
Abstract
Oxidative DNA damage to cells activates poly(ADP-ribose)polymerase-1 (PARP-1) and the poly(ADP-ribose) formed is rapidly degraded to ADP-ribose by poly(ADP-ribose)glycohydrolase (PARG). Here we show that PARP-1 and PARG control extracellular Ca2+ fluxes through melastatin-like transient receptor potential 2 channels (TRPM2) in a cell death signaling pathway. TRPM2 activation accounts for essentially the entire Ca2+ influx into the cytosol, activating caspases and causing the translocation of apoptosis inducing factor (AIF) from the inner mitochondrial membrane to the nucleus followed by cell death. Abrogation of PARP-1 or PARG function disrupts these signals and reduces cell death. ADP-ribose-loading of cells induces Ca2+ fluxes in the absence of oxidative damage, suggesting that ADP-ribose is the key metabolite of the PARP-1/PARG system regulating TRPM2. We conclude that PARP-1/PARG control a cell death signal pathway that operates between five different cell compartments and communicates via three types of chemical messengers: a nucleotide, a cation, and proteins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Oxidative stress may induce DNA damage signaling in cells. Within seconds, poly(ADP-ribose)polymerase-1 (PARP-1) reacts to DNA nicks arising from oxidative damage, activating PARP-1. It signals downstream into DNA repair and cell death pathways [1–3]. PARP-1 produces large amounts of long and branched poly(ADP-ribose) molecules primarily on itself in an automodification reaction [1, 3]. The local polymer concentration increases several 100-fold, but decreases rapidly due to degradation by poly(ADP-ribose)glycohydrolase (PARG). PARG produces primarily ADP-ribose, albeit a few polymers may arise from endoglycosidic enzyme activity [4, 5].
The biological role of this dynamic poly(ADP-ribose) turnover is poorly understood. In several cell types, PARP-1 activation induces the release of apoptosis-inducing factor (AIF) from the inner mitochondrial membrane and its translocation to the nucleus, where it triggers chromatin condensation and large-scale DNA fragmentation [6–11]. AIF release involves proteolytic cleavage, either by calpain or by caspases pending on the type of cytotoxic stress and the cell type [8, 12–14]. By contrast, AIF translocation apparently may proceed also in the absence of proteolytic cleavage [15].
In vitro and in vivo evidence combined from different cell types indicates that poly(ADP-ribose) metabolism is involved in Ca2+ gating. Patch clamp experiments in lymphocytes showed that ADP-ribose addition to the inner chamber may activate a calcium channel at the outer cell membrane, termed melastatin-like transient receptor potential 2 channel (TRPM2) [16]. In microglia cells, ADP-ribose added to the intracellular pipette of a whole-cell-patch clamp assay causes TRPM2-mediated Ca2+ currents [17]. Oxidant-induced TRPM2 activation is inhibited by genetic or chemical disruption of PARP-1 activity in lymphocytes [16] and in a kidney cell line overexpressing TRPM2 [18].
In the present study, we used genetic knockout, RNAi, and chemical inhibitors to manipulate PARP-1, PARG, and TRPM2 to pinpoint the role of poly(ADP-ribose) metabolism in triggering calcium fluxes, caspase activation, AIF translocation, and cell death in a well-defined cell system after H2O2. We found that TRPM2 is the major channel for generating the Ca2+ influx after oxidative stress. Furthermore, abrogation of PARP-1 or PARG function affected the signal for TRPM2 activation, Ca2+ fluxes, caspase activation, AIF translocation, and cell death. The results suggest that PARG acts as the immediate upstream regulator of TRPM2 by generating ADP-ribose. Loading cells with ADP-ribose triggered a Ca2+ influx in the absence of oxidative damage. Thus, PARP-1 and PARG are an integral part of a cell death pathway that operates between five different cell compartments and communicates via three types of chemical messengers: a nucleotide, a cation, and proteins.
Materials and methods
Cell culture
Immortalized mouse embryonic fibroblasts (MEFs) of wild-type and parp-1 −/− background were used. MEFs were cultured at 37°C in a water-saturated, 5% CO2/95% air atmosphere, in complete Dulbecco’s modified Eagle’s medium (DMEM) containing 1 g/l glucose and supplemented with 10% (v/v) FBS and antibiotics (Invitrogen).
Calcium measurements and visualization
Twenty thousand cells per well in 96-well plates (Costar Corning Incorporated) were washed twice with 49 parts of calcium-free HBSS (0.49 mM MgCl2, 0.41 mM MgSO4, 5.33 mM KCl, 0.44 mM KH2PO4, 4.17 mM NaHCO3, 137 mM NaCl, 0.34 mM Na2HPO4, 5.56 mM dextrose) supplemented with 1 part 1 M HEPES (pH 7.2) (assay buffer) containing CaCl2 or not. Then, 100 μl Fluo-4-NW-dye-mix from Molecular Probes (Invitrogen) was added and incubated for 30 min at 37°C, followed by 30-min incubation in the dark at room temperature. Basic relative fluorescence units (RFU) before challenging the cells are shown in Supplementary Figures S4A, S4B. Changes in fluorescence from the Fluo-4-NW-dye quantify changes in intracellular calcium concentrations (excitation/emission 485/535 nm; slits 10/15 nm) in LS55 luminescence spectrometer (Perkin-Elmer) after H2O2. Calcium was monitored for 30 min with a measure frequency of 0.1 s. Each 50th time-point was taken for calculation. Every Fluo-4 analysis was monitored in parallel by at least one positive or negative control at each day of measurement. The statistical analysis is summarized in Supplementary Figure S4E.
For Ca2+ visualization, Fluo-4 loaded cells were analyzed 3 h after H2O2 treatment in a fluorescence microscope (Nikon). This time point was chosen according the offscale time in fluospectroscopic Fluo-4 analysis.
Loading of cells with ADP-ribose
For the Fluo-4 assays with pure ADP-ribose as a trigger for intracytosolic Ca2+ shifts, MEFs were loaded with monomeric ADP-ribose using the transfection reagent siPORT amine (Ambion). A total of 50 μl of 10 mM ADP-ribose in Fluo-4 assay buffer was incubated with 3 μl siPORT amine for 20 min at 37°C. Afterwards, this mixture was given on Fluo-4-loaded cells and preincubated together with cells for 25 min in a final volume of 200 μl (ADP-ribose at 2.5 mM) before intracytosolic Ca2+ shifts were monitored for 30 min as described above. Control experiments were performed with ADP-ribose solution without transfectant and Fluo-4 assay buffer with siPORTamine.
Chemicals
H2O2, N-(N-((Phenylmethoxy)carbonyl)-l-valyl)-phenylalaninal (MDL 28170) and ADP-ribose from SIGMA. 3-aminobenzamide (3-AB), 2-Methyl-3,5,7,8-tetrahydrothiopyranol-(4,3-d)-pyrimidine-4-one (DR2313), N-(6-Oxo-5,6-dihydro-phenanthridin-2-yl)-N,N-dimethylacetamide.HCl (PJ-34) from Alexis Biochemicals. 1,2-bis-(o-Aminophenoxy)-ethane-N,N,N′,N′-tetraaceticacid-tetraacetoxymethylester (BAPTA-AM) from Enzo Life Sciences. N-(p-Amylcinnamoyl)-anthranilicacid (ACA), N-(2-Quinolyl)valyl-aspartyl-(2,6-difluorophenoxy)methylketone (Q-VD-OPh) from Calbiochem. 2-(4-Morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) was obtained from Biaffin.
Inhibitors were dissolved in DMSO. All other chemicals were from Applichem, Fluka, Merck, or SIGMA.
Viability assay
Cells were seeded in 96-well plates (15,000 cells/well). H2O2 was diluted in OptiMEM and cells were treated with 50 μl/well for 1 h before 150 μl DMEM was added. After 20 h, medium was replaced with 200 μl DMEM 10% (v/v) Alamar blue (Serotec). After 4 h, fluorescence was monitored at 530-nm excitation and 590-nm emission wavelength (LS55 luminescence spectrometer, Perkin-Elmer).
Western-blot analyses and PAR immunofluorescence
The following primary antibodies were used: anti-PARP-1 (Alexis; C2-10, 1:5,000), anti-AIF (Serotec; 1:500), anti-GAPDH (Ambion; 1:1,00,000), anti-MnSOD (BD Biosciences; 1:1,000), anti-Topo-1 (BD Biosciences; 1:5,000), anti-α-tubulin (Sigma; 1:50,000), anti-PAR (Alexis; 1:1,000), anti-α-fodrin (ENZO Life Sciences; 1:8,000); anti-cleaved PARP (Asp214, 1:1,000), anti-caspase 12 (1:1,000), anti-cleaved caspase 9 (Asp353, 1:1,000), anti-cleaved caspase 6 (Asp162, 1:1,000), anti-cleaved caspase 3 (Asp175, 1:1,000), anti-caspase 9 (1:1,000), anti-caspase 6 (1:1,000), anti-caspase 3 (1:1,000), anti-phospho-PERK (Thr 980, 1:1,000, all from Cell Signaling). Anti-p-CaMKIIα (Thr 286-R, 1:1,000) and anti-CaMKII (M-176, 1:1,000) were obtained from Santa Cruz. Anti-calmodulin (1:1,000) was from Epitomics. All secondary antibodies were from Sigma. PAR detection after H2O2 exposure was performed by immunofluorescence microscopy [7].
Cellular subfractionation
This was performed as described before [7]. Briefly, MEFs were harvested and washed twice with Tris–HCl (pH 7.0), 0.13 M NaCl, 5 mM KCl, 7.5 mM MgCl2. After incubation in 3.5 mM Tris–HCl (pH 7.8), 3 mM NaCl, 0.5 mM MgCl2 for 5 min, cells were mechanically broken. The homogenate was mixed with one-ninth of the cell-volume of 0.35 M Tris–HCl (pH 7.8), 0.2 M NaCl, 50 mM MgCl2, and spun for 3 min at 1,600 × g. The pellet (nuclei) was harvested directly in Laemmli buffer and the supernatant was recentrifuged under the same conditions. The final supernatant was spun at 13,000 × g for 1 min and the resulting mitochondrial pellet was washed once with 35 mM Tris–HCl (pH 7.8), 20 mM NaCl, 5 mM MgCl2, then twice with 10 mM Tris–HCl (pH 7.4), 1 mM EDTA, 0.32 M sucrose and finally resuspended in Laemmli buffer. The procedure was performed at 4°C and a protease-inhibitor-cocktail (CompleteTM, EDTA-free, Roche) was added to all Tris-buffers.
PCR analysis
cDNA was produced from cells using the cells-to-cDNA II kit (Ambion) and used as templates for GAPDH and TRPM2 amplification in quantitative real-time PCR, with primers from Assays-on-demand Gene Expression products, in a reaction mixture including TaqMan Universal PCR Master Mix (Applied Biosystems). Real-time PCR experiments were carried out on an ABI PRISM 7700 cycler (95°C, 15 s; 60°C, 1 min; 40 cycles). For quantification, ΔΔCt method was used. The TRPM2 primers were 5′-TACACTGACAGGCCACGGCT-3′ and 5′-CCAGCATGCACAGGATGATC-3′ (Microsynth). PCR analysis was performed in a reaction mixture including TaqMan Mix (Sigma) on Touchgene machine (TECHNE; 95°C, 5 min initial denaturation; 30 cycles of denaturation (95°C, 30 s), annealing (54°C, 30 s) and elongation (72°C, 1 min); 5 min of final extension at 70°C).
RNA interference
Poly(ADP-ribose)polymerase-1 targeting template sequences (siRNA-P1): 5′-AAACAGTAATAAGCGTCGCTCCCTTC-3′ (sense) and 5′-AAGAGCGACGCTTATTACTGTCCTGTCTC-3′ (antisense) [7]. The PARG targeting siRNA (siRNA-G) has been described earlier (Microsynth) [7, 19]. The siRNA against TRPM2 (siRNA-T2) was obtained from Applied Biosystems; 5′-GAGUCUACGUGGUCGAGUATT-3′ (sense) and 5′-UACUCGACCACGUAGACUCCA-3′ (antisense). All transfection experiments were performed with siPORT amine (Ambion) as transfection reagent under conditions described earlier [19].
NAD+ determination
Measurement of NAD+ was according to Jacobson and Jacobson [20]. Cells were in 96-well plates (25,000 cells/well). After treatment with 5 mM H2O2 in OptiMEM (50 μl/well), the cells were lysed with 50 μl ethanol supplemented with isonicotinic acid-hydrazide (20 mM, for 30 min at −80°C) at 10, 20, and 30 min. The assay volume of 300 μl/well contained 570 mM ethanol, 114 mM bicine pH 7.8, 4.8 mM EDTA and 1 mg/ml BSA. The color change of thiazolyl-blue-tetrazolium-bromide (MTT, 0.48 mM, SIGMA) was measured 15 min after ADH was added (6 μg/ml, Roche) in a UV spectrometer (Bio-Rad) at 570 nm. A calibration curve was performed in parallel.
Statistical analysis
Results are shown as mean ± SD of at least three independent experiments. The significance of differences was estimated by ANOVA followed by the method of Kolmogorov and Smirnov. p < 0.05 was considered significant. All statistical analyses were performed with InStat3 software (GraphPad Software).
Results
Oxidative stress induces PAR formation, Ca2+ fluxes, and cell death
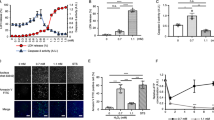
Cell death induced by H2O2 is preceded by PAR formation and Ca2+ fluxes in mouse embryonic fibroblasts (MEFs) (Fig. 1). Cell death was dose-dependent (Fig. 1a), reducing viable cells by 29.8 ± 7.4% at 0.5 mM and 68.5 ± 4.1% at 5 mM H2O2 within 20 h. PAR induction (Fig. 1b, c) and Ca2+ gating into the cytosol (Fig. 1d–f) were observed within min of oxidative insults. All cytosolic Ca2+ could be completely quenched by a pretreatment with the Ca2+ chelator BAPTA (Suppl. Fig. S1A) as tested with the Fluo-4 assay. Figure 1d shows the effects of 0.5 and 5 mM H2O2 on intracellular Ca2+ levels over 30 min. Figure 1e shows the impact of 5 mM H2O2, performed in presence or absence of 1 mM exogenous Ca2+. Moreover, 5 mM exogenous Ca2+ could not further promote the level of Ca2+ signals obtained with 1 mM Ca2+ (Suppl. Fig. S1D). Cytosolic Ca2+ also increased in the absence of exogenous Ca2+, suggesting that Ca2+ was released from endogenous Ca2+ stores. Ca2+-free conditions did not affect cell viability or baseline Ca2+ levels (Suppl. Fig. S1B, S1C). Figure 1f shows a comparison of Ca2+ from internal and external sources. Low levels of H2O2 (0.5 mM) cause exclusively Ca2+ influxes from the extracellular milieu, rising continuously up to 30 min. High levels of H2O2 (5 mM) lead to Ca2+ shifts from extra- and intracellular sources, producing a peak at 150 s, a trough at 650 s, followed by a continuous rise of Ca2+ (Fig. 1d–f). Compared to other cell organelles, the basal Ca2+ concentration of endoplasmic reticulum is 5,000-fold higher than cytosolic Ca2+, and hence is the most likely source of intracellular Ca2+ releases [21]. However, the Ca2+ peak at 150 s could be addressed to PI3K (phosphoinositide 3-kinase) activity as it could be reduced by the parallel incubation with its inhibitor LY294002 (50 μM, data not shown).

Effects of H2O2 on poly(ADP-ribose), Ca2+ levels, and cell survival. a Wild-type MEFs were treated with 0.5 or 5 mM of H2O2 for 20 h. Cell survival was monitored by Alamar blue assay. Results represent the mean ± SD for at least three independent experiments (# p < 0.0025; *p < 0.0001; t test). b Western-blot analysis of PAR after 15 min of H2O2 treatment. GAPDH is loading control. c PAR detection by immunofluorescence after 30 min of H2O2 treatment. HOECHST staining is also shown. d Fluo-4 assay of H2O2-challenged cells. Changes of cytosolic Ca2+ were monitored directly after treatment for 30 min by fluorescence spectrometry and are expressed as ΔRFU compared to basic levels. Results represent mean ± SD (# p < 0.005; *p < 0.0001; n = 4; t test). e Ca2+ determination after 5 mM H2O2 with and without extracellular Ca2+ ions. Shown are ΔRFUs and error bars (# p < 0.025; *p < 0.001; n = 4; t test). f Comparison of Ca2+ from intra- and extracellular sources after 0.5 or 5 mM of H2O2. Shown are ΔRFUs with or without Ca2+ in the buffer at characteristic time points (150, 650, and 1,700 s; n = 4)
PARP-1 inhibition by chemicals, RNAi and knockout reduce H2O2 cytotoxicity and cytosolic Ca2+ shifts
Using chemical inhibition, RNAi silencing or genetic knockout of parp-1, we determined the consequences on cytosolic Ca2+ shifts and cell survival (Fig. 2). Regardless of the type of PARP-1 abrogation, H2O2-induced cytotoxicity (Fig. 2a) and cytosolic Ca2+ increases (Fig. 2b–g) were reduced. In particular, PARP inhibitor 3-aminobenzamide (3-AB) decreased cytosolic Ca2+ shifts up to 30 min (Fig. 2b), and this was observed over a dose range of 1–10 mM (Suppl. Fig. S2C). Inhibition of PARP reduced Ca2+ levels after H2O2 (Fig. 2c). 3-AB was not toxic per se nor did it affect the expression of PARP-1 (Suppl. Fig. S2A, S2B). Moreover, the presence or absence of Ca2+ did not alter the baseline levels (Suppl. Figs. S2D, S2E, S4A). These results were confirmed with other PARP inhibitors: cells grown in the presence of 5 μM PJ-34 (Fig. 2d; Suppl. Fig. S4A) or 100 μM DR2313 (Fig. 2e; Suppl. Fig. S4A) showed similar Ca2+ shifts after H2O2 as those grown in 3 mM 3-AB (Fig. 2b).

Inhibition of PARP-1 protects cells against cytotoxicity and reduces Ca2+ levels after H2O2. a Wild-type (wt P1), parp-1 −/− MEFs (P1−/−) and cells incubated with 3 mM 3-AB overnight (3-AB) or silenced for the parp-1 gene (siRNA-P1) were challenged with 5 mM H2O2. After 20 h, cytotoxicity was determined by Alamar blue assay (mean ± SD; # p < 0.025; *p < 0.0025; n ≥ 3; t test). b Ca2+ assays of wild-type (wt P1) and 3-AB (3 mM) treated cells. MEFs were monitored for Ca2+ alterations after 5 mM H2O2 (mean ± SD; n ≥ 4). c Differences in cytosolic Ca2+ levels after 5 mM H2O2 in wild-type (wt P1), 3-AB (3 mM) treated cells and MEFs silenced with siRNA against parp-1 (siRNA-P1) as well as cells transfected with a control siRNA (siRNA-C) after 1,700 s (mean ± SD; *p < 0.025; n ≥ 3; t test). d Ca2+ assays of wild-type cells (wt P1) and cells with 5 μM of PARP-inhibitor PJ-34 (wt P1 + PJ-34) after 5 mM H2O2 (mean ± SD; n ≥ 4). Values of controls (wt P1 + PJ-34 untreated) are shown too (mean ± SD; n = 2). e Effect of PARP-inhibitor DR2313 (100 μM) on Ca2+ shifts after 5 mM H2O2 (mean ± SD; n ≥ 3). Ca2+ values of controls (wt P1 + DR2313 untreated) are presented (n = 1). f MEFs were silenced for the parp-1 gene (siRNA-P1) and subjected to Fluo-4 assay. Cells treated with a control RNA sequence (siRNA-C) were analyzed in parallel. Shown are the mean ± SD (n = 4). Silencing efficacy was determined by Western blot with GAPDH as loading control. g Comparison of Ca2+ levels from intra- and extracellular sources after 5 mM H2O2 in wild-type (wt P1) and parp-1 −/− (P1−/−). Shown are ΔRFUs (mean ± SD; n ≥ 4) of Ca2+ assays with or without Ca2+ in the buffer as indicated at 1,700 s after treatment. Western-blot analysis for PARP-1 protein is shown too with α-tubulin as loading control
As expected, RNAi silencing of parp-1 reduced PARP-1 protein to very low levels (Fig. 2f, Western blot) and caused a further reduction of Ca2+ signals from extracellular space after an oxidative insult (Fig. 2c, f). Using a third approach, we determined Ca2+ shifts in parp-1 −/− cells (Fig. 2g, 3a). The parp-1 gene knockout was verified by Western blot (Fig. 2g) and the inability to convert NAD+ into PAR after H2O2 (Suppl. Fig. S2G). The reduction of the basal NAD+ content in parp-1 −/− cells challenged with 5 mM H2O2 is likely due to PARP-2 activity [33–35]. Evidently, MEFs lacking PARP-1 are resistant to H2O2-induced cytotoxicity (Fig. 2a) and did not show any Ca2+ influx from the extracellular space (Fig. 2g; Suppl. Figs. S2F, S2H, S2I, S4A).

Effects of H2O2 on cytosolic Ca2+ and Ca2+ signaling. a Microscopic visualization of intracellular Ca2+ shifts after 5 mM H2O2 (3 h) using the Ca2+ indicator Fluo-4 in wild-type and parp-1 −/− cells. b Western-blot analysis of phosphorylated PERK protein with α-tubulin as loading control. Wild-type (wt P1) and parp-1 knockout MEFs (P1−/−) were treated for the indicated timepoints with 5 mM H2O2. c Western-blot analysis of phosphorylated CaMKII in H2O2 (5 mM) treated wild-type (wt P1) and parp-1 −/− MEFs (P1−/−) at the indicated timepoints. CaMKII is shown as a loading control. d Western-blot analysis of α-fodrin in wild-type (wt P1) and parp-1 knockout (P1−/−) cells. Cells were treated with 5 mM H2O2 for the indicated time points, harvested and subjected to Western-blot analysis. GAPDH is loading control. e Activation of caspases and appearance of cleaved PARP-1 after 5 mM H2O2 is shown in Western-blot analysis of whole cell lysates prepared from wild-type (wt P1) and parp-1 knockout (P1−/−) cells at the indicated timepoints. GAPDH and α-tubulin are shown as loading controls. f Translocation of AIF after H2O2 insult was determined in wild-type cells silenced for the parp-1 gene by RNAi (siRNA 1) and cells silenced with a scrambled control siRNA (siRNA C). After 6 h treatment with 5 mM H2O2, cells were subjected to subcellular fractionation and immunoblotting. Topo-1 and MnSOD protein are also shown
To further address the role of PARP-1 in Ca2+ signaling, Ca2+ levels were visualized in parp-1 −/− versus wild-type cells by fluorescence microscopy after 5 mM H2O2 (Fig. 3a). While detectable Ca2+ levels were seen in wild-type cells, parp-1 −/− failed to show a signal. The amount of cytosolic Ca2+ after H2O2 is of biological relevance because the Ca2+/calmodulin dependent protein kinase II (CaMKII) becomes phosphorylated only in wild-type cells (Fig. 3c). The expression level of calmodulin is similar in wild-type and parp-1 −/− cells (Suppl. Fig. S3A). Interestingly, the marginal PARP-1-independent Ca2+ released from intracellular sources displays endoplasmic reticulum stress as assayed by the phosphorylation of protein kinase-like endoplasmic reticulum kinase [22] (PERK, Fig. 3b). Our findings suggest that PARP-1 activity is required for the influx of Ca2+ from the extracellular space leading to Ca2+ signaling after an oxidative insult.
PARP-1-dependent Ca2+ gating initiates downstream factors in cell death
We next determined the PARP-1 dependency of extracellular Ca2+ influxes on the activation of calpain and caspases, which is a controversial issue in AIF release [8, 12–15, 23–25]. A total of 5 mM H2O2 induced activation of calpain as determined by α-fodrin cleavage [14]. However, this activation was independent of PARP-1 (Fig. 3d). By contrast, the activation of caspases 3, 6, and 9 was clearly reduced in parp-1 −/− cells (Fig. 3e), some of them being implicated in AIF release from the inner mitochondrial membrane [7, 12, 14]. Caspase 12 activation was identical in PARP-1-proficient and -deficient cells (Fig. 3e), which is in line with results of Fig. 3d and the literature demonstrating intracellular Ca2+ currents and calpain induction in various cell-death models connected with stress to endoplasmic reticulum [26–28]. The expression levels of caspases were identical in wild-type and parp-1 −/− cells (Suppl. Fig. S3C). A parallel inhibition of calpains (MDL 28170) or caspases (Q-VD-OPh) could reduce the specific proteolytic truncates (Suppl. Fig. S3B, S3D). Hence we conclude that the PARP-1 independent intracellular Ca2+ shifts are sufficient for calpain activation and that the activation of caspases requires PARP-1.
The next series of experiments examined AIF translocation in cells silenced for parp-1. While AIF migrates from mitochondria to the nucleus in H2O2-challenged control cells (siRNA C), the translocation was inhibited in cells silenced for PARP-1 (siRNA 1, Fig. 3f). As a final consequence, cells with an impaired PARP-1 system showed an increased survival rate after an oxidative challenge (Fig. 2a).
TRPM2 is the primary channel for PARP-1-dependent extracellular Ca2+ gating
In order to determine the type of channel involved in Ca2+ gating, RNAi experiments were performed. RNAi silencing of TRPM2 channel reduced Ca2+ to the levels resistant to PARP-1/PARG regulation (Fig. 4a; Suppl. Figs. S2N, S4B), suggesting a primary role of this channel in death signaling. Likewise, N-(p-Amylcinnamoyl)anthranilic acid (ACA), a broad spectrum TRP channel blocker with an IC50 of 1.7 μM for TRPM2 [29–31], confirmed this finding (Fig. 4b; Suppl. Fig. S4A). Thus, TRPM2 seems to account for the extracellular Ca2+ transients in cells exposed to 5 mM H2O2. We then examined the possibility that PARP-1 could affect the expression levels of TRPM2 in MEFs. Supplementary Figure S2L and S2M show that TRPM2 expression was identical in wild-type and parp-1 −/− MEFs as determined by RT-PCR and endpoint PCR analysis.

PARG and TRPM2 are required for Ca2+ gating. a Fluo-4 assay in cells silenced (siRNA-T2) or unsilenced (siRNA-C) for the trpm2 gene after 5 mM H2O2 (mean ± SD; n = 3). b Differences in extracellular Ca2+ entry after 5 mM H2O2 in control and ACA-treated cells (5 μM ACA with additional pretreatment of ACA for 5 min) after 1,700 s (mean ± SD; n ≥ 4; *p < 0.001; t test). c MEFs were silenced for the parg gene (siRNA-G) and subjected to Fluo-4 assay. Cells treated with a control RNA sequence (siRNA-C) were analyzed in parallel. Shown are the mean ± SD (n ≥ 3). d Western-blot analysis of AIF translocation from mitochondrial to the nuclear fraction. Control (siRNA-C) and parg silenced cells (siRNA-G) treated with 5 mM H2O2 or solvent controls were subjected to subcellular fractionation at 6 h. Topo-1 and MnSOD served as loading and fraction purity controls. e Control MEFs (siRNA-C) and parg-silenced cells (siRNA-G) were challenged with 5 mM H2O2. After 20 h, survival rates were determined by Alamar blue dye. Shown are the results as percent of the untreated control cells (mean ± SD; *p < 0.005; n = 3; t test). f MEFs were loaded with ADP-ribose (2.5 mM ADP-ribose in assay buffer + transfectant) before subjected to the Fluo-4 assay (mean ± SD; n = 3). ADP-ribose (2.5 mM) without transfectant was used as a control (mean ± SD; n = 3). g Control (siRNA-C) and trpm2-silenced cells (siRNA-T2) were loaded with ADP-ribose (2.5 mM ADP-ribose in assay buffer + transfectant) before subjected to the Fluo-4 assay (mean ± SD; n = 2). h Western-blot analysis of AIF translocation from mitochondrial to the nuclear fraction. Control (siRNA-C) and trpm2-silenced cells (siRNA-T2) treated with 5 mM H2O2 or solvent controls were subjected to subcellular fractionation at 6 h. Topo-1 and MnSOD served as loading controls
PARG generates ADP-ribose, the signal for TRPM2 activation and downstream events in oxidant-induced cell death
We next hypothesized that PARG could control Ca2+ shifts by converting poly(ADP-ribose) into ADP-ribose, a possible activator of TRPM2 channels in vitro. First, we analyzed Ca2+ in MEFs with a silenced parg gene (siRNA-G) after 5 mM H2O2 and compared them with cells transfected with control siRNA (siRNA-C, Fig. 4c; Suppl. Figs. S2J, S2K, S4B). We published earlier that parg silencing is characterized by a reduced level of parg mRNA, PARG protein as well as enzymatic activity and results in a transient accumulation of long PAR molecules after oxidative stress [19]. Ca2+ shifts obtained in parg silenced cells were similar to those previously described in cells with abrogated TRPM2 (Fig. 4a) or PARP-1 function (Fig. 2b–g). Thus, PARG is the major regulator of TRPM2-mediated Ca2+ fluxes in cells subjected to oxidative stress. As expected, the reduced Ca2+ shifts resulted in an impaired AIF translocation from mitochondria to nucleus (Fig. 4d) and in a reduced cytotoxicity after 5 mM (Fig. 4e). To directly demonstrate the role of ADP-ribose as TRPM2 activator, cells were loaded with the nucleoside and intracellular Ca2+ levels monitored (Fig. 4f). The results show that ADP-ribose per se can cause a rise in cytosolic Ca2+ in the complete absence of oxidative stress. This effect is not seen when ADP-ribose was added in the absence of transfection agent or with transfection agent and assay buffer (Suppl. Fig. S4D). In fact, when MEFs were silenced for the trpm2 gene, transfected ADP-ribose could not generate a cytosolic Ca2+ shift (Fig. 4g), indicating an important role of TRPM2 in Ca2+ gating through intracellular ADP-ribose as second messenger. Moreover, the translocation of AIF after oxidative stress was impaired after silencing of trpm2 (Fig. 4h). Interestingly, RNAi against trpm2 could not rescue the cells from cell death (data not shown), suggesting the involvement of an AIF-independent branch of the cell death pathway, as has been reported [57].
Discussion
Cell death following oxidative stress involves diverse ‘death signals’. It is not clear how these signals cooperate between several cell compartments to trigger cell death in a specific cell type [2, 28, 32]. Our results link PARP-1 and PARG activity, TRPM2-gated Ca2+, and caspase activation with AIF translocation and cell death. Figure 5 assembles the signaling elements into a schematic pathway. The first step is the generation of DNA damage by oxidative stress (H2O2). This activates PARP-1 generating a burst of PAR synthesis, a Ca2+ influx and cell death (Fig. 1). Besides PARP-1, PARP-2 has been shown to be stimulated by genotoxins in vitro and in vivo [33–35]. However, PARP-1 is the main enzyme producing PAR, accounting for >99% of PAR in cells after genotoxic insults [36–38]. Our results demonstrate that regardless of the method of PARP-1 abrogation, the Ca2+ influx and cell death was reduced (Fig. 2). Likewise, caspase activation and AIF translocation from mitochondria to the nucleus was suppressed (Fig. 3). Importantly, RNAi against TRPM2 or PARG blocked the Ca2+ influx suggesting that TRPM2 was the primary Ca2+ channel for cell death signaling under PARG control. PARG-silencing also prevented AIF translocation and cell death (Fig. 4). Finally, cells loaded with ADP-ribose, the enzymatic product of PARG activity, showed a distinct Ca2+ influx even in the absence of oxidative damage (Fig. 4f). We conclude that PARG acts as the immediate upstream regulator of TRPM2 by generating ADP-ribose. This is compatible with patch-clamp experiments showing activation of TRPM2 when ADP-ribose was included in the intracellular pipette [16, 39].

PARP-1 and PARG control TRPM2 in a multicompartment pathway to cell death. Oxidative DNA damage activates PARP-1, which converts NAD+ into PAR. PARG degrades PAR and produces free ADP-ribose monomers, which are released into the cytosol. There they activate intermembrane TRPM2 channels and induce a Ca2+ influx. Ca2+ activates calpains and subsequently caspases, but only the latter depends on PARP-1. Caspases become activated as a cascade (9 → 6 → 3) cleaving mitochondrial AIF, which then translocates to the nucleus to cause DNA fragmentation, chromatin condensation, and cell death
TRPM2 contains a NUDIX box sequence motif in the C-terminal domain with a binding site for ADP-ribose. The EC50 for ADP-ribose is 90 μM [40, 41], which is slightly above the intracellular ADP-ribose concentration (in the range of 5 and 90 μM) [42, 43]. Thus, TRPM2 gating is very sensitive to changes in intracellular ADP-ribose concentrations due to oxidant-induced stimulation of PARP-1 and PARG activity. In fact, when TRPM2 is silenced or inhibited, the gating activity for extracellular Ca2+ is abolished (Fig. 4a, b). Accordingly, both the level and the rate of Ca2+ increase depended on the dose of H2O2 (Fig. 1d). The resulting Ca2+ influx from the extracellular milieu (~1 mM Ca2+) merges with the low concentration of the cytosolic pool (~100 nM Ca2+) [21], which is in the detection range of the Ca2+ probe Fluo-4 (K D for Ca2+ 345 nM) [44] used in this study. A low dose of H2O2 is sufficient to induce extracellular Ca2+ shifts (Fig. 1f). Nevertheless, we detected an additional Ca2+ shift from intracellular stores in cells treated with high doses of H2O2 (Fig. 1e, f; Suppl. Fig. S4C). The trough after the initial rise (Figs. 1, 2, 4) could reflect the Ca2+ reuptake into intracellular Ca2+ stores [21]. Release of Ca2+ from intracellular Ca2+ depots has been reported in pancreatic acinar cells [45], neutrophils [46], hepatocytes [47], and in LPS-challenged microglia [17] cells after H2O2.
An elevated intracellular Ca2+ level was shown in relationship with two types of cell death proteases involved in AIF processing and translocation from the mitochondria to the nucleus: calpain [8, 13, 23, 24] and caspases [12, 14, 25]. Both types were activated (Fig. 3) but only the activation of caspases 3, 6, and 9—not 12—was PARP-1-dependent (Fig. 3e). Since AIF translocation from mitochondria to the nucleus was strictly PARP-1-dependent (Fig. 3f), we conclude that Ca2+ induces a signaling pathway leading to the consecutive activation of caspases 9, 6, and 3 (Fig. 3e) and this in turn releases AIF from the inner mitochondrial membrane to translocate to the nucleus (Fig. 3f) and to induce cell death. By contrast, calpain activation by H2O2-induced Ca2+ fluxes is not dependent on PARP-1 (Fig. 3d). Consistent with our observations, the PARP-1 dependency of AIF translocation has been demonstrated in other cells, but attributed to the action of polymeric and not monomeric ADP-ribose [6, 7, 10]. Accordingly, parg silencing prevented AIF translocation and cell death as well (Fig. 4d, e). This may explain why PARG inhibition or genetic disruption has a cytoprotective effect in various cell types [19, 48–50].
The H2O2-induced Ca2+ influx has been described before in cells overexpressing the TRPM2 channel [16, 41, 51] as well as in trpm2 −/− cells restored with TRPM2 [52]. Beside H2O2, cytoplasmic Ca2+ and ADP-ribose act as coactivators of TRPM2 gating [17, 40, 41, 53]. ADP-ribose may be converted into AMP by pyrophosphatase activity of the NUDT9 domain and thereby initiate Ca2+ gating via TRPM2 [40, 54]. Formentini et al. [55] recently described a functional role for NUDIX hydrolases after PARP-1 hyperactivation, resulting in accumulation of AMP. However, enhancing the enzymatic activity of the NUDIX box abolishes ADP-ribose gating of TRPM2, confirming that ADP-ribose itself, and not its degradation product, is involved in TRPM2 gating (reviewed by Hecquet et al. [56]).
Some of our observations seem to be in contrast to published results. Calpain has been implicated in AIF cleavage prior to its release from the inner mitochondrial membrane. While we could confirm calpain activation in H2O2-treated MEFs, which was inhibited by calpain inhibitor MDL 28170, it was unaffected by deletion of the parp-1 −/− gene (Fig. 3d; Suppl. Fig. S3B). The discrepancy could be explained by differences in cell death inducers, i.e., an alkylating agent [8] versus H2O2 (this study), and hence differences in cell death signaling. Moreover, a recent report confirms that calpain is not required for PARP-1- and AIF-dependent cell death [25]. Likewise, PARP-1-dependent cell death may also progress in an AIF-independent manner as shown by Tang et al. [57] in cells deficient in base excision repair. More studies are needed to understand the complexity of molecular interactions of PAR in cell death.
We conclude that the two major PAR metabolizing enzymes PARP-1 and PARG control a cell death pathway that operates between five different cell compartments and communicates via three types of chemical messengers: a nucleotide, a cation, and proteins. PARP-1 is producing the upstream death signal poly(ADP-ribose), which is converted by PARG into ADP-ribose, to specifically activate TRPM2 with concomitant cytosolic Ca2+ shifts that trigger caspases, AIF translocation, and cell death.
Abbreviations
- AIF:
-
Apoptosis-inducing factor
- PAR:
-
Poly(ADP-ribose)
- PARG:
-
PAR glycohydrolase
- PARP:
-
PAR polymerase
- TRPM2:
-
Melastatin-like transient receptor potential 2 channel
References
Ame JC, Spenlehauer C, de Murcia G (2004) The PARP superfamily. Bioessays 26:882–893
Hassa PO, Haenni SS, Elser M, Hottiger MO (2006) Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev 70:789–829
Schreiber V, Dantzer F, Ame JC, de Murcia G (2006) Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 7:517–528
Brochu G, Duchaine C, Thibeault L, Lagueux J, Shah GM, Poirier GG (1994) Mode of action of poly(ADP-ribose) glycohydrolase. Biochim Biophys Acta 1219:342–350
Davidovic L, Vodenicharov M, Affar EB, Poirier GG (2001) Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp Cell Res 268:7–13
Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, Hurn PD, Poirier GG, Dawson VL, Dawson TM (2006) Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci USA 103:18308–18313
Cohausz O, Blenn C, Malanga M, Althaus FR (2008) The roles of poly(ADP-ribose)-metabolizing enzymes in alkylation-induced cell death. Cell Mol Life Sci 65:644–655
Moubarak RS, Yuste VJ, Artus C, Bouharrour A, Greer PA, Menissier-de Murcia J, Susin SA (2007) Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol Cell Biol 27:4844–4862
Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397:441–446
Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL (2006) Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci USA 103:18314–18319
Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL (2002) Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297:259–263
Arnoult D, Parone P, Martinou JC, Antonsson B, Estaquier J, Ameisen JC (2002) Mitochondrial release of apoptosis-inducing factor occurs downstream of cytochrome c release in response to several proapoptotic stimuli. J Cell Biol 159:923–929
Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM, Clark RS, Graham SH, Chen J (2007) Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J Neurosci 27:9278–9293
Diwakarla S, Nagley P, Hughes ML, Chen B, Beart PM (2009) Differential insult-dependent recruitment of the intrinsic mitochondrial pathway during neuronal programmed cell death. Cell Mol Life Sci 66:156–172
Yu SW, Wang Y, Frydenlund DS, Ottersen OP, Dawson VL, Dawson TM (2009) Outer mitochondrial membrane localization of apoptosis-inducing factor: mechanistic implications for release. ASN Neuro. doi: 10.1042/AN20090046
Buelow B, Song Y, Scharenberg AM (2008) The Poly(ADP-ribose) polymerase PARP-1 is required for oxidative stress-induced TRPM2 activation in lymphocytes. J Biol Chem 283:24571–24583
Kraft R, Grimm C, Grosse K, Hoffmann A, Sauerbruch S, Kettenmann H, Schultz G, Harteneck C (2004) Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am J Physiol Cell Physiol 286:C129–C137
Fonfria E, Marshall IC, Benham CD, Boyfield I, Brown JD, Hill K, Hughes JP, Skaper SD, McNulty S (2004) TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br J Pharmacol 143:186–192
Blenn C, Althaus FR, Malanga M (2006) Poly(ADP-ribose) glycohydrolase silencing protects against H2O2-induced cell death. Biochem J 396:419–429
Jacobson EL, Jacobson MK (1997) Tissue NAD as a biochemical measure of niacin status in humans. Methods Enzymol 280:221–230
Szabadkai G, Duchen MR (2008) Mitochondria: the hub of cellular Ca2+ signaling. Physiology (Bethesda) 23:84–94
Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397:271–274
Churbanova IY, Sevrioukova IF (2008) Redox-dependent changes in molecular properties of mitochondrial apoptosis-inducing factor. J Biol Chem 283:5622–5631
Norberg E, Gogvadze V, Ott M, Horn M, Uhlen P, Orrenius S, Zhivotovsky B (2008) An increase in intracellular Ca2+ is required for the activation of mitochondrial calpain to release AIF during cell death. Cell Death Differ 15:1857–1864
Wang Y, Kim NS, Li X, Greer PA, Koehler RC, Dawson VL, Dawson TM (2009) Calpain activation is not required for AIF translocation in Parp-1-dependent cell death (Parthanatos). J Neurochem 110:687–696
Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y (2002) An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem 277:34287–34294
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403:98–103
Szegezdi E, Logue SE, Gorman AM, Samali A (2006) Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 7:880–885
Bari MR, Akbar S, Eweida M, Kuhn FJ, Gustafsson AJ, Luckhoff A, Islam MS (2009) H2O2-induced Ca2+ influx and its inhibition by N-(p-amylcinnamoyl)anthranilic acid in the beta-cells: involvement of TRPM2 channels. J Cell Mol Med 13:3260–3267
Kraft R, Grimm C, Frenzel H, Harteneck C (2006) Inhibition of TRPM2 cation channels by N-(p-amylcinnamoyl)anthranilic acid. Br J Pharmacol 148:264–273
Pantaler E, Luckhoff A (2009) Inhibitors of TRP channels reveal stimulus-dependent differential activation of Ca2+ influx pathways in human neutrophil granulocytes. Naunyn Schmiedebergs Arch Pharmacol 360:497–507
Orrenius S, Gogvadze V, Zhivotovsky B (2007) Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol 47:143–183
Ame JC, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, Muller S, Hoger T, Menissier-de Murcia J, de Murcia G (1999) PARP-2, a novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem 274:17860–17868
Ame JC, Schreiber V, Fraulob V, Dolle P, de Murcia G, Niedergang CP (2001) A bidirectional promoter connects the poly(ADP-ribose) polymerase 2 (PARP-2) gene to the gene for RNase P RNA. Structure and expression of the mouse PARP-2 gene. J Biol Chem 276:11092–11099
Schreiber V, Ame JC, Dolle P, Schultz I, Rinaldi B, Fraulob V, Menissier-de Murcia J, de Murcia G (2002) Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J Biol Chem 277:23028–23036
Althaus FR, Richter C (1987) ADP-ribosylation of proteins. Enzymology and biological significance. Mol Biol Biochem Biophys 37:1–237
Sallmann FR, Vodenicharov MD, Wang ZQ, Poirier GG (2000) Characterization of sPARP-1. An alternative product of PARP-1 gene with poly(ADP-ribose) polymerase activity independent of DNA strand breaks. J Biol Chem 275:15504–15511
Shieh WM, Ame JC, Wilson MV, Wang ZQ, Koh DW, Jacobson MK, Jacobson EL (1998) Poly(ADP-ribose) polymerase null mouse cells synthesize ADP-ribose polymers. J Biol Chem 273:30069–30072
Buelow B, Uzunparmak B, Paddock M, Scharenberg AM (2009) Structure/function analysis of PARP-1 in oxidative and nitrosative stress-induced monomeric ADPR formation. PLoS One 4:e6339. doi:10.1371/journal.pone.0006339
Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, Kinet JP, Scharenberg AM (2001) ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 411:595–599
Wehage E, Eisfeld J, Heiner I, Jungling E, Zitt C, Luckhoff A (2002) Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J Biol Chem 277:23150–23156
Gasser A, Glassmeier G, Fliegert R, Langhorst MF, Meinke S, Hein D, Kruger S, Weber K, Heiner I, Oppenheimer N, Schwarz JR, Guse AH (2006) Activation of T cell calcium influx by the second messenger ADP-ribose. J Biol Chem 281:2489–2496
Gasser A, Guse AH (2005) Determination of intracellular concentrations of the TRPM2 agonist ADP-ribose by reversed-phase HPLC. J Chromatogr B Analyt Technol Biomed Life Sci 821:181–187
Hansen KB, Brauner-Osborne H (2009) FLIPR assays of intracellular calcium in GPCR drug discovery. Methods Mol Biol 552:269–278
Klonowski-Stumpe H, Schreiber R, Grolik M, Schulz HU, Haussinger D, Niederau C (1997) Effect of oxidative stress on cellular functions and cytosolic free calcium of rat pancreatic acinar cells. Am J Physiol 272:G1489–G1498
Kilpatrick LE, Jakabovics E, McCawley LJ, Kane LH, Korchak HM (1995) Cromolyn inhibits assembly of the NADPH oxidase and superoxide anion generation by human neutrophils. J Immunol 154:3429–3436
Sato H, Takeo T, Liu Q, Nakano K, Osanai T, Suga S, Wakui M, Wu J (2009) Hydrogen peroxide mobilizes Ca2+ through two distinct mechanisms in rat hepatocytes. Acta Pharmacol Sin 30:78–89
Burns DM, Ying W, Kauppinen TM, Zhu K, Swanson RA (2009) Selective down-regulation of nuclear poly(ADP-ribose) glycohydrolase. PLoS One 4:e4896
Erdelyi K, Bai P, Kovacs I, Szabo E, Mocsar G, Kakuk A, Szabo C, Gergely P, Virag L (2009) Dual role of poly(ADP-ribose) glycohydrolase in the regulation of cell death in oxidatively stressed A549 cells. Faseb J 23:3553–3563
Formentini L, Arapistas P, Pittelli M, Jacomelli M, Pitozzi V, Menichetti S, Romani A, Giovannelli L, Moroni F, Chiarugi A (2008) Mono-galloyl glucose derivatives are potent poly(ADP-ribose) glycohydrolase (PARG) inhibitors and partially reduce PARP-1-dependent cell death. Br J Pharmacol 155:1235–1249
Zhang W, Hirschler-Laszkiewicz I, Tong Q, Conrad K, Sun SC, Penn L, Barber DL, Stahl R, Carey DJ, Cheung JY, Miller BA (2006) TRPM2 is an ion channel that modulates hematopoietic cell death through activation of caspases and PARP cleavage. Am J Physiol Cell Physiol 290:C1146–C1159
Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, Negoro T, Hiroi T, Kiuchi Y, Okada T, Kaneko S, Lange I, Fleig A, Penner R, Nishi M, Takeshima H, Mori Y (2008) TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med 14:738–747
Naziroglu M, Luckhoff A (2008) A calcium influx pathway regulated separately by oxidative stress and ADP-Ribose in TRPM2 channels: single channel events. Neurochem Res 33:1256–1262
Kuhn FJ, Luckhoff A (2004) Sites of the NUDT9-H domain critical for ADP-ribose activation of the cation channel TRPM2. J Biol Chem 279:46431–46437
Formentini L, Macchiarulo A, Cipriani G, Camaioni E, Rapizzi E, Pellicciari R, Moroni F, Chiarugi A (2009) Poly(ADP-ribose) catabolism triggers AMP-dependent mitochondrial energy failure. J Biol Chem 284:17668–17676
Hecquet CM, Malik AB (2009) Role of H2O2-activated TRPM2 calcium channel in oxidant-induced endothelial injury. Thromb Haemost 101:619–625
Tang JB, Goellner EM, Wang XH, Trivedi RN, St Croix CM, Jelezcova E, Svilar D, Brown AR, Sobol RW (2010) Bioenergetic metabolites regulate base excision repair-dependent cell death in response to DNA damage. Mol Cancer Res 8:67–79
Acknowledgements
This work was supported by the Vetsuisse Faculty, the Swiss National Science Foundation, and a grant from the Lotte and Adolf Hotz-Sprenger Foundation, Zurich, awarded to F.R.A.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
C. Blenn and P. Wyrsch contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Blenn, C., Wyrsch, P., Bader, J. et al. Poly(ADP-ribose)glycohydrolase is an upstream regulator of Ca2+ fluxes in oxidative cell death. Cell. Mol. Life Sci. 68, 1455–1466 (2011). https://doi.org/10.1007/s00018-010-0533-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-010-0533-1