
Abstract
Effectiveness of immune surveillance of intracellular viruses and bacteria depends upon a functioning antigen presentation pathway that allows infected cells to reveal the presence of an intracellular pathogen. The antigen presentation pathway uses virtually all endogenous polypeptides as a source to produce antigenic peptides that are eventually chaperoned to the cell surface by MHC class I molecules. Intriguingly, MHC I molecules present peptides encoded not only in the primary open reading frames but also those encoded in alternate reading frames. Here, we review recent studies on the generation of cryptic pMHC I. We focus on the immunological significance of cryptic pMHC I, and the novel translational mechanisms that allow production of these antigenic peptides from unconventional sources.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nearly all vertebrate cells express molecules of the major histocompatibility complex class I (MHC) on their surface. Each MHC I molecule contains a peptide derived from one of various endogenous proteins. Thus, the peptide repertoire presented by MHC I reflects the intracellular protein milieu and, in infected or transformed cells, includes additional proteins such as those derived from intracellular pathogens. Because the pMHC I serve as unique ligands for the CD8+ cytotoxic T cell receptors, the generation of pMHC I by the antigen presentation pathway allows the CD8+ T cells to detect and eventually eliminate infected or transformed cells.
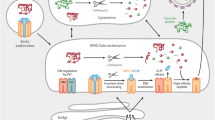
The antigen presentation pathway begins in the cytoplasm (Fig. 1) [1, 2]. Most polypeptide precursors are fragmented by the proteasome into proteolytic intermediates in the cytoplasm. The intermediates are then transported into the endoplasmic reticulum (ER) by the TAP heterodimer. Once in the ER, the MHC I molecules in the peptide-loading complex (PLC) are loaded with appropriate peptides with the help of ER aminopeptidases and housekeeping chaperones. After being loaded with the appropriate peptide cargo, MHC I carry the bound peptides to the cell surface for presentation to CD8+ T cells. Because neither the MHC I molecules nor the CD8+ T cells discriminate among sources of precursor polypeptides, immune surveillance can use pMHC I that arise from normal as well as non-conventional translational mechanisms.

Schematic diagram of the MHC class I antigen processing pathway that culminates in display of peptide bound MHC I on the cell surface. Most antigenic peptides are derived from polypeptides synthesized in the cytoplasm by translation of open reading frames (ORF). However, many other peptides arise as defective ribosomal polypeptides (DRiPs) by translation of ORF or cryptic reading frames (RF). The polypeptides undergo proteolysis in the same compartment and are then transported into the endoplasmic reticulum (ER). In the ER, many peptides are further trimmed by aminopeptidases and assembled with the resident MHC molecules. The MHC I molecules then chaperone the peptides to the cell surface where they serve as potential ligands for the killer CD8+ T cell repertoire
New protein synthesis supplies a significant portion of peptides presented on the cell surface as pMHC I [3, 4]. Indeed, it has been argued that a ribosome-based mechanism may have evolved to use a fraction of newly translated polypeptides exclusively as substrates for antigen processing and presentation [5]. In addition, evidence has accumulated showing that non-conventional translational mechanisms also serve as a unique source of peptides for presentation by MHC class I [1, 6, 7]. Here, we review recent immunological and biochemical studies of non-conventional sources of MHC I ligands that arise during unexpected ribosome decoding events on endogenous as well as viral mRNAs.
Naturally processed cryptic peptide ligands for T cells
Since the original observations of Boon and colleagues describing presentation of antigenic peptides referred to as “peptons” that seemed to arise from unexpected sources [8], the list of cryptic peptides recognized by T cells has grown considerably (reviewed in [6, 7]. The examples (Table 1) include peptides that arise from untranslated regions (UTRs or introns) of the mRNA—challenging the very definition of an open reading frame (ORF)—as well as peptides encoded in alternate translational reading frames (ARF) and from non-AUG start codon initiation on both endogenous and viral mRNAs. That cryptic pMHC I arise from endogenous as well as virally-encoded mRNAs suggests that immune surveillance has evolved to exploit highly conserved aspects of protein translation.
The examples listed in Table 1 suggest that while mammalian ribosomes must carefully select the correct start codon of the ORF for synthesis of functional proteins they also promiscuously translate other regions of the mRNA irrespective of the ORF location for MHC class I presentation. On one hand, it may be tempting to dismiss these translational events simply as mistakes without any physiological relevance. On the other hand, the fact that cryptic peptides effectively compete with the hundreds of thousands of other peptides presented by MHC class I on the cell surface and elicit T cell responses argues that cryptic peptides are immunologically relevant for immune surveillance.
Immunological significance of cryptic translation
In the last few years, the identification of non-conventional epitopes arising from ARF translation on viral mRNAs has highlighted the functional significance of cryptic epitopes during immune surveillance (Table 1). In addition to murine AIDS which was found to elicit protective cytotoxic CD8+ T lymphocyte responses (CTLs) towards an ARF peptide encoded in the LP-BM5 gag gene [9, 10], simian immunodeficiency virus (SIV) mac239-infected rhesus macaques mounted a strong CTL response towards an epitope that was translated from the +2 reading frame relative to the env ORF [11]. Further, using HLA-B*07 transgenic mice, Cardinaud et al. [12] showed that CTLs target an HLA-B*07-restricted ARF epitope translated from the +2 ORF within the gag gene and that this ARF epitope was recognized by CTLs in HIV-infected individuals. More recently, Maness et al. [13] have determined that nearly one-quarter of the anti-SIV CD8+ T cell responses in SIV-infected rhesus macaques are directed towards cryptic epitopes generated from ARF translation.
To assess the global impact of these cryptic epitopes for both immune surveillance and viral evolution, several groups have used bioinformatic prediction methods to identify potential non-conventional epitopes in HIV-infected individuals [14–16]. Berger et al. analyzed HIV gag, pol, and nef sequences from a cohort of 765 individuals to identify HLA allele-associated viral polymorphisms. A total of 64 HLA-associated viral polymorphisms translated in the +2 or +3 reading frames were identified. One particular polymorphism showed that individuals expressing the HLA-A*03 allele were significantly less likely to have serine at position 241 in the +2 reading frame of integrase compared with individuals lacking this HLA type. Indeed, epitope mapping showed the optimal sequence to be RR9 (RTSKASLER) with serine at position 6 (underlined) which potently inhibited viral replication in vitro. As predicted from the identified polymorphism within this sequence, the most common escape variant RPR9 (RTSKAPLER) contained proline at position 6 (underlined). Not only was this sequence translated in an alternate reading frame, but no upstream in-frame AUG start codons were present. Instead, a codon normally encoding leucine was identified as a likely start site. Bansal et al. also determined HLA-associated polymorphisms in the six possible translational reading frames of HIV-1 gag, pol, and nef which include antisense transcripts. They found both sense- and antisense-encoded ARF epitopes were immunogenic during primary and chronic infections and that these ARF epitopes were often mutated during the first year of infection. This observation is consistent with an important role for ARF epitopes in immune responses that inhibit viral replication and can thus select for escape variants.
A different escape variant was recently identified by Cardinaud et al. who characterized a mutation within the HIV-1 gag ARF (QPRSDTHVF, Q9VF) (personal communication). They found that patients eliciting CTL responses against Q9VF did not contain provirus encoding this epitope, but rather Q9VF/5 N (QPRSNTHVF), a parental epitope with a D to N mutation. Apparently, this point mutation inhibits presentation of Q9VF/5 N by a proteasome degradation mechanism. This is an interesting example of how ARF peptides providing a cellular defense against infection allow viruses to evolve, within a short period of time, to subvert effectiveness of immune surveillance by CD8+ cytotoxic T cells.
These examples underscore the possibility that vaccine design could be enhanced by the inclusion of non-conventional epitopes, including ARF epitopes described from HIV-1. Indeed, Maness et al. [17] show that ARF epitopes in DNA vaccines elicit a much stronger response in rhesus macaques than normal infection. However, presentation of immunogenic peptides from ARFs can also have deleterious effects as shown by Samulski’s group, where a DNA cassette being used for human gene therapy trials generated unexpected ARF polypeptides [18]. The targeting of these pMHC I by cytotoxic T cells caused a loss of genetically modified cells and failure of their therapeutic potential. Certainly, it is desirable to understand the fundamental mechanisms which direct non-conventional translation on both endogenous and viral message in order to harness the utility and prevent unwanted effects of non-conventional epitopes.
Mechanisms for producing cryptic peptides for presentation
Non-conventional epitopes are derived from polypeptide precursors that serve no obvious function in the cell but are nevertheless presented on MHC class I molecules. By definition, these precursors could be described as DRiPs (defective ribosomal products), destined for automatic entry into the MHC class I presentation pathway [19]. If such precursors were readily available for antigen presentation, they could serve to efficiently report the status of the cell (infected or transformed) to the immune system. This would be particularly beneficial during viral infection since many viral proteins have evolved to be extremely stable [half life (t½) > 24 h] and recalcitrant to proteasomal degradation [e.g., EBNA1 from Epstein-Barr virus (EBV) and LANA1 from Kaposi’s sarcoma-associated herpesvirus (KHSV)] [20–23]. The different translational mechanisms for producing cryptic polypeptide precursors could circumvent the delay in presentation of stable viral proteins through protein turn-over. Indeed, Cardinaud et al. show that truncated antigenic precursors are generated when ribosomes terminate translation of EBNA1 prematurely [24]. Other mechanisms include translation from non-coding regions and alternate reading frames of the mRNA as well as from non-AUG start codons within any region of the mRNA. The flexibility to use a variety of translation mechanisms might relieve the bottlenecks of conventional translation such as when translation initiation factors are limiting or even missing (Fig. 2).

Schematic view of translational products of a typical mRNA containing untranslated regions (UTR) and the translated open reading frame (ORF). The polypeptides arise from translation of the ORF (RF0), or alternate reading frames (RF1 or RF2) by ribosomes initiating at the conventional AUG or non-conventional CUG codons. The AUG and the CUG initiation codons can be translated as the canonical methionine (M) or the leucine (L) residues, respectively. UAG is a translation termination codon. The final peptides presented by MHC molecules are shown as colored circles
Translation of “non-coding” mRNA
The recent discovery of an HLA-B*2705 bound peptide translated from the 5′-UTR of VEGF, likely through initiation at a CUG start codon, suggests cryptic translation products are novel targets for tumor therapy [25]. This peptide is abundantly presented by HLA-B*2705 molecules in tumor tissues as a result of VEGF mRNA overexpression, and draws attention to the existence of tumor antigens within non-coding regions of the mRNA. Translation of “untranslated” regions indicates that ribosomes are capable of initiating translation upstream or downstream of the normal ORF start codon (including introns from unspliced mRNA). These findings challenge the model of translation where ribosomes scan for and initiate only at the first AUG start codon of an ORF in a proper Kozak context [26].
In the conventional model for translation, ribosomes preloaded with initiator methionine-tRNA (Met-tRNA Meti ), the only tRNA thought to initiate translation, bind to the 5′-end of the mRNA and scan linearly in the 3′ direction for the start codon. Initiation of protein synthesis is a tightly regulated step requiring at least ten initiator factors that comprise more than 26 polypeptides [27] and is the target of many cellular pathways during normal and stress conditions [28]. How frequently non-coding regions of the mRNA are translated is presently unknown. The analysis is made especially difficult by our reliance on current gene annotations, including the specified 5′- and 3′-UTR sequences. Indeed, construction and sequencing of human 5′-UTRs show that many annotated genes are missing on average 45 bases of the 5′-UTR and nearly 30% contain upstream AUG starts (uAUG), many with a suitable Kozak context [29, 30]. Given that ribosomes enter the mRNA at the 5′ end, use of uAUGs or non-uAUGs could be an important source of cryptic epitopes. Accordingly, UTRs should be considered during epitope discovery, especially when short uORFs yield precursors that require minimal processing prior to entry into the antigen presentation pathway.
Translation of alternate reading frames
More than half the documented non-conventional MHC class I epitopes arise from ARF translation in a variety of human diseases such as influenza virus infections, cancer, and autoimmunity (Table 1). In addition to initiation at the primary ORF start codon, ribosomes can also translate the mRNA in the two other reading frames on both endogenous and viral messages. Furthermore, many ARF epitopes arise from precursors that are <50 amino acids [12, 31–33], which should require minimal degradation in the cytosol prior to entry into the ER and loading onto MHC class I molecules. A reduced requirement for proteolytic processing places many of these ARF epitopes at a kinetic advantage compared to the hundreds of thousands of peptides that are thought to compete for eventual loading onto MHC class I molecules either before or after arriving into the ER.
Several distinct translational mechanisms could account for the generation of ARF epitopes: (1) initiation codon read-through, (2) frame-shifting, and (3) re-initiation. Bullock and Eisenlohr [34] showed that, despite a primary ORF start codon in an ideal Kozak context, ribosomes often bypassed this codon and initiated translation further downstream to generate antigenic precursors. Since this seminal discovery, many examples have been described for both endogenous and viral mRNAs where the primary ORF start codon is bypassed for downstream initiation of cryptic epitopes (Table 1).
Frame-shifting is a phenomena whereby ribosomes may initiate at the primary ORF start codon but ‘slip’ either forward (+1 frame-shift) or backwards (−1 frame-shift) and continue translation in an alternate reading frame. This event produces an N-terminal primary ORF polypeptide fused to a polypeptide decoded from an alternate reading frame. Indeed, Saulquin et al. [35] showed that +1 frame-shifting in the IL-10 mRNA sequence generated a cryptic epitope derived partly from ORF1 and partly from ORF2. Likewise, another epitope within the thymidine kinase gene was generated only when ribosomal frame-shifting occurred and was effective in eliciting protective CD8+ T cells in vivo [36]. While frame-shifting has been best characterized in lower organisms [37], human mitochondrial (mt) ribosomes undergo frame-shifting at AGA and AGG codons due to lack of mt-tRNAs that recognize these codons [38]. These so-called ‘hungry’ codons signal the ribosome to frame-shift in the −1 direction which highlights the possibility that ribosomes on cellular messages may also be subject to frame-shifting during a reduced supply of tRNA due to either fluctuations in natural abundance or reduction in steady-state levels of aminoacylated-tRNAs.
Re-initiation is an additional mechanism that could give rise to ARF epitopes. This mechanism has been best characterized from translation of Saccharomyces cerevisiae transcriptional activator GCN4 whereby amino acid starvation-induced phosphorylation of the initiation factor eIF2 causes ribosomes to scan past inhibitory uORFs and re-initiate at the primary GCN4 ORF [39]. Initiation factor eIF2, recruits initiator Met-tRNA Meti to the small ribosomal subunit in a GTP-dependent manner, prior to entry at the 5′-end of mRNAs [40]. Different stresses activate cellular kinases (PKR, PERK, HRI, and GCN2) which phosphorylate Ser51 on the α subunit of eIF2 and limit its availability for initiation at AUG start codons. Interestingly, in mammalian cells, stress-induced phosphorylation of eIF2α causes ribosomes to bypass translation of uORFs and re-initiate at the transcriptional regulator activating transcription factor, ATF4-coding region [41].
Recently, Ingolia et al. [42] developed an elegant technique to globally monitor the position of ribosomes as thousands of mRNAs are translated in budding yeast under both rich and starvation conditions. Since initiation is a rate-limiting step of translation, this approach allows a direct determination of the translational reading frame utilized by the ribosomes anywhere along an mRNA sequence. Interestingly, during starvation conditions when eIF2α phosphorylation is enhanced, ribosome initiation at non-AUG start codons in uORFs dramatically increased. This suggests that translation of non-conventional regions of mRNAs in mammalian cells may also be upregulated during cellular stress. Interestingly, some viral infections activate PKR-mediated phosphorylation of eIF2α [43], which may encourage re-initiation not only at uORFs but other regions of the mRNA generating additional sources of non-conventional epitopes.
Non-AUG translation initiation
Initiation at non-AUG start codons on cellular and viral messages (CUG, ACG, etc.) is another mechanism to generate non-conventional antigenic peptides whether in the primary or alternate ORF (Table 1) [44]. In the scanning model of translation, the small ribosomal subunit containing initiator Met-tRNA Meti along with eIF4F and a whole host of initiation factors scans linearly until the first AUG in a good Kozak context (CC(A/G)CCAUGG) is encountered [26, 45] This mechanism predisposes initiation to commence only with methionine. Even non-AUG start codons, such as CUG, are believed to be decoded only with Met-tRNA Meti through ‘wobble’ interactions between the codon and anticodon of the tRNA. Interestingly, regulation of the innate immune response through type-I IFN has recently been shown to be linked to the phosphorylation state of eIF4E [46]. The precise connections between translational control and innate immunity are outside the scope of this review but highlight the emerging intersection between translational control and immune regulation [47].
Recently, we showed that the CUG start codon either in the primary ORF or within a 3′-untranslated region (3′-UTR) is a substrate for an unusual alternative translational mechanism using leucine as the initiating amino acid [48, 49]. To study this mechanism in vivo, Schwab et al. [49] generated mice with a transgene encoding a cryptic peptide initiated by CUG in the 3′-UTR of a conventional antigenic peptide ORF. T cell assays were used to detect the CUG/leucyl-initiated antigen and, despite its low abundance, the cryptic pMHC I was fully capable of inducing tolerance in transgenic mice as well as eliciting CD8+ T cell responses in wild-type mice. This suggests that even low levels of CUG/leucyl initiation, regardless of the reading frame, directs the generation of precursors which can efficiently compete for loading onto MHC I molecules. Further, the existence of a non-competing initiation event with leucine offers the cell a distinct advantage for antigen presentation when methionine and/or associated initiation factors are limiting. To what extent leucine initiation contributes to the pool of cryptic pMHC I awaits discovery of the set of proteins translated using the unconventional mechanism of CUG/leucine initiation, such as human trypsinogen [50].
The only other example of initiation without methionine is during IRES-mediated translation of the downstream capsid protein coding sequence of the cricket paralysis (CrPV) or the Plautia stali intestine viruses [51, 52]. Initiation at these non-AUG start codons using alanine (GCU) or glutamine (CAA) residues in mammalian cells suggests that highly conserved features of the ribosome are amenable to a range of non-canonical initiation events [53]. In contrast to translation on these viral messages, CUG/leucine initiation is not directed by a specific sequence or IRES element [49]. Furthermore, initiation at a CUG start codon, similar to AUG, was enhanced by the Kozak context [26]. This observation indicates that leucine insertion occurred during initiation as opposed to a post-translational modification. In contrast, CUG/leucine initiation was inhibited by insertion of upstream, out-of-frame CUG codons but not AUG start codons [54], suggesting that a distinct ribosome initiation mechanism specifically scans for and decodes CUG start codons with the alternative amino acid leucine.
The cellular response to viral infection is often manifested by shut-down of host-translation (reviewed in [55]). This antiviral translational response can induce phosphorylation of eIF2α and limit initiator Met-tRNA Meti recruitment to the ribosome [40], which ultimately favors translation of viral mRNAs. Interestingly, chemical mimicry of the antiviral response using sodium arsenite to phosphorylate eIF2α showed that while conventional translation at AUG was inhibited cryptic translation from the CUG start codon was resistant [54]. This suggests that during viral infection a switch from AUG/methionine to CUG/leucine would continue to supply a source of epitopes for immune surveillance by the CD8+ T cell repertoire.
Evidence that cryptic pMHC I is generated by a distinct initiation mechanism was recently investigated by analyzing ribosome initiation complexes at CUG start codons directly. Starck et al. used a technique called primer extension inhibition analysis or ‘toeprinting’ to monitor initiation complex assembly at CUG start codons in a cell-free extract. This approach showed that ribosome initiation complexes do assemble at cryptic CUG with up to 1/5th the efficiency of canonical initiation at AUG [56]. Further, these cryptic CUG initiation complexes were fully dependent on recognition of the mRNA m7GpppN cap structure by eIF4E and the Kozak context, which further indicates that the cryptic translation product arises from a novel initiation mechanism in contrast to post-translational alterations of the epitope.
Using a series of protein synthesis inhibitors, Starck et al. further defined the requirements for cryptic translation at CUG. For example, both methionine-sulfamide and edeine, which inhibit initiator Met-tRNA Meti -eIF2 activity [57, 58], compromised ribosome assembly at AUG while CUG complexes were resistant. These data extend the measurements from Schwab et al. [54] which show eIF2-independent translation of cryptic pMHC and indicate that the functional differences are intrinsic to the ribosome initiation complex. Independently, Yewdell and Nicchitta [59] have argued in favor of an “immunoribosome” dedicated to the efficient supply of polypeptides for presentation by MHC I.
The series of inhibitors which distinguish canonical initiation from cryptic CUG initiation all act within the P site of the ribosome, the site where initiator Met-tRNA Meti assembles for addition of the first amino acid [56]. This suggests that structural features of the ribosome initiation complex such as a unique initiator tRNA and/or ribosomal RNA directs CUG initiation. While the precise molecular mechanism awaits further characterization, we propose that CUG/leucine initiation increases the complexity of the proteome especially under certain cellular stress conditions that inhibit canonical initiation with Met-tRNA Meti . These cryptic initiation events would continue to supply an important source of peptides for antigen presentation during cellular stress when conventional translation mechanisms are subverted.
Conclusions and future perspectives
More than 20 years have elapsed since CD8+ T cells were discovered to recognize cryptic peptides presented by MHC class I molecules. Although initially considered a curiosity, we now know that cryptic peptides can arise from polypeptides translated from many endogenous and viral mRNAs. These peptides are immunologically significant and can play a protective role in viral infections. Like the large fraction of antigenic peptides presented by MHC class I molecules that are derived from DRiPs, cryptic peptides arise from a variety of mechanisms also tied to protein translation. Recent studies have provided tantalizing hints that ribosomes that carry out the synthesis of antigenic precursors for cryptic peptides are distinct from those responsible for conventional translation. We expect characterization of these novel ribosomes as well as the pathways that regulate their activity will reveal further insights into the as yet mysterious sources of antigenic peptides that make immune surveillance possible.
References
Shastri N, Cardinaud S, Schwab SR et al (2005) All the peptides that fit: the beginning, the middle, and the end of the MHC class I antigen-processing pathway. Immunol Rev 207:31–41
Townsend A, Bodmer H (1989) Antigen recognition by class I-restricted T lymphocytes. Annu Rev Immunol 7:601–624
Reits EAJ, Vos JC, Gromme M, Neefjes J (2000) The major substrates for TAP in vivo are derived from newly synthesized proteins. Nature 404(6779):774–778
Schubert U, Ott DE, Chertova EN et al (2000) Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc Natl Acad Sci USA 97(24):13057–13062
Yewdell JW (2007) Plumbing the sources of endogenous MHC class I peptide ligands. Curr Opin Immunol 19(1):79–86
Shastri N, Schwab S, Serwold T (2002) Producing nature’s gene-chips, the generation of peptides for display by MHC class I molecules. Annu Rev Immunol 20:463–493
Ho O, Green WR (2006) Alternative translational products and cryptic T cell epitopes: expecting the unexpected. J Immunol 177(12):8283–8289
Boon T, Van Pel A (1989) T cell-recognized antigenic peptides derived from the cellular genome are not protein degradation products but can be generated directly by transcription and translation of short subgenic regions. A hypothesis. Immunogenetics 29(2):75–79
Mayrand SM, Schwarz DA, Green WR (1998) An alternative translational reading frame encodes an immunodominant retroviral CTL determinant expressed by an immunodeficiency-causing retrovirus. J Immunol 160(1):39–50
Ho O, Green WR (2006) Cytolytic CD8+ T cells directed against a cryptic epitope derived from a retroviral alternative reading frame confer disease protection. J Immunol 176(4):2470–2475
Maness NJ, Valentine LE, May GE et al (2007) AIDS virus specific CD8+ T lymphocytes against an immunodominant cryptic epitope select for viral escape. J Exp Med 204(11):2505–2512
Cardinaud S, Moris A, Fevrier M et al (2004) Identification of cryptic MHC I-restricted epitopes encoded by HIV-1 alternative reading frames. J Exp Med 199(8):1053–1063
Maness NJ, Walsh AD, Piaskowski SM et al (2010) CD8+ T cell recognition of cryptic epitopes is a ubiquitous feature of AIDS virus infection. J Virol 84(21):11569–11574
Garrison KE, Champiat S, York VA et al (2009) Transcriptional errors in human immunodeficiency virus type 1 generate targets for T-cell responses. Clin Vaccine Immunol 16(9):1369–1371
Berger CT, Carlson JM, Brumme CJ et al (2010) Viral adaptation to immune selection pressure by HLA class I-restricted CTL responses targeting epitopes in HIV frameshift sequences. J Exp Med 207(1):61–75
Bansal A, Carlson J, Yan J et al (2010) CD8 T cell response and evolutionary pressure to HIV-1 cryptic epitopes derived from antisense transcription. J Exp Med 207(1):51–59
Maness NJ, Wilson NA, Reed JS et al (2010) Robust, vaccine-induced CD8(+) T lymphocyte response against an out-of-frame epitope. J Immunol 184(1):67–72
Li C, Goudy K, Hirsch M et al (2009) Cellular immune response to cryptic epitopes during therapeutic gene transfer. Proc Natl Acad Sci USA 106(26):10770–10774
Yewdell JW, Anton LC, Bennink JR (1996) Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? J Immunol 157(5):1823–1826
Levitskaya J, Coram M, Levitsky V et al (1995) Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature 375(6533):685–688
Levitskaya J, Sharipo A, Leonchiks A et al (1997) Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc Natl Acad Sci USA 94(23):12616–12621
Davenport MG, Pagano JS (1999) Expression of EBNA-1 mRNA is regulated by cell cycle during Epstein-Barr virus type I latency. J Virol 73(4):3154–3161
Kwun HJ, da Silva SR, Shah IM et al (2007) Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mimics Epstein-Barr virus EBNA1 immune evasion through central repeat domain effects on protein processing. J Virol 81(15):8225–8235
Cardinaud S, Starck SR, Chandra P, Shastri N (2010) The synthesis of truncated polypeptides for immune surveillance and viral evasion. PLoS One 5(1):e8692
Weinzierl AO, Maurer D, Altenberend F et al (2008) A cryptic vascular endothelial growth factor T-cell epitope: identification and characterization by mass spectrometry and T-cell assays. Cancer Res 68(7):2447–2454
Kozak M (1987) At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J Mol Biol 196(4):947–950
Pestova TV, Hellen CU (2000) The structure and function of initiation factors in eukaryotic protein synthesis. Cell Mol Life Sci 57(4):651–674
Holcik M, Sonenberg N (2005) Translational control in stress and apoptosis. Nat Rev Mol Cell Biol 6(4):318–327
Suzuki Y, Ishihara D, Sasaki M et al (2000) Statistical analysis of the 5′ untranslated region of human mRNA using “oligo-capped” cDNA libraries. Genomics 64(3):286–297
Rogozin IB, Kochetov AV, Kondrashov FA et al (2001) Presence of ATG triplets in 5′ untranslated regions of eukaryotic cDNAs correlates with a ‘weak’ context of the start codon. Bioinformatics 17(10):890–900
Rosenberg SA, Tong-On P, Li Y et al (2002) Identification of BING-4 cancer antigen translated from an alternative open reading frame of a gene in the extended MHC class II region using lymphocytes from a patient with a durable complete regression following immunotherapy. J Immunol 168(5):2402–2407
Graddis TJ, Diegel ML, McMahan CJ et al (2004) Tumor immunotherapy with alternative reading frame peptide antigens. Immunobiology 209(7):535–544
Tykodi SS, Fujii N, Vigneron N et al (2008) C19orf48 encodes a minor histocompatibility antigen recognized by CD8+ cytotoxic T cells from renal cell carcinoma patients. Clin Cancer Res 14(16):5260–5269
Bullock TNJ, Eisenlohr LC (1996) Ribosomal scanning past the primary initiation codon as a mechanism for expression of CTL epitopes encoded in alternative reading frames. J Exp Med 184:1319–1329
Saulquin X, Scotet E, Trautmann L et al (2002) +1 Frameshifting as a novel mechanism to generate a cryptic cytotoxic T lymphocyte epitope derived from human interleukin 10. J Exp Med 195(3):353–358
Zook MB, Howard MT, Sinnathamby G et al (2006) Epitopes derived by incidental translational frameshifting give rise to a protective CTL response. J Immunol 176(11):6928–6934
Atkins JF, Bjork GR (2009) A gripping tale of ribosomal frameshifting: extragenic suppressors of frameshift mutations spotlight P-site realignment. Microbiol Mol Biol Rev 73(1):178–210
Temperley R, Richter R, Dennerlein S et al (2010) Hungry codons promote frameshifting in human mitochondrial ribosomes. Science 327(5963):301
Hinnebusch AG, Dever TE, Asano K (2007) Mechanism of translation initiation in the yeast Saccharomyces cerevisiae translational control in biology and medicine. In: Mathews M, Sonenberg N, Hershey JWB (eds) Translational control in biology and medicine. Cold Spring Harbor Laboratory, New York, pp 225–268
Hershey JWB, Merrick WC (2000) The pathway and mechanism of initiation of protein synthesis. In: Mathews M, Sonenberg N, Hershey JWB (eds) Translational control of gene expression. Cold Spring Harbor Laboratory, New York, pp 33–88
Vattem KM, Wek RC (2004) Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci USA 101(31):11269–11274
Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS (2009) Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324(5924):218–223
Garcia MA, Gil J, Ventoso I et al (2006) Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev 70(4):1032–1060
Malarkannan S, Horng T, Shih PP et al (1999) Presentation of out-of-frame peptide/MHC class I complexes by a novel translation initiation mechanism. Immunity 10(6):681–690
Pestova TV, Borukhov SI, Hellen CUT (1998) Eukaryotic ribosomes require initiation factors 1 and 1A to locate initiation codons. Nature 394(6696):854–859
Colina R, Costa-Mattioli M, Dowling RJ et al (2008) Translational control of the innate immune response through IRF-7. Nature 452(7185):323–328
Pierre P (2009) Immunity and the regulation of protein synthesis: surprising connections. Curr Opin Immunol 21(1):70–77
Malarkannan S, Horng T, Shih P et al (1999) Presentation of out-of-frame peptide/MHC class I complexes by a novel translation initiation mechanism. Immunity 10:681–690
Schwab S, Li KC, Kang C, Shastri N (2003) MHC I molecules consititutively display cryptic translation products. Science 301:1367–1371
Nemeth AL, Medveczky P, Toth J et al (2007) Unconventional translation initiation of human trypsinogen 4 at a CUG codon with an N-terminal leucine. A possible means to regulate gene expression. Febs J 274(6):1610–1620
Wilson JE, Pestova TV, Hellen CUT, Sarnow P (2000) Initiation of protein synthesis from the A site of the ribosome. Cell 102(4):511–520
Sasaki J, Nakashima N (2000) Methionine-independent initiation of translation in the capsid protein of an insect RNA virus. Proc Natl Acad Sci USA 97(4):1512–1515
Pisarev AV, Shirokikh NE, Hellen CU (2005) Translation initiation by factor-independent binding of eukaryotic ribosomes to internal ribosomal entry sites. CR Biol 328(7):589–605
Schwab SR, Shugart JA, Horng T et al (2004) Unanticipated antigens: translation initiation at CUG with leucine. PLoS Biol 2(11):e366
Aranda M, Maule A (1998) Virus-induced host gene shutoff in animals and plants. Virology 243(2):261–267
Starck SR, Ow Y, Jiang V et al (2008) A distinct translation initiation mechanism generates cryptic peptides for immune surveillance. PLoS One 3(10):e3460
Lee J, Kang SU, Kang MK et al (1999) Methionyl adenylate analogues as inhibitors of methionyl-tRNA synthetase. Bioorg Med Chem Lett 9(10):1365–1370
Kozak M, Shatkin AJ (1978) Migration of 40 S ribosomal subunits on messenger RNA in the presence of edeine. J Biol Chem 253(18):6568–6577
Yewdell JW, Nicchitta CV (2006) The DRiP hypothesis decennial: support, controversy, refinement and extension. Trends Immunol 27(8):368–373
Uenaka A, Ono T, Akisawa T et al (1994) Identification of a unique antigen peptide pRL1 on BALB-c RL1 leukemia recognized by cytotoxic T lymphocytes and its relation to the Akt oncogene. J Exp Med 180(5):1599–1607
Coulie PG, Lehmann F, Lethe B et al (1995) A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc Natl Acad Sci USA 92:7976–7980
Guilloux Y, Lucas S, Brichard VG et al (1996) A peptide recognized by human cytolytic T lymphocytes on HLA-A2 melanomas is encoded by an intron sequence of the N-acetylglucosaminyltransferase V gene. J Exp Med 183(3):1173–1183
Robbins PF, El-Gamil M, Li YF et al (1997) The intronic region of an incompletely spliced gp100 gene transcript encodes an epitope recognized by melanoma-reactive tumor-infiltrating lymphocytes. J Immunol 159(1):303–308
Lupetti R, Pisarra P, Verrecchia A et al (1998) Translation of a retained intron in tyrosinase-related protein (TRP) 2 mRNA generates a new cytotoxic T lymphocyte (CTL)-defined and shared human melanoma antigen not expressed in normal cells of the melanocytic lineage. J Exp Med 188(6):1005–1016
Wang R-F, Parkhurst MR, Kawakami Y et al (1996) Utilization of an alternative open reading frame of a normal gene in generating a novel human cancer antigen. J Exp Med 183:1131–1140
Shichijo S, Nakao M, Imai Y et al (1998) A gene encoding antigenic peptides of human squamous cell carcinoma recognized by cytotoxic T lymphocytes. J Exp Med 187(3):277–288
Wang R-F, Johnston SL, Zeng G et al (1998) A breast and melanoma-shared tumor antigen: T cell responses to antigenic peptides translated from different open reading frames. J Immunol 161:3596–3606
Aarnoudse CA, van den Doel PB, Heemskerk B, Schrier PI (1999) Interleukin-2-induced, melanoma-specific T cells recognize CAMEL, an unexpected translation product of LAGE-1. Int J Cancer 82(3):442–448
Ronsin C, Chung-Scott V, Poullion I et al (1999) A non-AUG-defined alternative open reading frame of the intestinal carboxyl esterase mRNA generates an epitope recognized by renal cell carcinoma-reactive tumor-infiltrating lymphocytes in situ. J Immunol 163(1):483–490
Probst-Kepper M, Stroobant V, Kridel R et al (2001) An alternative open reading frame of the human macrophage colony-stimulating factor gene is independently translated and codes for an antigenic peptide of 14 amino acids recognized by tumor-infiltrating CD8 T lymphocytes. J Exp Med 193(10):1189–1198
Casnici C, Volpe G, Lattuada D et al (2009) Out of frame peptides from BCR/ABL alternative splicing are immunogenic in HLA A2.1 transgenic mice. Cancer Lett 276(1):61–67
Bullock TNJ, Patterson AE, Franlin LL et al (1997) Initiation codon scanthrough versus termination codon readthrough demonstrates strong potential for major histocompatibility complex class I-restricted cryptic epitope expression. J Exp Med 186(7):1051–1058
Schirmbeck R, Riedl P, Fissolo N et al (2005) Translation from cryptic reading frames of DNA vaccines generates an extended repertoire of immunogenic, MHC class I-restricted epitopes. J Immunol 174(8):4647–4656
Maness NJ, Walsh AD, Piaskowski SM et al (2010) CD8+ T cell recognition of cryptic epitopes is a ubiquitous feature of AIDS virus infection. J Virol 84(21):11569–11574
Dolstra H, Fredrix H, Maas F et al (1999) A human minor histocompatibility antigen specific for B cell acute lymphoblastic leukemia. J Exp Med 189:301–308
Acknowledgements
S.R.S. was supported by the Cancer Research Institute and the NRSA fellowship from the National Institutes of Health. We thank Sylvain Cardinaud for sharing findings before publication. Research in this laboratory is supported by the National Institutes of Health and the International AIDS Vaccine Initiative (IAVI).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Starck, S.R., Shastri, N. Non-conventional sources of peptides presented by MHC class I. Cell. Mol. Life Sci. 68, 1471–1479 (2011). https://doi.org/10.1007/s00018-011-0655-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-011-0655-0