of human pre-miRNA structural diversity has been estimated by the analysis of predicted structures of 460 pre-miRNAs whose sequences were reconstructed from mature miRNA sequences [108, 109]. The lowest energy structures of pre-miRNAs [110] were analyzed for the presence of various secondary structure motifs (mismatches, bulges, symmetrical and asymmetrical internal loops). All motifs identified were cataloged according to their type, size, position and orientation. Of the 1,243 motifs, 631 were symmetrical (1- to 5-nt-long mismatches and internal loops) and 612 were asymmetrical (bulges and internal loops). Single nucleotide mismatches and bulges accounted for most of the findings (Fig. 3a). The number of structural motifs in the pre-miRNA structures analyzed ranged from zero to seven, with an average of 2.7 motifs per precursor. Based on the distribution, localization and sequence content of the structural motifs, the following interesting observations were reported: (1) the frequency of symmetrical motifs tended to increase and that of asymmetrical motifs decreased when proceeding from the pre-miRNA hairpin base to its terminal loop; (2) bulges were significantly overrepresented in the 5′ arm of the precursor (262) compared to the 3′ arm (172); (3) there were no strongly overrepresented specific sequences within the structural motifs analyzed. The predicted structures of many pre-miRNAs [109] showed great variation, possibly explaining the length diversity of mature miRNAs. To determine the specific structural motifs that could predominantly account for miRNA length diversity, a large number of synthetic pre-miRNAs selected to contain various structural motifs at specific locations were subjected to a cleavage assay with recombinant Dicer [96]. Prior to Dicer cleavage, the structures of nearly 20 pre-miRNAs were determined experimentally by chemical and biochemical methods. The set of metal ions used in that study (Fig. 3b), namely Pb, Ca, Mg and Mn ions [111, 112], was previously used to demonstrate the structural diversity of extended pre-miRNAs [43]. The ions mapped the single-stranded fragments, i.e. the terminal and internal symmetrical and asymmetrical loops. When the structures of the terminal loops were probed, the best results were obtained with S1 and T1 nucleases, but V1 nuclease was used for mapping well-paired stem portions of the secondary structure. The small differences between the predicted and experimentally determined structures were localized mainly in the hairpin terminal loops [96].

Structure and dicing of miRNA precursors. a Structural diversity of miRNA precursors based on bioinformatics analysis of the predicted structures of 460 human pre-miRNAs [109]. The frequencies of various secondary structure motifs are presented in a pie chart. b Structural probing of the 5′-end labeled pre-miR-31 using the indicated probes. Lanes: Ci Incubation control (no probe), F formamide ladder, T guanine-specific ladder. S1, T1, T2 Nucleases. The positions of selected G residues are shown. On the right is the experimentally determined pre-miRNA structure. The cleavage sites and intensities for the selected probes are indicated by the symbols described in the inset. c Results of a Dicer cleavage assay for pre-miR-31. The precursor was incubated with human recombinant Dicer for 1, 2 and 5 min as described by Starega–Roslan [96]. The Dicer cleavage sites (black arrowheads), are shown in the secondary structure model; the reported miRNA sequences are marked in red. The values of the weighted average length of diced RNA (WALDI) parameter for miRNAs generated from the precursor 5′ and 3′ arms are indicated. Other designations are as in 3b
The Dicer cleavage assay (Fig. 3c) of 19 pre-miRNAs and several pre-miRNA mutants revealed the important role of the structure of the precursor in determining the cleavage position. In particular, the presence of asymmetrical structural motifs was found to be a major determinant of miRNA length diversity. Precursor arms harboring excessive unpaired nucleotides gave rise to longer miRNAs [96]. To make the analysis more straightforward and quantitative, a new parameter was introduced in that study, namely, the weighted average length of diced RNA–WALDI, which facilitated finding the correlation between the pre-miRNA structure and the lengths of products excised by Dicer. The results from the Dicer cleavage assay of pre-miRNAs were confirmed by bioinformatic analyses of miRNAs deposited in miRBase [73]. The results of both approaches confirmed that the presence of “excessive” nucleotides in any pre-miRNA arm results in the generation of longer miRNAs from this arm by Dicer [96].
Most of the other information currently available on Dicer cleavage of dsRNA comes from biochemical studies in which the requirements for Dicer binding and cleavage were determined [31, 113–115]. The preferences toward dsRNA over single-stranded RNA were shown, and the binding of the PAZ domain to the 3′ terminal single-stranded overhang was demonstrated [114]. Dicer binding was not only shown to the typical 2-nt 3′ overhang, but also to 1-nt and 3- to 5-nt protruding ends [113, 116] as well as less efficient binding to blunt ends of RNA [114, 117]. Not only the overhang structure itself, but also the base composition of the overhanging nucleotides influence the efficiency of Dicer binding and cleavage [113, 118]. The pre-miRNA terminal loop structure has also recently been shown to influence Dicer’s cleavage efficiency [119], and the existence of some local sequence preferences at Dicer cleavage sites have been postulated [27] (our unpublished data). Additionally, the RNase IIIB Dicer domain was shown to prefer cleaving phosphodiester bonds adjacent to the structural distortions that occur in the 5′ arm of the RNA substrate, therefore determining the Dicer cleavage site [27]. Nevertheless, the 3′ terminus of pre-miRNAs is the major determinant of Dicer’s cleavage position as Dicer measures the distance (two helical turns) from the 3′ end to its cleavage site [30].
Dicer partners in pre-miRNA cleavage
As mentioned earlier in this review, in cells, Dicer functions within the RLC, which comprises the two components TRBP and AGO2, as well as some additional proteins [35]. The role of Dicer’s protein partners in pre-miRNA (dsRNA) dicing has been addressed in several studies, but to date it has not been satisfactorily resolved. In this section, the protein components of the human dicing complex are characterized, and the results of studies aimed at resolving their role in pre-miRNA processing are summarized.
There are four argonaute proteins (AGO1–4) in human cells, but only AGO2 has RNA slicing activity. AGO2 is a major protein involved in the RNAi mechanism whose main function is to induce the guide strand-mediated cleavage of target mRNA by the catalytically competent RISC. The approximately 100-kDa AGO2 protein contains three domains. The PAZ domain is responsible for the binding of the 2-nt overhangs of miRNA and siRNA duplexes [120–123], the MID domain interacts with the 5′-phosphate group of RNA [124] and the PIWI domain, located at the C-terminus, possesses the RNase H activity required for the endonucleolytic cleavage of the miRNA passenger strand and subsequently the target mRNA [121, 124].
TRBP is an approximately 45-kDa RNA binding protein containing three dsRBDs. Two of the dsRBDs can homodimerize or bind to the interferon (IFN)-induced protein kinase R (PKR) and the protein activator of PKR kinase (PACT). The third dsRBD interacts with the N-terminal helicase domain of Dicer [125]. The exact function of TRBP in miRNA biogenesis has not yet been determined and remains controversial [37, 38]. This protein is believed to somehow cooperate with Dicer to facilitate miRNA/siRNA production [126], probably by enhancing the stability of Dicer–substrate complexes [127]. Alternatively, TRBP may assist in recruiting substrates to Dicer [127–129]. Based on the biochemical results [130] and data from electron microscopy imaging of the RLC complex [131], TRBP is the sensor for proper strand loading to RISC, which can proofread incorrect strand loading. TRBP mutations observed in human cancers have been shown to cause defects in miRNA biogenesis [132].
The localization and function of other accessory proteins, such as PACT, in the complex are still poorly understood; however, it has been shown that the loss of PACT expression impairs miRNA biogenesis [34]. PACT is a protein that contains three dsRBDs. Since TRBP and PACT are very similar in terms of their domain composition, it is likely that they compete for the same binding site in Dicer and that their functions may be mutually compensated [34]. TRBP and PACT may interact with each other and also associate with Dicer to facilitate the cleavage of dsRNA; therefore, both proteins play a stimulatory role [126]. The following proteins may also be involved in the regulation of processing selected miRNAs by Dicer: ARS2 [133], FMRP [134], KSRP [135], Lin 28 [103] or ADAR [136]. These proteins act by stimulating pre-miRNA processing by Dicer or by repressing miRNA maturation (reviewed in [67]).
Several experimental systems have been used to date to gain insight into the role of Dicer’s partners in its cleavage efficiency and specificity. Initially, the silencing of endogenous Dicer partners resulted in decreased pre-miRNA processing efficiency rather than in changed cleavage specificity. Even with regard to cleavage efficiency, the results obtained in independent studies were rather discordant [37, 38]. Synthetic precursors were also injected or transfected to cells in culture to follow their cleavage by the endogenous dicing complex [137] (Koscianska et al., submitted). The processing products of synthetic pre-miRNAs in cells were found to correspond rather well to the cleavage products generated by the recombinant Dicer; however, the cleavage efficiency differed between these systems. The experimental systems also included pre-miRNA cleaved by an immunoprecipitate containing Dicer with its protein partners [33, 35] and pre-miRNAs incubated in cellular extracts [138, 139]. Both exogenous stimulators [140] and endogenous inhibitors [141] of dicing were shown to exist. Human RLC was reconstituted in a 1:1:1 stoichiometry from recombinant Dicer, AGO2 and TRBP proteins, and this RLC was used in a cleavage assay with synthetic pre-let-7 [36]. Dicer’s partners showed little or no effect on cleavage specificity compared to Dicer’s activity alone [36] (Koscianska et al., submitted).
Structure and dynamics of the dicing complex
The successful reconstitution of RLC activity from recombinant proteins [36] has prompted researchers to aim at acquiring deeper insights into the molecular architecture of the human Dicer in the complex generated with its protein partners. The high-resolution crystal structures of Giardia intestinalis Dicer [142, 143], Thermus thermophilus AGO2 [144] and the DEAD-box helicase domain [145] have provided useful information for developing the human Dicer, Dicer–TRBP and RLC models based on single particle electron microscopy images [131, 146]. These analyses yielded low-resolution (20 Å) information on the mutual arrangement and possible interactions between the proteins within the binary and ternary complex. Multiple images obtained from electron microscopy suggest that RLC forms an L-shaped structure with an active RNase III center of Dicer in the back portion of the L-structure and in N-terminal domains localized at the base of the L-structure [146]. Using its PIWI and MID domains, AGO2 interacts with the Dicer platform formed by the C-terminal region. The N-terminal domain of AGO2, together with TRBP, interacts with Dicer’s DEAD-box domain localized at the base of the L-structure. AGO2 transiently interacts with TRBP to form a closed complex with Dicer. AGO2’s position in the RLC complex is flexible; it can move upon the binding of RNA and may play the role in increasing pre-miRNA access to Dicer [131, 147]. TRBP increases the affinity of AGO2 for Dicer, thus stabilizing the whole complex. The three-component complex forms a stable, triangle-like architecture [131], with an inside channel with a diameter of about 20 Å and a length of >100 Å. This channel, which runs along the long edge of the L-shaped portion, may be used to bind and position the pre-miRNA for catalysis. Attempts have been made to fit a hairpin structure into the cleft of the reconstructed Dicer–TRBP complex [146]. Most human pre-miRNAs range from 57 to 66 nt [108] and are approximately 78–90 Å long; they can, therefore, be accommodated within the channel. The “catalytic valley” formed by the two RNase III domains in Giardia Dicer is about 20 Å wide (which is similar to the diameter of the RNA-A helix) and 50 Å long [30] and covers about two-thirds of the length of a typical pre-miRNA (Fig. 4). A more in-depth understanding of the RLC and Dicer–TRBP structures [131, 146] and better insight into pre-miRNA structure and dicing [96] will provide answers to the intriguing question of whether the formation of the complex with pre-miRNA requires the structural adaptation of both the RNA and protein components or whether structural changes in only one of them would be sufficient to provide an induced fit [30, 143]. Previous studies that addressed this question focused mainly on the adaptive features in Dicer’s structure [142, 148]. Protein flexibility was proposed to be a critical factor, allowing Dicer to adjust its shape to accommodate the structural diversity of its pre-miRNA substrates [142, 148]. To excise the 20-nt miRNAs from the pre-miRNA hairpin with a fully base-paired stem in the RNA-A conformation, the catalytic site of the RNase III domain has to be located approximately 56 Å from the pre-miRNA 3′-base. To excise 24-nt miRNAs, the distance needs to be approximately 67 Å. Thus, the amplitude of motion of the Dicer catalytic center has to be at least 10 Å, i.e. approximately one-tenth the entire length of the substrate channel. However, the movement of the Dicer structure does not need to be as great. Only a few known human pre-miRNAs have perfectly paired hairpin stems, and their derived miRNAs vary in length from 21 to 22 nt [73, 108]. The stems of other human pre-miRNAs are mosaics of base pairs and internal loops of various types and sizes (Fig. 3a). The unmatched bases of asymmetrical motifs probably bulge out of the helix when the pre-miRNA is accommodated within the substrate channel (Fig. 4); these bases are therefore not counted by Dicer when it sets the distance to its cleavage site [96]. The accumulation of structural imperfections in pre-miRNA hairpins results in a higher plasticity of the structures of the precursor; thus, the pre-miRNA may also contribute to the induced fit required for active complex formation.

A hypothetical model highlighting the role of structural plasticity of the precursors in the dynamics of the pre-miRNA dicing complex. The pre-miRNA hairpin is forced to enter a narrow substrate channel formed by the Dicer portion of RISC loading complex (RLC) [131]. Excessive nucleotides present in any precursor arm bulge out and are not counted by Dicer, which measures the double-stranded RNA (dsRNA) distance from its anchoring site (PAZ domain) to the cleavage site [96]. RIIIA and RIIIB domains: RNase IIIA and RNase IIIB Dicer domains
Concluding remarks
Over the past several years numerous important advances have been made in the miRNA field, and research focusing on miRNA biogenesis is one of the most rapidly progressing areas in this field. The discovery that many miRNA variants containing slightly shifted sequences and differing in lengths are generated from individual miRNA genes has brought an extra interest to the mechanism of miRNA biogenesis and to the sources of miRNA variety. Results of miRNA deep sequencing studies have provided new insights into the specificity of miRNA processing steps triggered by Drosha and Dicer and into the nature of post-cleavage modifications. On the other hand, insightful information regarding the structures of miRNA precursors and their processing enzymes has also been obtained by the conventional molecular and structural biology methods. In this review, we have discussed structural aspects of mammalian microRNA biogenesis, placing special emphasis on the cytoplasmic step triggered by RNase Dicer and the role of precursor structure in generating miRNA length variety. It may be concluded from this article that the mechanism of miRNA precursor cleavages induced by Dicer is now fairly well established and that structural sources of miRNA length diversity are well recognized. There remains, however, a number of unresolved or poorly understood issues. Among these are the questions of sequence preferences for Dicer-induced cleavages and the exact role of Dicer protein partners in the dicing process. The low-resolution structures of the dicing complex provide first insights into its functioning, but further refinement of these structures will be needed to learn more about its dynamics. The recent identification of numerous auxiliary proteins implicated in the nuclear step of miRNA maturation sets the stage for more challenging studies on the structure and dynamics of the Microprocessor complex.
Abbreviations
- AGO:
-
Argonaute
- miRNA:
-
MicroRNA
- PACT:
-
Protein activator of PKR kinase
- RISC:
-
RNA induced silencing complex
- RIIIA and RIIIB:
-
RNase IIIA and RNase IIIB Dicer domains
- RLC:
-
RISC loading complex
- siRNA:
-
Small interfering RNA
- TRBP:
-
HIV-1 transactivation response (TAR) RNA-binding protein
- WALDI:
-
Weighted average length of diced RNA
References
Filipowicz W, Bhattacharyya SN, Sonenberg N (2008) Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 9(2):102–114. doi:10.1038/nrg2290
Ambros V (2004) The functions of animal microRNAs. Nature 431(7006):350–355. doi:10.1038/nature02871
Friedman JM, Jones PA (2009) microRNAs: critical mediators of differentiation, development and disease. Swiss Med Wkly 139(33–34):466–472. doi:smw-12794
Gomase VS, Parundekar AN (2009) microRNA: human disease and development. Int J Bioinform Res Appl 5(5):479–500
Bushati N, Cohen SM (2007) microRNA functions. Annu Rev Cell Dev Biol 23:175–205
Croce CM (2009) Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet 10(10):704–714. doi:10.1038/nrg2634
Ventura A, Jacks T (2009) microRNAs and cancer: short RNAs go a long way. Cell 136(4):586–591
Melo SA, Esteller M (2010) Dysregulation of microRNAs in cancer: Playing with fire. FEBS Lett. doi:10.1016/j.febslet.2010.08.009
Hebert SS, De Strooper B (2009) Alterations of the microRNA network cause neurodegenerative disease. Trends Neurosci 32(4):199–206. doi:10.1016/j.tins.2008.12.003
Lau P, de Strooper B (2010) Dysregulated microRNAs in neurodegenerative disorders. Semin Cell Dev Biol 21(7):768–773. doi:10.1016/j.semcdb.2010.01.009
Cai X, Hagedorn CH, Cullen BR (2004) Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 10(12):1957–1966. doi:10.1261/rna.7135204
Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN (2004) microRNA genes are transcribed by RNA polymerase II. EMBO J 23(20):4051–4060. doi:10.1038/sj.emboj.7600385
Borchert GM, Lanier W, Davidson BL (2006) RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol 13(12):1097–1101. doi:10.1038/nsmb1167
Zeng Y, Cullen BR (2005) Efficient processing of primary microRNA hairpins by Drosha requires flanking nonstructured RNA sequences. J Biol Chem 280(30):27595–27603. doi:10.1074/jbc.M504714200
Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, Sohn SY, Cho Y, Zhang BT, Kim VN (2006) Molecular basis for the recognition of primary microRNAs by the Drosha–DGCR8 complex. Cell 125(5):887–901. doi:10.1016/j.cell.2006.03.043
Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ (2004) Processing of primary microRNAs by the Microprocessor complex. Nature 432(7014):231–235. doi:10.1038/nature03049
Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R (2004) The Microprocessor complex mediates the genesis of microRNAs. Nature 432(7014):235–240. doi:10.1038/nature03120
Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S et al (2003) The nuclear RNase III Drosha initiates microRNA processing. Nature 425(6956):415–419. doi:10.1038/nature01957
Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN (2004) The Drosha–DGCR8 complex in primary microRNA processing. Genes Dev 18(24):3016–3027
Morlando M, Ballarino M, Gromak N, Pagano F, Bozzoni I, Proudfoot NJ (2008) Primary microRNA transcripts are processed co-transcriptionally. Nat Struct Mol Biol 15(9):902–909
Kim YK, Kim VN (2007) Processing of intronic microRNAs. EMBO J 26(3):775–783. doi:10.1038/sj.emboj.7601512
Basyuk E, Suavet F, Doglio A, Bordonne R, Bertrand E (2003) Human let-7 stem-loop precursors harbor features of RNase III cleavage products. Nucleic Acids Res 31(22):6593–6597
Zeng Y, Cullen BR (2004) Structural requirements for pre-microRNA binding and nuclear export by Exportin 5. Nucleic Acids Res 32(16):4776–4785
Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U (2004) Nuclear export of microRNA precursors. Science 303(5654):95–98. doi:10.1126/science.1090599
Yi R, Qin Y, Macara IG, Cullen BR (2003) Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev 17(24):3011–3016. doi:10.1101/gad.1158803
Okada C, Yamashita E, Lee SJ, Shibata S, Katahira J, Nakagawa A, Yoneda Y, Tsukihara T (2009) A high-resolution structure of the pre-microRNA nuclear export machinery. Science 326(5957):1275–1279
Warf MB, Johnson WE, Bass BL (2011) Improved annotation of C. elegans microRNAs by deep sequencing reveals structures associated with processing by Drosha and Dicer. RNA 17(4):563–577
Bernstein E, Caudy AA, Hammond SM, Hannon GJ (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409(6818):363–366. doi:10.1038/35053110
Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD (2001) A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293(5531):834–838
MacRae IJ, Doudna JA (2007) Ribonuclease revisited: structural insights into ribonuclease III family enzymes. Curr Opin Struct Biol 17(1):138–145. doi:10.1016/j.sbi.2006.12.002
Zhang H, Kolb FA, Jaskiewicz L, Westhof E, Filipowicz W (2004) Single processing center models for human Dicer and bacterial RNase III. Cell 118(1):57–68. doi:10.1016/j.cell.2004.06.017
Takeshita D, Zenno S, Lee WC, Nagata K, Saigo K, Tanokura M (2007) Homodimeric structure and double-stranded RNA cleavage activity of the C-terminal RNase III domain of human dicer. J Mol Biol 374(1):106–120. doi:10.1016/j.jmb.2007.08.069
Gregory RI, Chendrimada TP, Cooch N, Shiekhattar R (2005) Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell 123(4):631–640
Lee Y, Hur I, Park SY, Kim YK, Suh MR, Kim VN (2006) The role of PACT in the RNA silencing pathway. EMBO J 25(3):522–532
Maniataki E, Mourelatos Z (2005) A human, ATP-independent, RISC assembly machine fueled by pre-miRNA. Genes Dev 19(24):2979–2990. doi:10.1101/gad.1384005
MacRae IJ, Ma E, Zhou M, Robinson CV, Doudna JA (2008) In vitro reconstitution of the human RISC-loading complex. Proc Natl Acad Sci USA 105(2):512–517
Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, Shiekhattar R (2005) TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 436(7051):740–744. doi:10.1038/nature03868
Haase AD, Jaskiewicz L, Zhang H, Laine S, Sack R, Gatignol A, Filipowicz W (2005) TRBP, a regulator of cellular PKR and HIV-1 virus expression, interacts with Dicer and functions in RNA silencing. EMBO Rep 6(10):961–967. doi:10.1038/sj.embor.7400509
Mourelatos Z, Dostie J, Paushkin S, Sharma A, Charroux B, Abel L, Rappsilber J, Mann M, Dreyfuss G (2002) miRNPs: a novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev 16(6):720–728
Kawamata T, Tomari Y (2010) Making RISC. Trends Biochem Sci 35(7):368–376
Khvorova A, Reynolds A, Jayasena SD (2003) Functional siRNAs and miRNAs exhibit strand bias. Cell 115(2):209–216
Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD (2003) Asymmetry in the assembly of the RNAi enzyme complex. Cell 115(2):199–208
Krol J, Sobczak K, Wilczynska U, Drath M, Jasinska A, Kaczynska D, Krzyzosiak WJ (2004) Structural features of microRNA (miRNA) precursors and their relevance to miRNA biogenesis and small interfering RNA/short hairpin RNA design. J Biol Chem 279(40):42230–42239. doi:10.1074/jbc.M404931200
Okamura K, Liu N, Lai EC (2009) Distinct mechanisms for microRNA strand selection by Drosophila Argonautes. Mol Cell 36(3):431–444. doi:10.1016/j.molcel.2009.09.027
Czech B, Zhou R, Erlich Y, Brennecke J, Binari R, Villalta C, Gordon A, Perrimon N, Hannon GJ (2009) Hierarchical rules for Argonaute loading in Drosophila. Mol Cell 36(3):445–456. doi:10.1016/j.molcel.2009.09.028
Ghildiyal M, Xu J, Seitz H, Weng Z, Zamore PD (2010) Sorting of Drosophila small silencing RNAs partitions microRNA* strands into the RNA interference pathway. RNA 16(1):43–56
Hu HY, Yan Z, Xu Y, Hu H, Menzel C, Zhou YH, Chen W, Khaitovich P (2009) Sequence features associated with microRNA strand selection in humans and flies. BMC Genomics 10:413
Okamura K, Phillips MD, Tyler DM, Duan H, Chou YT, Lai EC (2008) The regulatory activity of microRNA* species has substantial influence on microRNA and 3′ UTR evolution. Nat Struct Mol Biol 15(4):354–363
Ameres SL, Martinez J, Schroeder R (2007) Molecular basis for target RNA recognition and cleavage by human RISC. Cell 130(1):101–112. doi:10.1016/j.cell.2007.04.037
Bartel DP (2009) microRNAs: target recognition and regulatory functions. Cell 136(2):215–233. doi:10.1016/j.cell.2009.01.002
Brennecke J, Stark A, Russell RB, Cohen SM (2005) Principles of microRNA-target recognition. PLoS Biol 3(3):e85
Witkos TM, Koscianska E, Krzyzosiak WJ (2011) Practical aspects of microRNA target prediction. Curr Mol Med 11(2):93–109
Guo H, Ingolia NT, Weissman JS, Bartel DP (2010) Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466(7308):835–840
Miyoshi K, Miyoshi T, Siomi H (2010) Many ways to generate microRNA-like small RNAs: non-canonical pathways for microRNA production. Mol Genet Genomics 284(2):95–103
Berezikov E, Chung WJ, Willis J, Cuppen E, Lai EC (2007) Mammalian mirtron genes. Mol Cell 28(2):328–336
Okamura K, Hagen JW, Duan H, Tyler DM, Lai EC (2007) The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell 130(1):89–100. doi:10.1016/j.cell.2007.06.028
Ruby JG, Jan CH, Bartel DP (2007) Intronic microRNA precursors that bypass Drosha processing. Nature 448(7149):83–86. doi:10.1038/nature05983
Chung WJ, Agius P, Westholm JO, Chen M, Okamura K, Robine N, Leslie CS, Lai EC (2011) Computational and experimental identification of mirtrons in Drosophila melanogaster and Caenorhabditis elegans. Genome Res 21(2):286–300
Yang JS, Maurin T, Robine N, Rasmussen KD, Jeffrey KL, Chandwani R, Papapetrou EP, Sadelain M, O’Carroll D, Lai EC (2010) Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proc Natl Acad Sci USA 107(34):15163–15168
Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ (2010) A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 465(7298):584–589. doi:10.1038/nature09092
Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, Ma E, Mane S, Hannon GJ, Lawson ND et al (2010) A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science 328(5986):1694–1698
Krol J, Busskamp V, Markiewicz I, Stadler MB, Ribi S, Richter J, Duebel J, Bicker S, Fehling HJ, Schubeler D et al (2010) Characterizing light-regulated retinal microRNAs reveals rapid turnover as a common property of neuronal microRNAs. Cell 141(4):618–631
Hwang HW, Wentzel EA, Mendell JT (2007) A hexanucleotide element directs microRNA nuclear import. Science 315(5808):97–100
van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN (2007) Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 316(5824):575–579
Gatfield D, Le Martelot G, Vejnar CE, Gerlach D, Schaad O, Fleury-Olela F, Ruskeepaa AL, Oresic M, Esau CC, Zdobnov EM et al (2009) Integration of microRNA miR-122 in hepatic circadian gene expression. Genes Dev 23(11):1313–1326
Sethi P, Lukiw WJ (2009) micro-RNA abundance and stability in human brain: specific alterations in Alzheimer’s disease temporal lobe neocortex. Neurosci Lett 459(2):100–104
Krol J, Loedige I, Filipowicz W (2010) The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 11(9):597–610
Chekulaeva M, Filipowicz W (2009) Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr Opin Cell Biol 21(3):452–460
Kim VN, Han J, Siomi MC (2009) Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10(2):126–139. doi:10.1038/nrm2632
Winter J, Jung S, Keller S, Gregory RI, Diederichs S (2009) Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol 11(3):228–234
Siomi H, Siomi MC (2010) Posttranscriptional regulation of microRNA biogenesis in animals. Mol Cell 38(3):323–332. doi:10.1016/j.molcel.2010.03.013
Berezikov E, Guryev V, van de Belt J, Wienholds E, Plasterk RH, Cuppen E (2005) Phylogenetic shadowing and computational identification of human microRNA genes. Cell 120(1):21–24
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ (2008) miRBase: tools for microRNA genomics. Nucleic Acids Res 36(Database issue):D154–D158. doi:10.1093/nar/gkm952
Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T (2002) Identification of tissue-specific microRNAs from mouse. Curr Biol 12(9):735–739
Morin RD, O’Connor MD, Griffith M, Kuchenbauer F, Delaney A, Prabhu AL, Zhao Y, McDonald H, Zeng T, Hirst M et al (2008) Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res 18(4):610–621. doi:10.1101/gr.7179508
Marti E, Pantano L, Banez-Coronel M, Llorens F, Minones-Moyano E, Porta S, Sumoy L, Ferrer I, Estivill X (2010) A myriad of miRNA variants in control and Huntington’s disease brain regions detected by massively parallel sequencing. Nucleic Acids Res. doi:10.1093/nar/gkq575
Wu H, Neilson JR, Kumar P, Manocha M, Shankar P, Sharp PA, Manjunath N (2007) miRNA profiling of naive, effector and memory CD8 T cells. PLoS One 2(10):e1020. doi:10.1371/journal.pone.0001020
Seitz H, Ghildiyal M, Zamore PD (2008) Argonaute loading improves the 5′ precision of both microRNAs and their miRNA* strands in flies. Curr Biol 18(2):147–151. doi:10.1016/j.cub.2007.12.049
Azuma-Mukai A, Oguri H, Mituyama T, Qian ZR, Asai K, Siomi H, Siomi MC (2008) Characterization of endogenous human Argonautes and their miRNA partners in RNA silencing. Proc Natl Acad Sci USA 105(23):7964–7969. doi:10.1073/pnas.0800334105
Nygaard S, Jacobsen A, Lindow M, Eriksen J, Balslev E, Flyger H, Tolstrup N, Moller S, Krogh A, Litman T (2009) Identification and analysis of miRNAs in human breast cancer and teratoma samples using deep sequencing. BMC Med Genomics 2:35. doi:10.1186/1755-8794-2-35
Ruby JG, Jan C, Player C, Axtell MJ, Lee W, Nusbaum C, Ge H, Bartel DP (2006) Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell 127(6):1193–1207. doi:10.1016/j.cell.2006.10.040
Berezikov E, Robine N, Samsonova A, Westholm JO, Naqvi A, Hung JH, Okamura K, Dai Q, Bortolamiol-Becet D, Martin R et al (2011) Deep annotation of Drosophila melanogaster microRNAs yields insights into their processing, modification, and emergence. Genome Res 21(2):203–215
Huse SM, Huber JA, Morrison HG, Sogin ML, Welch DM (2007) Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol 8(7):R143
Dohm JC, Lottaz C, Borodina T, Himmelbauer H (2008) Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res 36(16):e105. doi:10.1093/nar/gkn425
Tian G, Yin X, Luo H, Xu X, Bolund L, Zhang X (2010) Sequencing bias: comparison of different protocols of microRNA library construction. BMC Biotechnol 10:64
Wu H, Ye C, Ramirez D, Manjunath N (2009) Alternative processing of primary microRNA transcripts by Drosha generates 5′ end variation of mature microRNA. PLoS One 4(10):e7566. doi:10.1371/journal.pone.0007566
Calabrese JM, Seila AC, Yeo GW, Sharp PA (2007) RNA sequence analysis defines Dicer’s role in mouse embryonic stem cells. Proc Natl Acad Sci USA 104(46):18097–18102. doi:10.1073/pnas.0709193104
Palakodeti D, Smielewska M, Graveley BR (2006) microRNAs from the Planarian Schmidtea mediterranea: a model system for stem cell biology. RNA 12(9):1640–1649
Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M et al (2007) A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129(7):1401–1414. doi:10.1016/j.cell.2007.04.040
Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB (2003) Prediction of mammalian microRNA targets. Cell 115(7):787–798
Frank F, Sonenberg N, Nagar B (2010) Structural basis for 5′-nucleotide base-specific recognition of guide RNA by human AGO2. Nature 465(7299):818–822
Houbaviy HB, Murray MF, Sharp PA (2003) Embryonic stem cell-specific microRNAs. Dev Cell 5(2):351–358
Miska EA, Alvarez-Saavedra E, Townsend M, Yoshii A, Sestan N, Rakic P, Constantine-Paton M, Horvitz HR (2004) Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol 5(9):R68
Kim J, Krichevsky A, Grad Y, Hayes GD, Kosik KS, Church GM, Ruvkun G (2004) Identification of many microRNAs that copurify with polyribosomes in mammalian neurons. Proc Natl Acad Sci USA 101(1):360–365
Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V (2004) Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol 5(3):R13
Starega-Roslan J, Krol J, Koscianska E, Kozlowski P, Szlachcic WJ, Sobczak K, Krzyzosiak WJ (2011) Structural basis of microRNA length variety. Nucleic Acids Res 39(1):257–268
Koscianska E, Starega-Roslan J, Czubala K, Krzyzosiak WJ (2011) High-resolution northern blot for a reliable analysis of microRNAs and their precursors. ScientificWorldJournal 11:102–117
Koscianska E, Starega-Roslan J, Sznajder LJ, Olejniczak M, Galka-Marciniak P, Krzyzosiak WJ (2011) Northern blotting analysis of microRNAs, their precursors and RNA interference triggers. BMC Mol Biol 12(1):14. doi:10.1186/1471-2199-12-14
Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, Johnston WK, Russ C, Luo S, Babiarz JE et al (2010) Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev 24(10):992–1009
Kawahara Y, Megraw M, Kreider E, Iizasa H, Valente L, Hatzigeorgiou AG, Nishikura K (2008) Frequency and fate of microRNA editing in human brain. Nucleic Acids Res 36(16):5270–5280
Burroughs AM, Ando Y, Hoon ML, Tomaru Y, Suzuki H, Hayashizaki Y, Daub CO (2011) Deep-sequencing of human Argonaute-associated small RNAs provides insight into miRNA sorting and reveals Argonaute association with RNA fragments of diverse origin. RNA Biol 8 (1)
Burroughs AM, Ando Y, de Hoon MJ, Tomaru Y, Nishibu T, Ukekawa R, Funakoshi T, Kurokawa T, Suzuki H, Hayashizaki Y et al (2010) A comprehensive survey of 3′ animal miRNA modification events and a possible role for 3′ adenylation in modulating miRNA targeting effectiveness. Genome Res 20(10):1398–1410
Heo I, Joo C, Cho J, Ha M, Han J, Kim VN (2008) Lin28 mediates the terminal uridylation of let-7 precursor microRNA. Mol Cell 32(2):276–284
Jones MR, Quinton LJ, Blahna MT, Neilson JR, Fu S, Ivanov AR, Wolf DA, Mizgerd JP (2009) Zcchc11-dependent uridylation of microRNA directs cytokine expression. Nat Cell Biol 11(9):1157–1163
Katoh T, Sakaguchi Y, Miyauchi K, Suzuki T, Kashiwabara S, Baba T, Suzuki T (2009) Selective stabilization of mammalian microRNAs by 3′ adenylation mediated by the cytoplasmic poly(A) polymerase GLD-2. Genes Dev 23(4):433–438
Kai ZS, Pasquinelli AE (2010) microRNA assassins: factors that regulate the disappearance of miRNAs. Nat Struct Mol Biol 17(1):5–10
Heo I, Joo C, Kim YK, Ha M, Yoon MJ, Cho J, Yeom KH, Han J, Kim VN (2009) TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell 138(4):696–708
Krol J, Starega-Roslan J, Milanowska K, Nowak D, Kubiaczyk E, Nowak M, Majorek K, Kaminska K, Krzyzosiak WJ (2006) Structural features of microRNAs and their precursors In: Clarke N, Sanseau P (eds) microRNA: biology, function & expression. DNA Press, Eagleville, pp 95–110
Kozlowski P, Starega-Roslan J, Legacz M, Magnus M, Krzyzosiak WJ (2008) Structures of microRNA precursors. In: Ying S-Y (ed) Current perspectives in microRNAs (miRNA). Springer SBM, Dordrecht, pp 1–16. doi:10.1007/978-1-4020-8533-8_1
Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31(13):3406–3415
Ciesiolka J, Michalowski D, Wrzesinski J, Krajewski J, Krzyzosiak WJ (1998) Patterns of cleavages induced by lead ions in defined RNA secondary structure motifs. J Mol Biol 275(2):211–220
Krzyzosiak WJ, Marciniec T, Wiewiorowski M, Romby P, Ebel JP, Giege R (1988) Characterization of the lead(II)-induced cleavages in tRNAs in solution and effect of the Y-base removal in yeast tRNAPhe. Biochemistry 27(15):5771–5777
Vermeulen A, Behlen L, Reynolds A, Wolfson A, Marshall WS, Karpilow J, Khvorova A (2005) The contributions of dsRNA structure to Dicer specificity and efficiency. RNA 11(5):674–682. doi:10.1261/rna.7272305
Provost P, Dishart D, Doucet J, Frendewey D, Samuelsson B, Radmark O (2002) Ribonuclease activity and RNA binding of recombinant human Dicer. EMBO J 21(21):5864–5874
Zhang H, Kolb FA, Brondani V, Billy E, Filipowicz W (2002) Human Dicer preferentially cleaves dsRNAs at their termini without a requirement for ATP. EMBO J 21(21):5875–5885
Ando Y, Maida Y, Morinaga A, Burroughs AM, Kimura R, Chiba J, Suzuki H, Masutomi K, Hayashizaki Y (2011) Two-step cleavage of hairpin RNA with 5′ overhangs by human DICER. BMC Mol Biol 12(1):6. doi:10.1186/1471-2199-12-6
MacRae IJ, Zhou K, Doudna JA (2007) Structural determinants of RNA recognition and cleavage by Dicer. Nat Struct Mol Biol 14(10):934–940
DiNitto JP, Wang L, Wu JC (2010) Continuous fluorescence-based method for assessing dicer cleavage efficiency reveals 3′ overhang nucleotide preference. Biotechniques 48(4):303–311
Zhang X, Zeng Y (2010) The terminal loop region controls microRNA processing by Drosha and Dicer. Nucleic Acids Res 38:7689–7697. doi: 10.1093/nar/gkq645
Yan KS, Yan S, Farooq A, Han A, Zeng L, Zhou MM (2003) Structure and conserved RNA binding of the PAZ domain. Nature 426(6965):468–474
Song JJ, Smith SK, Hannon GJ, Joshua-Tor L (2004) Crystal structure of Argonaute and its implications for RISC slicer activity. Science 305(5689):1434–1437
Ma JB, Ye K, Patel DJ (2004) Structural basis for overhang-specific small interfering RNA recognition by the PAZ domain. Nature 429(6989):318–322
Lingel A, Simon B, Izaurralde E, Sattler M (2003) Structure and nucleic-acid binding of the Drosophila Argonaute 2 PAZ domain. Nature 426(6965):465–469
Parker JS, Roe SM, Barford D (2004) Crystal structure of a PIWI protein suggests mechanisms for siRNA recognition and slicer activity. EMBO J 23(24):4727–4737
Laraki G, Clerzius G, Daher A, Melendez-Pena C, Daniels S, Gatignol A (2008) Interactions between the double-stranded RNA-binding proteins TRBP and PACT define the Medipal domain that mediates protein–protein interactions. RNA Biol 5(2):92–103
Kok KH, Ng MH, Ching YP, Jin DY (2007) Human TRBP and PACT directly interact with each other and associate with dicer to facilitate the production of small interfering RNA. J Biol Chem 282(24):17649–17657
Chakravarthy S, Sternberg SH, Kellenberger CA, Doudna JA (2010) Substrate-specific kinetics of Dicer-catalyzed RNA processing. J Mol Biol 404(3):392–402
Parker GS, Eckert DM, Bass BL (2006) RDE-4 preferentially binds long dsRNA and its dimerization is necessary for cleavage of dsRNA to siRNA. Rna 12(5):807–818
Parker GS, Maity TS, Bass BL (2008) dsRNA binding properties of RDE-4 and TRBP reflect their distinct roles in RNAi. J Mol Biol 384(4):967–979
Castanotto D, Sakurai K, Lingeman R, Li H, Shively L, Aagaard L, Soifer H, Gatignol A, Riggs A, Rossi JJ (2007) Combinatorial delivery of small interfering RNAs reduces RNAi efficacy by selective incorporation into RISC. Nucleic Acids Res 35(15):5154–5164
Wang HW, Noland C, Siridechadilok B, Taylor DW, Ma E, Felderer K, Doudna JA, Nogales E (2009) Structural insights into RNA processing by the human RISC-loading complex. Nat Struct Mol Biol 16(11):1148–1153. doi:10.1038/nsmb.1673
Melo SA, Ropero S, Moutinho C, Aaltonen LA, Yamamoto H, Calin GA, Rossi S, Fernandez AF, Carneiro F, Oliveira C et al (2009) A TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function. Nat Genet 41(3):365–370
Gruber JJ, Zatechka DS, Sabin LR, Yong J, Lum JJ, Kong M, Zong WX, Zhang Z, Lau CK, Rawlings J et al (2009) Ars2 links the nuclear cap-binding complex to RNA interference and cell proliferation. Cell 138(2):328–339
Xu XL, Li Y, Wang F, Gao FB (2008) The steady-state level of the nervous-system-specific microRNA-124a is regulated by dFMR1 in Drosophila. J Neurosci 28(46):11883–11889
Trabucchi M, Briata P, Garcia-Mayoral M, Haase AD, Filipowicz W, Ramos A, Gherzi R, Rosenfeld MG (2009) The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature 459(7249):1010–1014
Kawahara Y, Zinshteyn B, Chendrimada TP, Shiekhattar R, Nishikura K (2007) RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer–TRBP complex. EMBO Rep 8(8):763–769
Lund E, Dahlberg JE (2006) Substrate selectivity of exportin 5 and Dicer in the biogenesis of microRNAs. Cold Spring Harb Symp Quant Biol 71:59–66
Flores-Jasso CF, Arenas-Huertero C, Reyes JL, Contreras-Cubas C, Covarrubias A, Vaca L (2009) First step in pre-miRNAs processing by human Dicer. Acta Pharmacol Sin 30(8):1177–1185. doi:10.1038/aps.2009.108
Leuschner PJ, Martinez J (2007) In vitro analysis of microRNA processing using recombinant Dicer and cytoplasmic extracts of HeLa cells. Methods 43(2):105–109. doi:10.1016/j.ymeth.2007.04.005
Shan G, Li Y, Zhang J, Li W, Szulwach KE, Duan R, Faghihi MA, Khalil AM, Lu L, Paroo Z et al (2008) A small molecule enhances RNA interference and promotes microRNA processing. Nat Biotechnol 26(8):933–940
Obernosterer G, Leuschner PJ, Alenius M, Martinez J (2006) Post-transcriptional regulation of microRNA expression. RNA 12(7):1161–1167
MacRae IJ, Li F, Zhou K, Cande WZ, Doudna JA (2006) Structure of Dicer and mechanistic implications for RNAi. Cold Spring Harb Symp Quant Biol 71:73–80. doi:10.1101/sqb.2006.71.042
MacRae IJ, Zhou K, Li F, Repic A, Brooks AN, Cande WZ, Adams PD, Doudna JA (2006) Structural basis for double-stranded RNA processing by Dicer. Science 311(5758):195–198. doi:10.1126/science.1121638
Wang Y, Juranek S, Li H, Sheng G, Tuschl T, Patel DJ (2008) Structure of an argonaute silencing complex with a seed-containing guide DNA and target RNA duplex. Nature 456(7224):921–926
Hogbom M, Collins R, van den Berg S, Jenvert RM, Karlberg T, Kotenyova T, Flores A, Karlsson Hedestam GB, Schiavone LH (2007) Crystal structure of conserved domains 1 and 2 of the human DEAD-box helicase DDX3X in complex with the mononucleotide AMP. J Mol Biol 372(1):150–159
Lau PW, Potter CS, Carragher B, MacRae IJ (2009) Structure of the human Dicer–TRBP complex by electron microscopy. Structure 17(10):1326–1332. doi:10.1016/j.str.2009.08.013
Tan GS, Garchow BG, Liu X, Yeung J, Morris JPt, Cuellar TL, McManus MT, Kiriakidou M (2009) Expanded RNA-binding activities of mammalian Argonaute 2. Nucleic Acids Res 37(22):7533–7545
Gan J, Shaw G, Tropea JE, Waugh DS, Court DL, Ji X (2008) A stepwise model for double-stranded RNA processing by ribonuclease III. Mol Microbiol 67(1):143–154. doi:10.1111/j.1365-2958.2007.06032.x
Bar M, Wyman SK, Fritz BR, Qi J, Garg KS, Parkin RK, Kroh EM, Bendoraite A, Mitchell PS, Nelson AM et al (2008) microRNA discovery and profiling in human embryonic stem cells by deep sequencing of small RNA libraries. Stem Cells 26(10):2496–2505. doi:10.1634/stemcells.2008-0356
Acknowledgments
This work was supported by the European Regional Development Fund within the Innovative Economy Programme [Grant No POIG.01.03.01-00-098/08], the Polish Ministry of Science and Higher Education [Grant No. NN301-523038, NN302-278937], the European Union under the European Social Fund (8.2.2 Human Capital Operational Programme to J.S.-R.).
Conflict of interest
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Starega-Roslan, J., Koscianska, E., Kozlowski, P. et al. The role of the precursor structure in the biogenesis of microRNA. Cell. Mol. Life Sci. 68, 2859–2871 (2011). https://doi.org/10.1007/s00018-011-0726-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-011-0726-2